Introduction
Diabetes mellitus (DM), as a form of chronic
metabolic disease, is characterized by the lack of insulin and/or
insulin resistance that results in hyperglycemia and abnormal
metabolism (1,2). Long-term hyperglycemia can lead to
various complications, including cardiovascular disease (CVD) that
accounts for approximately a third of all mortalities worldwide
(2,3). In addition, DM is considered as an
independent risk factor for CVD, excluding other factors such as
obesity, hypertension and age (4,5).
Vascular smooth muscle cells (VSMCs) serve a key role in vascular
remodeling (6). Accumulating
evidence has indicated that the proliferation and migration of
VSMCs were important features of numerous types of CVD, including
atherosclerosis, hypertension and restenosis (7–9). It
has been reported that high glucose (HG) can induce the excessive
proliferation and migration of VSMCs, which promote the progression
of diabetic vascular diseases (10,11).
Thus, understanding the mechanism of HG-mediated VSMC proliferation
and migration is important for improving current intervention
strategies for the treatment of vascular diseases in diabetes.
MicroRNAs (miRNAs/miRs) are a type of small
noncoding single-stranded RNAs (~21–23 nucleotides) that negatively
regulate the expression of their target genes by complete or
partial complementary binding with the 3′-untranslated region
(3′UTR) of these target genes, to modulate various physiological
and pathological processes, including cancer, DM, CVD and other
diseases (12–14). Several studies have demonstrated
that miRNAs are involved in regulating the dysfunction of VSMC
behavior (15–17), including miR-145 (18), miR-155 (19), miR-146 (20), miR-504 (21), miR-24 (22) and miR-126 (23).
miR-132 originates from the miR-212/132 cluster that
is located on chromosome 17 in humans (24). The majority of studies of miR-132
revealed its roles in cancer and the regulation of neurons
(24–26). A recent study indicated that
miR-132 regulated brain vascular integrity (27). Additionally, the expression levels
of miR-132 are downregulated in VSMCs in diabetic rat models
(21); however, the role and
mechanism of miR-132 in diabetes-induced VSMC dysfunction are not
clear. The present study aimed to investigate the effects of
miR-132 on the proliferation and migration of VSMCs under HG
conditions to mimic diabetes, which may provide insight into the
biological mechanism for the development of novel approaches to
treat diabetic vascular complications.
Materials and methods
Animals
The diabetic animal model was generated as
previously described (28). In
total, 24 male Sprague-Dawley rats (age, 4–6 weeks; weight, 180–200
g) were obtained from Beijing Vital River Laboratory Animal
Technology Co., Ltd. (Beijing, China). A single dose of
streptozotocin (STZ; 65 mg/kg, citrate buffer) was injected into 12
rats. An equal volume of citrate buffer was used as the control
treatment (n=12). When blood glucose levels were >250 mg/dl, the
animals were defined as diabetic rats. After 2 days following
treatment, the animals became hyperglycemic. All rats were
sacrificed by cervical dislocation under ketamine administration.
Thoracic aorta samples from non-diabetic and diabetic rats were
collected and isolated; mRNA and protein were extracted for further
analysis. All the procedures and experimental protocols were
approved by the Institutional Animal Care and Use Committee of
Qianfoshan Hospital of Shandong (Jinan, China).
Cell culture and treatment
The isolation of primary VSMCs from the thoracic
aorta of non-diabetic and diabetic rats was performed according to
a previous study (29).
Morphological and immunohistochemical analyses of VSMCs were
performed as previously described (29). The 293 cells were purchased from
the Shanghai Institute of Chinese Academy of Sciences (Shanghai,
China). VSMCs and 293 cells were maintained in DMEM (HyClone;
Thermo Fisher Scientific, Inc.) with 10% FBS (HyClone; Thermo
Fisher Scientific, Inc.), as well as penicillin (100 U/ml;
Bio-sciences Ltd., Dublin, Ireland) and streptomycin (100 U/ml;
Bio-sciences, Ltd.) at 37°C within 5% CO2. Experiments
were conducted on transduced and non-transduced VSMCs of passages
3–8. For HG treatment, VSMCs were cultured in normal media (5 mM
glucose) for 24 h, and then incubated with HG (25 mM glucose) or 20
mM mannitol plus 5 mM glucose (normal glucose, NG) for 24 h at 37°C
with 5% CO2.
Plasmid construction and
infection
miR-132 precursor and the corresponding miR-control
were designed and synthesized by Shanghai GeneChem Co., Ltd.
(Shanghai, China), and were cloned into pCDH-CMV-MCS-EF1-coGFP
(System Biosciences, LLC, Palo Alto, CA, USA). The gene encoding
E2F transcription factor 5 (E2F5) lacking the 3′UTR was amplified
from rat genomic DNA by PCR using the Phusion High-Fidelity PCR Kit
(Thermo Fisher Scientific, Inc., Waltham, MA, USA) according to the
manufacturer's protocol. PCR thermocycling conditions were as
follows: Initial denaturation at 95°C for 30 sec, followed by 40
cycles of 95°C for 15 sec, 60°C for 30 sec, 72°C for 1 min and 4°C
for 1 min. The following primers were used: E2F5 forward,
AGAATTCATGGCGGCGGCGGAGCCCA and reverse,
ATGGATCCCTAATAATTTAGTATCTGAACATCAA. Then, the PCR product was
cloned into pCDH-CMV-MCS-EF1-Puro (Addgene). The production and
purification of lentivirus and cell infection (multiplicity of
infection, 100) were performed as previously described (30,31).
Empty lentiviral vector pCDH-CMV-MCS-EF1-Puro (vector) was used as
the control for lenti-E2F5 transduction. VSMCs from non-diabetic
rats were infected with lenti-E2F5 or vector. Small interfering
(si)RNAs targeting two different sites (cat. nos. RSS350386 and
RSS350386) of the rat E2F5 gene and the negative control (cat. no.
12935100; scramble) were purchased from Thermo Fisher Scientific
Inc. Cells were transfected with E2F5 siRNAs (100 nM) or scramble
(100 nM) using Lipofectamine® 2000 (Thermo Fisher
Scientific Inc.) according to the manufacturer's protocols. miR-132
inhibitor (cat. no. 4464084) and corresponding negative control
(inhibitor-NC; cat. no. 4464076) as well as miR-132 mimic (cat. no.
4464066) and corresponding negative control (miR-NC; cat. no.
4464058) were obtained from Thermo Fisher Scientific Inc. miR-132
mimic (100 nM) and miR-NC (100 nM) were used to transfect diabetic
VSMCs. Normal VSMCs were transfected with miR-132 inhibitor (100
nM) and inhibitor-NC (100 nM) using Lipofectamine 2000 (Thermo
Fisher Scientific Inc.) according to the manufacturer's protocol.
Cells were collected and used for analysis at 72 h after infection
or transfection. All constructions were confirmed by plasmid DNA
sequencing.
Dual luciferase assays
miR-132 mimic, miR-132 inhibitor and corresponding
negative control (miR-NC and inhibitor-NC, respectively) were
obtained from Shanghai GenePharma Co., Ltd. (Shanghai, China). The
wild-type E2F5 3′UTR and mutant-type E2F5 3′UTR were cloned into
the reporter plasmid pGL3 (Promega Corporation, Madison, WI, USA)
downstream of the luciferase reporter gene according to as
previously described (26). For
the dual luciferase assay, 293 cells were seeded into a 24-well
plate at a density of 2×104 cells per well. Cells were
co-transfected with 1 µg of wild-type or mutant luciferase vector
and miR-132mimics (100 nM), miR-132 inhibitor (100 nM) or their
NCs. After co-transfection for 48 h, luciferase activity was
detected using the dual luciferase Reporter Assay System (Promega
Corporation) according to the manufacturer's instructions. Firefly
luciferase activity was normalized to Renilla luciferase
activity.
VSMC proliferation assay
A bromodeoxyuridine (BrdU) incorporation assay was
employed to detect the proliferation of VSMCs (22). VSMCs were transfected with E2F5
siRNAs (100 nM) or scramble (100 nM) using Lipofectamine 2000
(Thermo Fisher Scientific Inc.) according to the manufacturer's
protocols. miR-132 mimic was used to transfect diabetic VSMCs.
Normal VSMCs were transfected with miR-132 inhibitor using
Lipofectamine 2000 (Thermo Fisher Scientific Inc.) according to the
manufacturer's protocols. Following transfection, stable infection
or co-infection of miR-132, miR-NC, lenti-E2F5 and lenti-vector,
cells were seeded into 96-well plates at 3×103
cells/well. Cells were cultured with NG or HG media for 24 h, and
were then starved in FBS-free DMEM containing the aforementioned
concentrations of glucose (NG or HG) for 24 h at 37°C with 5%
CO2. Subsequently, BrdU (10 µmol/l) was added into each
well and incubated at 37°C for 30 min. The cells were fixed in 4%
paraformaldehyde for 15 min at room temperature, and then detected
using the cell proliferation ELISA kit (Roche Diagnostics,
Mannheim, Germany) according to the manufacturer's instructions.
Absorbance values were detected at a wavelength of 450 nm using a
microplate reader (Thermo Fisher Scientific, Inc.).
VSMC migration assay
VSMC migration was detected by Transwell migration
assay (22). VSMCs from
non-diabetic rats were infected with lenti-E2F5 or vector.
Following transfection, stable infection or co-infection of
miR-132, miR-control, lenti-E2F5 and lenti-vector, cells were
treated with NG or HG media for 48 h. Cells (5×104) were
plated into the upper chamber with 8-mm pore size (BD Bioscience,
Franklin Lakes, NJ, USA), while 800 µl DMEM containing 1% FBS was
placed in the lower chamber. After incubation for 6 h at 37°C, the
cells in the upper membrane were removed with cotton swabs, and
cells on the lower membrane were fixed in 4% paraformaldehyde for
15 min at room temperature and stained with crystal violet (1%) for
1 h at room temperature (Beyotime Institute of Biotechnology,
Haimen, China). Cells were analyzed in 10 random fields per well
(magnification, ×100) using a phase contrast light microscope.
Protein extraction and western
blotting
Thoracic aorta tissue or VSMC was lysed with
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology) containing protease inhibitors (Roche Diagnostics)
according to the manufacturer's instructions. The protein
concentrations were measured with a BCA Protein Assay kit (Applygen
Technologies, Inc., Beijing, China) according to the manufacturer's
protocols. Protein samples (20 µg for each sample) were
fractionated by SDS-PAGE on 10% gels, and then transferred to a
polyvinylidene fluoride membrane (EMD Millipore, Bedford, MA, USA).
The PVDF membranes were incubated with anti-E2F5 antibody (1:1,500;
cat. no. ab176017; Abcam, Cambridge, UK) or anti-β-actin antibody
(1:6,000; cat. no. ab8226; Abcam) at 4°C overnight. Then, the
membranes were washed and incubated with horseradish
peroxidase-conjugated secondary antibodies (1:4,000; cat. no.
ab205718; Abcam) for 1 h at room temperature. Finally, the protein
bands were detected using an enhanced chemiluminescence detection
kit (Pierce; Thermo Fisher Scientific, Inc.) and a ChemoDoc XRS
detection system (Bio-Rad Laboratories, Inc.). The results were
analyzed with ImageJ software (version 1.34e; National Institutes
of Health, Bethesda, MD, USA).
RNA extraction and
reverse-transcription-quantitative polymerase chain reaction
(RT-qPCR)
Total RNA from thoracic aorta tissue or VSMCs was
isolated using TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocols.
miRNAs were reversely transcribed into cDNA using the miScript
Reverse Transcription Kit (Qiagen GmbH; cat. no. 218060) according
to the manufacturer's protocol. For mRNA detection, cDNA was
generated using the PrimeScript RT reagent kit (Takara Bio, Inc.)
according to the manufacturer's protocol. miR-132 and E2F5
expression levels were quantified using the SYBR Green PCR Master
Mix kit (Applied Biosystems; cat. no. 4309155). The sequences of
the primers used are as follows: miR-132 forward,
5′-GGGTAACAGTCTACAGCCAT-3′ and miR-132 reverse,
5′-GGCAATTGCACTGGATAC-3′; U6 forward,
5′-GCTTCGGCAGCACATATACTAAAAT-3′ and U6 reverse,
5′-CGCTTCACGAATTTGCGTGTCAT-3′; rat E2F5 forward,
5′-GTACTTCCTTTGGCCTTAGTTT-3′ and reverse,
5′-CTGGCACCATCACACCGACATA-3′; rat β-actin forward,
5′-TGTCACCAACTGGGACGATATG-3′ and reverse,
5′-GGCTGGGGTGTTGAAGGTCTC-3′. The expression levels of miR-132 and
E2F5 were quantified using the 2−ΔΔCq method (32). RT-qPCR experiments were performed
in triplicate.
Databases and bioinformatics
analysis
The predicted target genes of miR-132 were
identified using TargetScan (version 7.2; http://www.targetscan.org).
Statistical analyses
Data were expressed as the mean ± SD. One-way ANOVA
followed by a Tukey's post-hoc test was conducted to assess
significant differences. All statistical calculations were
performed using SPSS 19.0 software (IBM Corp. Armonk, NY, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
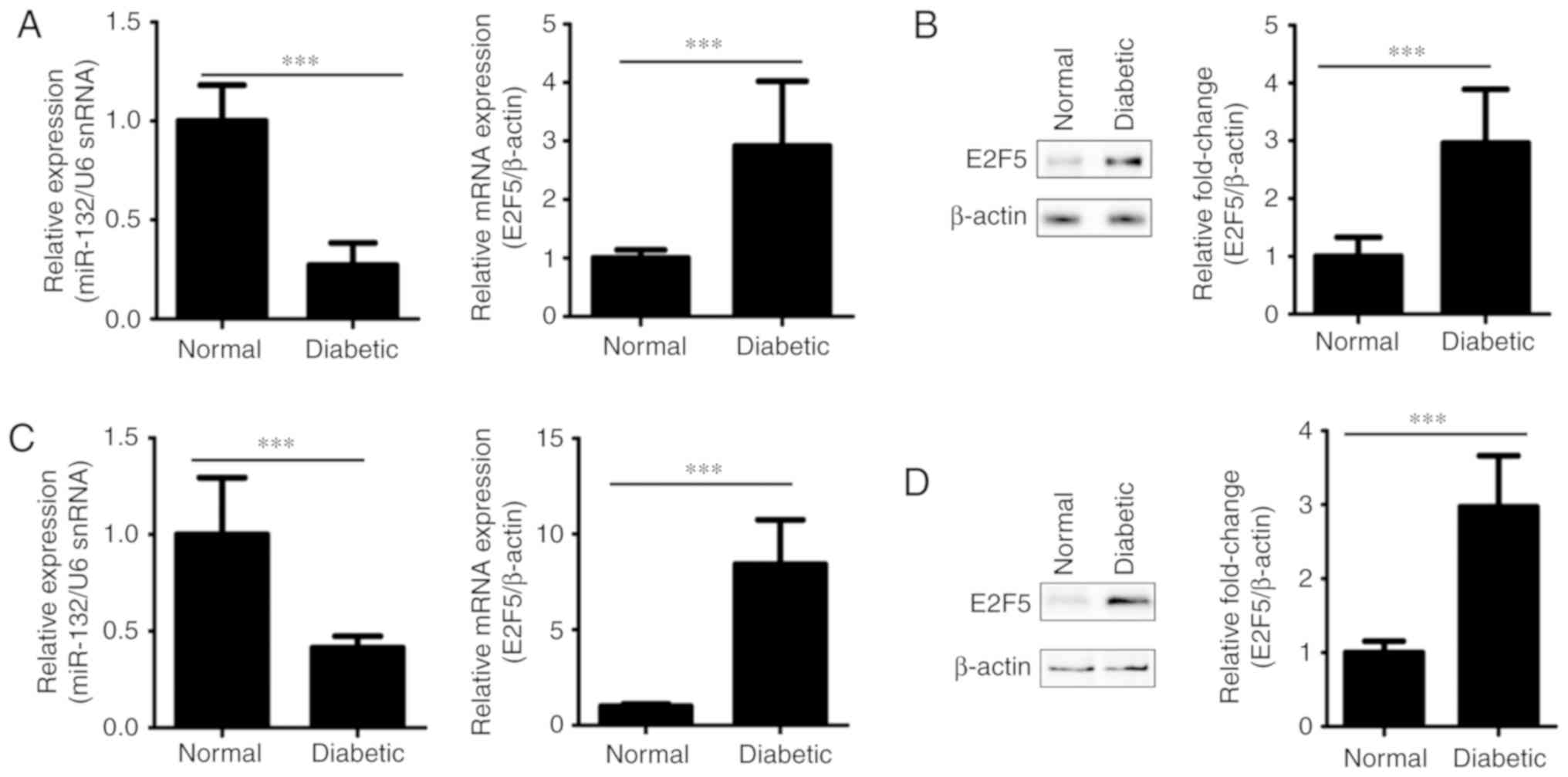
Expression of miR-132 and E2F5 in
thoracic aorta samples and VSMCs from non-diabetic and diabetic
rats
To investigate the potential roles of miR-132 and
E2F5 in VSMCs under diabetic conditions, the expression of miR-132
and E2F5 in thoracic aorta samples and VSMCs non-diabetic and
diabetic rats was detected by RT-qPCR and western blotting.
Compared with the non-diabetic control rats, the expression levels
of miR-132 in thoracic aorta samples and VSMCs of diabetic rats
were significantly decreased compared with the control (P<0.001;
Fig. 1A and C). Conversely, the
mRNA and protein expression levels of E2F5 in thoracic aorta
samples and VSMCs of diabetic rats were upregulated than in
non-diabetic control rats (P<0.001; Fig. 1A-D). Taken together, our results
revealed the opposing expression profiles of miR-132 and E2F5,
which suggested their conflicting roles in the abnormal behavior of
VSMCs mediated by diabetic conditions.
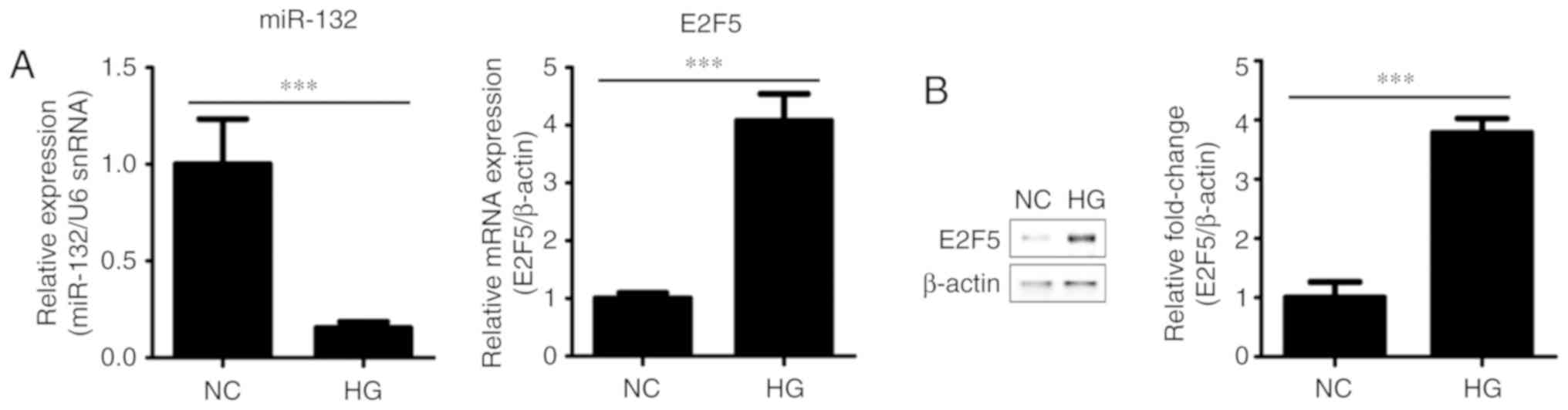
Expression of miR-132 and E2F5 in
HG-treated VSMCs
We investigated whether HG stimulation altered
expression of miR-132 and E2F5. The results revealed that miR-132
was significantly downregulated (P<0.001), while the mRNA and
protein expression levels of E2F5 were increased (P<0.001) in
VSMCs incubated with HG media for 24 h compared with the control
(Fig. 2). These findings further
suggested the opposing expression profiles of miR-132 and E2F5 in
HG-treated VSMCs.
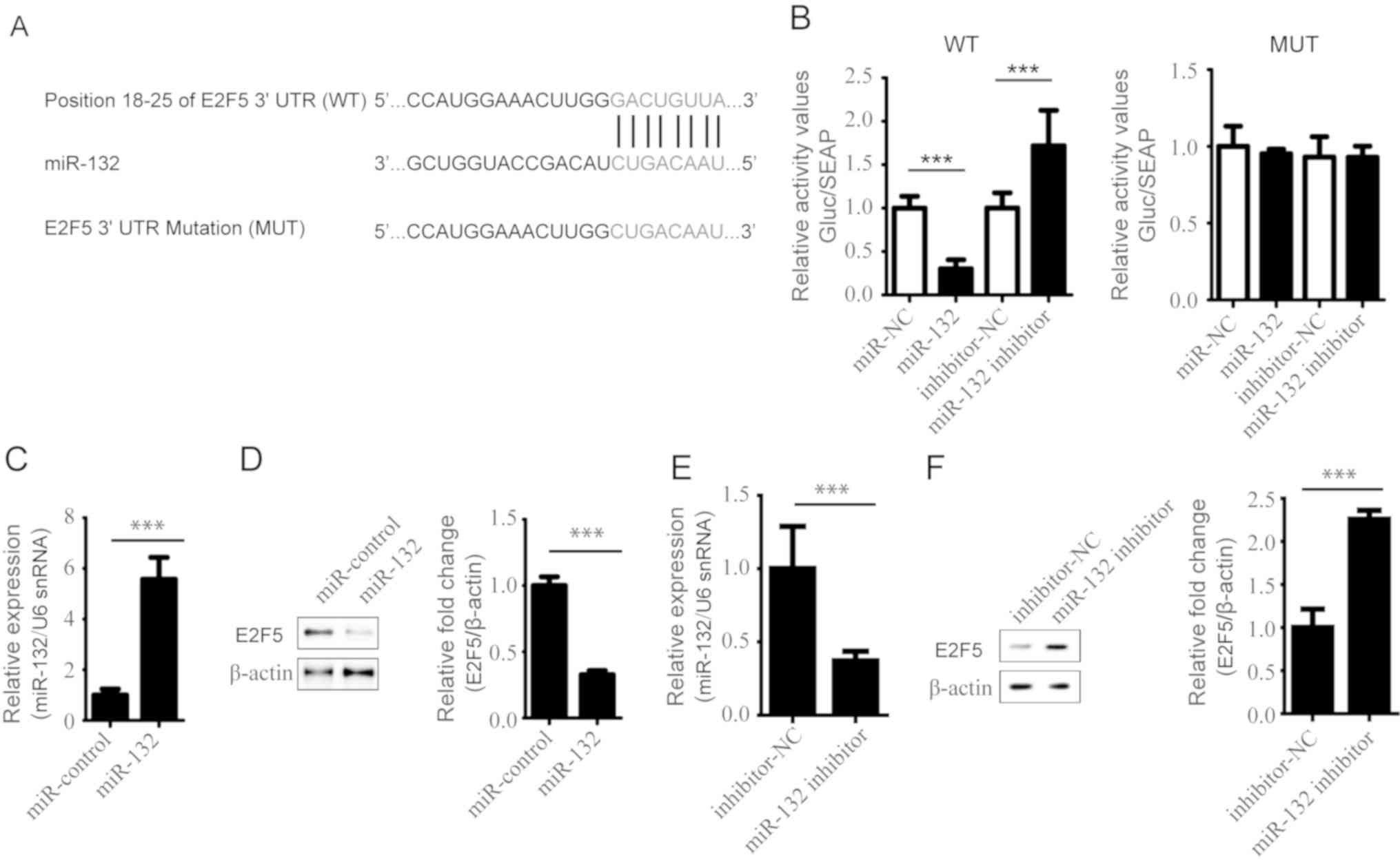
E2F5 is a target of miR-132
Generally, miRNAs regulate gene expression by
targeting the 3′UTR of these genes. Using TargetScan (http://www.targetscan.org), it was predicted that
miR-132 binds to the 3′UTR of E2F5. As presented in Fig. 3A, the paired sequences between
miR-132 and E2F5 3′UTR, and the mutant were determined. This
prediction was confirmed by a dual luciferase reporter assay. The
relative luciferase activity of the reporter containing the
wild-type 3′UTR was significantly decreased by miR-132
overexpression compared with the control, but increased following
transfection with miR-132 inhibitor (P<0.001; Fig. 3B). However, the luciferase activity
of the mutant reporter was markedly affected by miR-132
overexpression and knockdown (Fig.
3B). These results suggested that miR-132 could directly bind
to the 3′UTRs of E2F5 which were mutated in this study. We also
overexpressed miR-132 in VSCMs from diabetic rats. The expression
levels of miR-132 and E2F5 were detected by RT-qPCR and western
blotting, respectively. The results showed the successful
transfection of cells with miR-132 mimics (Fig. 3C); the expression of E2F5 was
significantly decreased by miR-132 overexpression in the VSCMs from
diabetic rats compared with the control (Fig. 3D). Additionally, downregulation of
miR-132 (Fig. 3E) significantly
increased the expression of E2F5 in VSCMs compared with the control
(Fig. 3F). Our findings suggested
that E2F5 was a target of miR-132.
Effects of miR-132 and E2F5 on VSMC
proliferation and migration
Excessive cell proliferation and migration of VSMCs
are fundamental mechanisms underlying CVDs in diabetes (10). This prompted us to investigate the
functions of miR-132 and E2F5 in the proliferation and migration of
VSMCs from diabetic rats by the nuclear incorporation of BrdU (DNA
synthesis) and Transwell migration assays. We reported that the
proliferation and migration of VSMCs from diabetic rats were
significantly increased than that of VSMCs of non-diabetic rats
(P<0.001; Fig. 4A and B).
Furthermore, miR-132 was overexpressed in VSMCs from diabetic rats
(Fig. 3C and D), then the
proliferation and migration of cells were analyzed. The results
showed that diabetic rat-derived VSMCs transfected with miR-132
exhibited significantly reduced proliferation and migration
compared with the control group (P<0.001; Fig. 4C and D). In addition, VSMCs from
non-diabetic rats were infected with lenti-E2F5 or vector. The
results demonstrated that the expression of E2F5 significantly
increased 5.2-fold in the lenti-E2F5 infection group compared the
control group (P<0.001; Fig.
4E). Overexpression of E2F5 significantly increased the
proliferative and migration abilities of normal rat VSMCs compared
with the control (P<0.001; Fig. 4F
and G). Thus, these data suggested that miR-132 and E2F5 serve
opposing roles in the proliferation and migration of VSMCs.
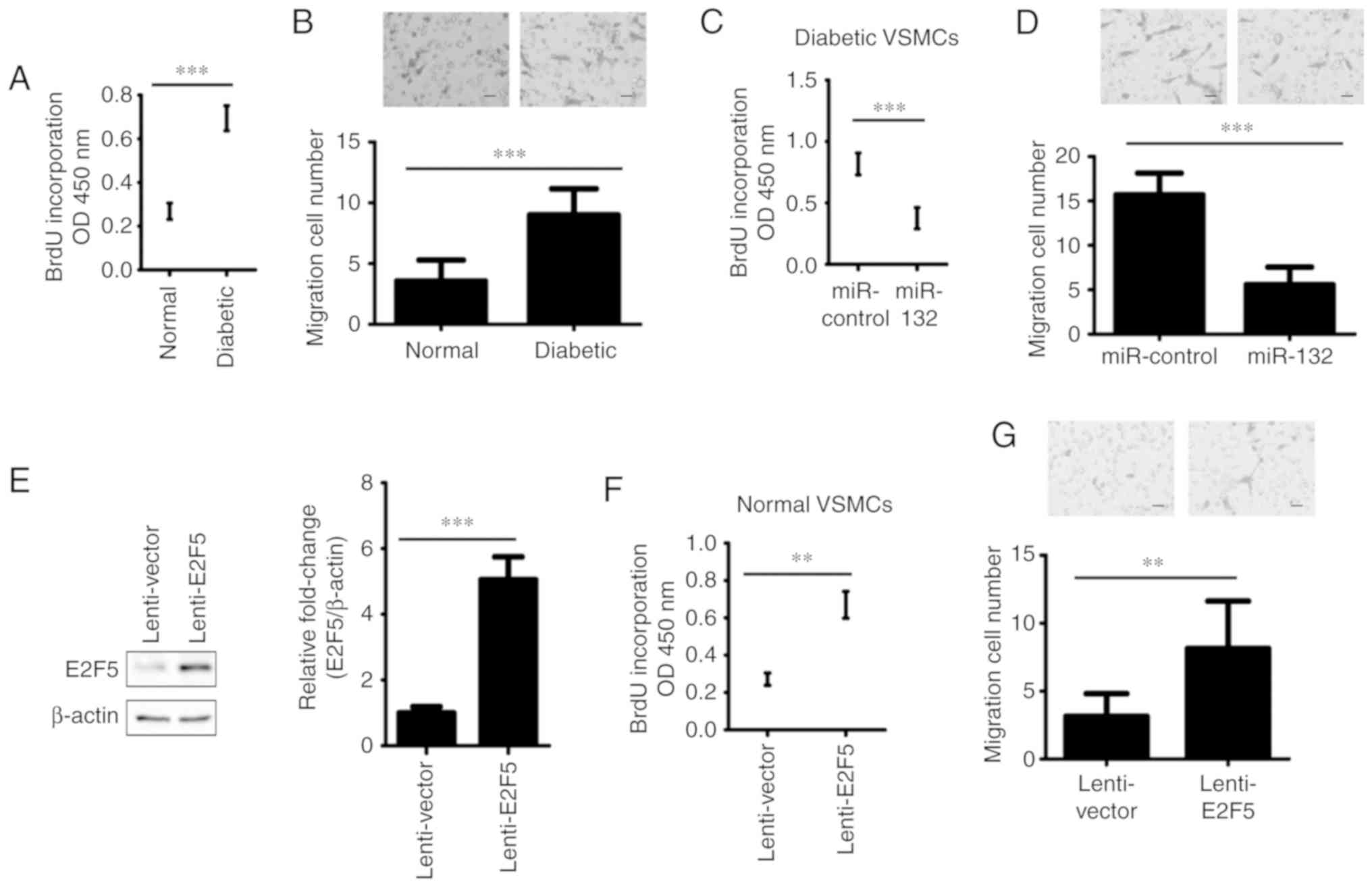 | Figure 4.Effects of miR-132 and E2F5 on VSMC
proliferation and migration. (A) The proliferation of VSMCs from
diabetic and non-diabetic rats were detected by an BrdU
incorporation assay. (B) The migration of VSMCs from diabetic and
non-diabetic rats was analyzed by a Transwell migration assay.
Magnification, ×100. VSMCs from diabetic rats were infected with
lentivirus containing miR-132 or miR-control, and non-diabetic rat
VSMCs were infected with lenti-E2F5 or lenti-vector for 72 h. (C)
The proliferation of VSMCs was detected by a BrdU incorporation
assay. (D) A Transwell migration assay was conducted to analyze the
migration of VSMCs. Magnification, ×100. (E) The protein expression
levels of E2F5 were determined by western blotting; left panel,
representative results and right panel, the quantified data for
western blotting. (F) After infection for 72 h, a BrdU
incorporation assay was performed to examine the proliferation of
VSMCs. (G) After infection for 72 h, a Transwell migration assay
was conducted to analyze the migration of VSMCs. Magnification,
×100. All data were presented as the mean ± standard deviation.
**P<0.001, ***P<0.001. The differences were tested by one-way
ANOVA followed by a Tukey's post hoc test. BrdU, bromodeoxyuridine;
E2F transcription factor 5; miR, microRNA; OD, optical density;
VSMCs, vascular smooth muscle cells. |
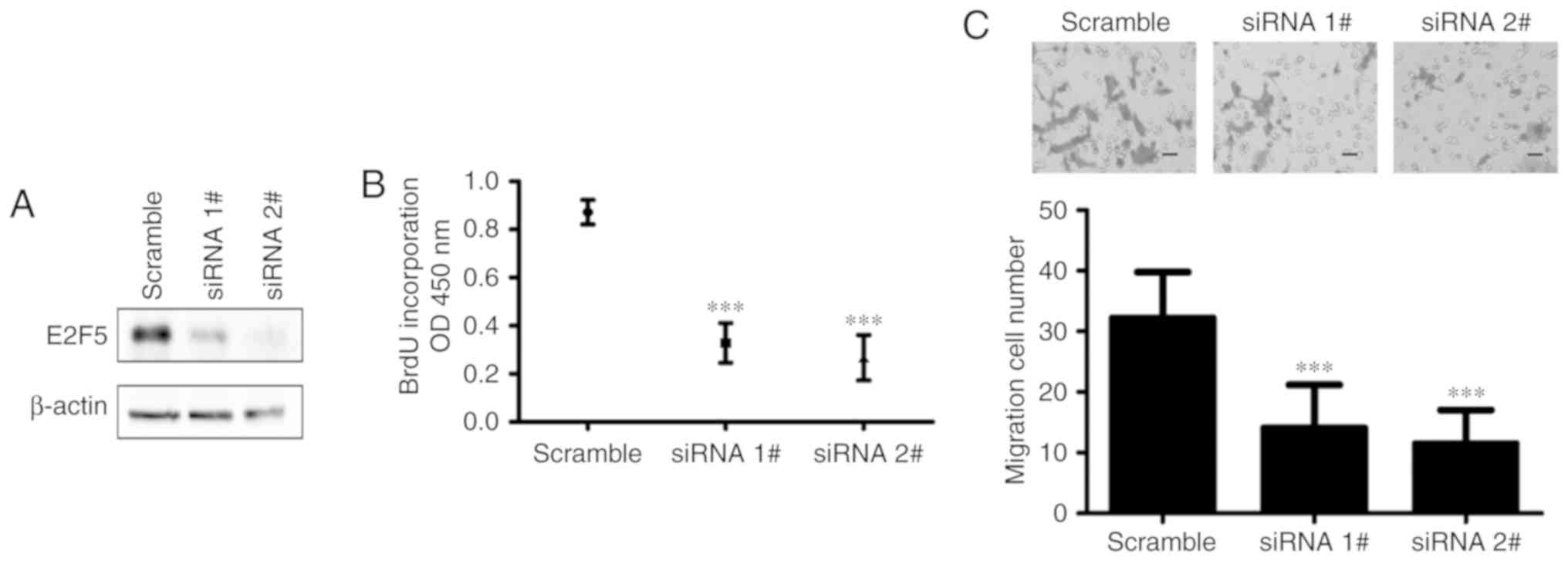
Downregulation of E2F5 inhibits
diabetic rat VSMC proliferation and migration
To further confirm the function of E2F5 in VSMCs
from diabetic rats, E2F5 expression was knocked down by delivery of
E2F5 siRNAs, and the proliferation and migration of these VSMCs
were analyzed. As determined by western blot analysis, the
expression of E2F5 was significantly knocked down compared with the
control (Fig. 5A). Of note,
downregulation of E2F5 significantly inhibited the proliferation of
diabetic rat VSMCs compared with those transfected with scramble
siRNA (Fig. 5B). Additionally,
compared with the control, downregulation of E2F5 significantly
suppressed migration of diabetic VSMCs (Fig. 5C). Taken together, these results
suggested that downregulation of E2F5 inhibited VSMC proliferation
and migration similar to miR-132 overexpression.
E2F5 overexpression rescues the
inhibitory effects of miR-132 on the proliferation and migration of
VSMCs treated with HG
We further examined whether E2F5 was involved in the
inhibitory effects of miR-132 on the HG-induced proliferation and
migration of VSMCs, overexpression of E2F5 was conducted by
infecting miR-132 overexpressed VSMCs; cell proliferation and
migration were detected by the nuclear incorporation of BrdU and a
Transwell migration assay, respectively. The results showed that
miR-132 was successfully overexpressed under HG conditions compared
with the corresponding control (Fig.
6A). In addition, miR-132 inhibited HG-induced upregulation of
E2F5 in VSMCs (Fig. 6B).
Furthermore, transduction with lenti-E2F5 exhibited non-significant
effect on the expression of miR-132, but could rescue downregulated
E2F5 expression induced by miR-132 (Fig. 6A and B). Furthermore, our results
showed that E2F5 overexpression could significantly rescue the
inhibitory effects of miR-132 on the proliferation and migration of
VSMCs, compared with VSMCs that were co-infected with miR-132 and
vector (P<0.001; Fig. 6C-E).
Thus, our findings suggested that miR-132 suppressed the
proliferation and migration of HG-treated VSMCs by targeting
E2F5.
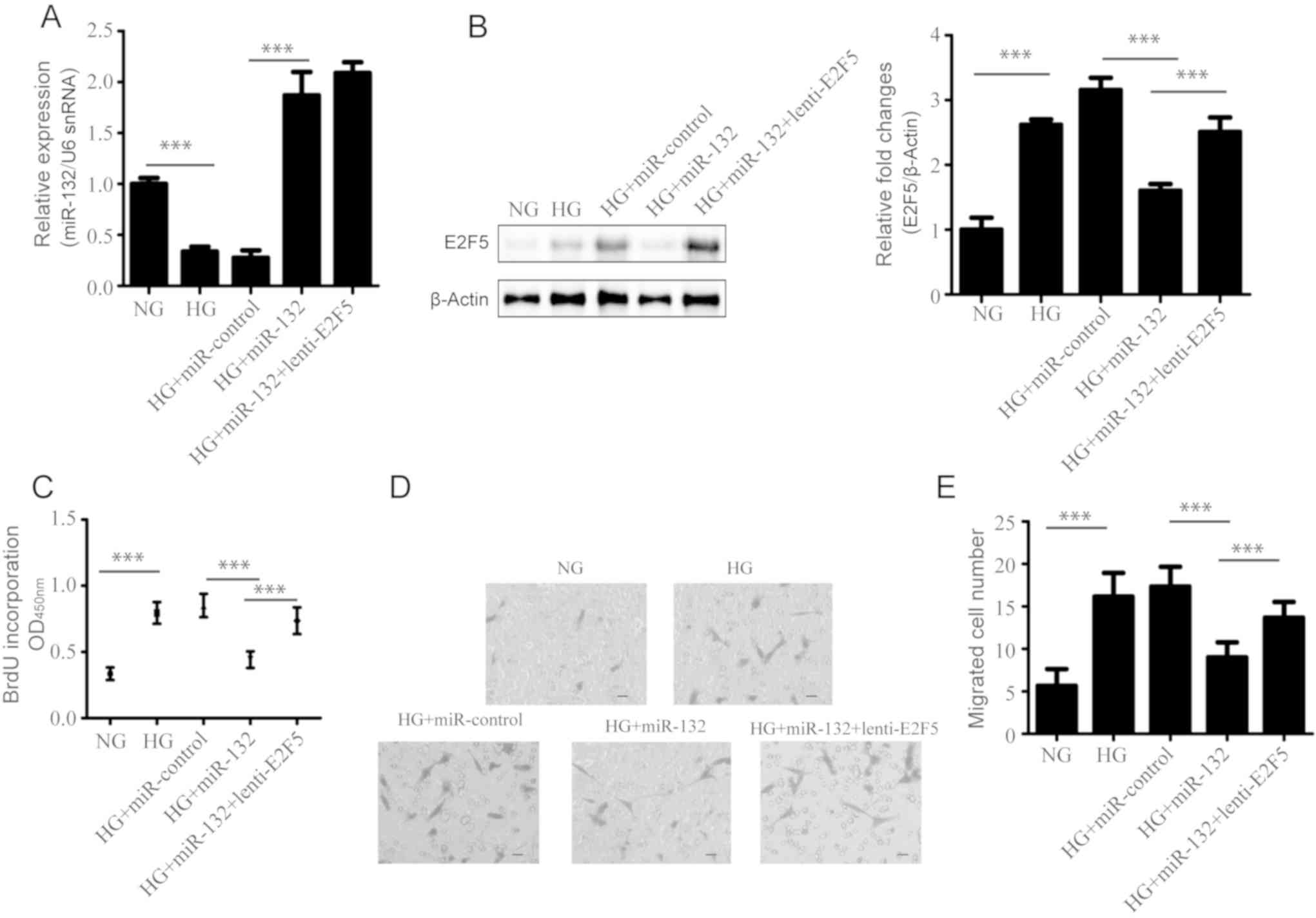 | Figure 6.Effects of miR-132 and E2F5 on
HG-induced VSMC proliferation and migration. VSMCs were co-infected
with miR-132 and lenti-vector, or lenti-E2F5 for 72 h, and were
then incubated under HG conditions for 48 h. (A) The expression of
miR-132 was detected by reverse transcription-quantitative
polymerase chain reaction. (B) The protein expression levels of
E2F5 were determined by western blotting; left panel,
representative results and right panel, quantified data for western
blotting. (C) The proliferation of these VSMCs was detected by a
BrdU incorporation assay. The migration of these VSMCs was measured
by a Transwell migration assay; (D) representative images and (E)
the quantified data. Magnification, ×100. All data were presented
as the mean ± standard deviation. ***P<0.001. The differences
were tested by one-way ANOVA followed by a Tukey's post hoc test.
BrdU, bromodeoxyuridine; E2F transcription factor 5; HG, high
glucose; NG, normal glucose; miR, microRNA; snRNA, small nuclear
RNA; VSMCs, vascular smooth muscle cells. |
Discussion
CVD-associated complications, such as restenosis,
hypertension and atherosclerosis are the leading causes of
mortality in patients with diabetes (2). The pathological development of
diabetes-related CVDs is characterized by dysfunction of VSMCs
(10). Previous studies have
reported that miRNAs exhibit a pathophysiological role in VSMCs in
diabetic CVD (33). It was
demonstrated that miR-132 was downregulated in VSMCs from diabetic
mice (21). Accordingly, our
results showed that the expression levels of miR-132 were decreased
in thoracic aorta tissue and VSMCs of diabetic rats. Furthermore,
the expression of miR-132 was suppressed in HG-treated VSMCs. These
findings suggested that miR-132 may serve a regulatory role in the
dysfunction of VSMCs in diabetes.
It has been reported that miR-132 is involved in
diabetic complications and CVDs, including diabetic cardiac
microangiopathy, neointimal hyperplasia, cardiac hypertrophy
endothelial cell function and angiogenesis (34–36).
Rawal et al (36) have
reported downregulation of proangiogenic miRNA-132 as an early
modulator of diabetic cardiac microangiopathy. In addition, miR-132
improved the repair of infarcted heart tissue via angiogenic
activation (37). A previous study
(37) reported that miR-132
promotes the proliferation of endothelial cells and facilitates
pathological angiogenesis. Additionally, miR-132 induced
myofibroblast proliferation (38);
however, accumulating evidence has indicated that miR-132 inhibited
the proliferation of VSMC (34,39).
These opposing effects of miR-132 on cell proliferation may be
associated with the various function of miRNAs in different cell
types and microenvironments. Hyperglycemia is harmful to VSMCs
(40); the present study reported
that HG-treatment induced the proliferation and migration of VSMCs,
which is consistent with recent studies (22,28,41).
VSMC proliferation and migration are fundamental processes of
vascular dysfunction in diabetes. Therefore, CVD-associated VSMC
dysfunction could be disrupted by regulating their proliferation
and migration (11,42). In the present study, it was
reported that miR-132 significantly inhibited the proliferation and
migration of diabetic and HG-treated VSMCs.
miR-132 regulates biological functions by binding to
the 3′UTR of target genes, inhibiting their expression. Previous
studies have identified many downstream mRNA targets of miR-132,
including p120 RasGAP in endothelial cells, RING finger protein 51
in cervical cancer cells and LRR binding FLII interacting protein 1
in VSMCs (34,43,44).
TargetScan analysis revealed E2F5 as a potential target of miR-132.
Furthermore, our results indicated that miR-132 interacted with the
3′UTR of E2F5, inhibiting its expression. In line with our
observations, it has been reported that miR-132 suppresses ovarian
cancer cell proliferation and migration by targeting E2F5 (26). E2F5 is a key member of the E2F
family that controls the transcription of proliferation-related
regulatory genes (45,46). In the present study, diabetic
rat-derived and HG-treated VSMCs exhibited high proliferative and
migration potentials. In addition, E2F5 was observed to be
upregulated in VSMCs from diabetic rats of and HG-treated VSMCs.
VSMCs from non-diabetic rats exhibited increased proliferation and
migration associated with E2F5 overexpression. In addition, the
anti-proliferative and anti-migratory effects of miR-132 in
HG-induced or diabetic rat VSMCs could be reversed by E2F5
overexpression. Numerous studies have proposed that E2F5 suppresses
the migration of several cancer cells, including breast cancer,
ovarian cancer and prostate cancer cells (26,47,48).
A previous study (48) has
reported that E2F5 binds to the promoter of heme oxygenase 1
(HMOX1) and inhibits its expression. Of note, it has been
demonstrated that HMOX1 inhibits VSMC proliferation and migration
by inhibiting the MAPK and AKT signaling pathways (49). Combined with the findings of these
previous studies, we proposed that the anti-proliferative and
anti-migratory effects of miR-132 may be associated with its
inhibitory effects on E2F5 expression, which may activate the MAPK
and AKT signaling pathways via the suppression of HMOX1; however,
further investigation is required.
In summary, our results revealed that miR-132 was
downregulated in diabetic rat-derived and HG-treated VSMCs.
Furthermore, miR-132 inhibited the proliferation and migration of
diabetic rat-derived and HG-treated VSMCs by targeting E2F5. The
findings of the present study suggest miR-132 and E2F5 as
potentially effective therapeutic targets for treating VSMC
dysfunction and CVDs in diabetes.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 81470404), Natural Science
Foundation of Shandong Province, China (grant no. ZR2014HM107) and
Projects of Medical and Health Technology Development Program in
Shandong province (grant no. 2017WSB04092).
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
QX, GY and ZG contributed to the design of the
experiment. CZ, XJL, HL, JL, QX, YL and XQL performed the
experiments, analyzed the data and wrote the manuscript. All of the
authors read and approved the final manuscript.
Ethics approval and consent to
participate
All the procedures and experimental protocols were
approved by the Institutional Animal Care and Use Committee of
Qianfoshan Hospital of Shandong Province. All the mice were treated
in accordance with the Guidelines for the Care and Use of
Laboratory Animals published by the U.S. National Institutes of
Health (50).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kahn SE: Clinical review 135: The
importance of beta-cell failure in the development and progression
of type 2 diabetes. J Clin Endocrinol Metab. 86:4047–4058. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ding Y, Sun X and Shan PF: MicroRNAs and
cardiovascular disease in diabetes mellitus. Biomed Res Int.
2017:40803642017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Quan A, Pan Y, Singh KK, Polemidiotis J,
Teoh H, Leong-Poi H and Verma S: Cardiovascular inflammation is
reduced with methotrexate in diabetes. Mol Cell Biochem.
432:159–167. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vamos EP, Millett C, Parsons C, Aylin P,
Majeed A and Bottle A: Nationwide study on trends in hospital
admissions for major cardiovascular events and procedures among
people with and without diabetes in England, 2004–2009. Diabetes
Care. 35:265–272. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nichols GA, Hillier TA, Erbey JR and Brown
JB: Congestive heart failure in type 2 diabetes: Prevalence,
incidence, and risk factors. Diabetes Care. 24:1614–1619. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li SL, Reddy MA, Cai Q, Meng L, Yuan H,
Lanting L and Natarajan R: Enhanced proatherogenic responses in
macrophages and vascular smooth muscle cells derived from diabetic
db/db mice. Diabetes. 55:2611–2619. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Doran AC, Meller N and McNamara CA: Role
of smooth muscle cells in the initiation and early progression of
atherosclerosis. Arterioscler Thromb Vasc Biol. 28:812–819. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gomez D and Owens GK: Smooth muscle cell
phenotypic switching in atherosclerosis. Cardiovasc Res.
95:156–164. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wall VZ and Bornfeldt KE: Arterial smooth
muscle. Arterioscler Thromb Vasc Biol. 34:2175–2179. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu WY, Yan H, Wang XB, Gui YZ, Gao F, Tang
XL, Qin YL, Su M, Chen T and Wang YP: Sodium tanshinone IIA silate
inhibits high glucose-induced vascular smooth muscle cell
proliferation and migration through activation of AMP-activated
protein kinase. PLoS One. 9:e949572014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shi L, Ji Y, Jiang X, Zhou L, Xu Y, Li Y,
Jiang W, Meng P and Liu X: Liraglutide attenuates high
glucose-induced abnormal cell migration, proliferation, and
apoptosis of vascular smooth muscle cells by activating the GLP-1
receptor, and inhibiting ERK1/2 and PI3K/Akt signaling pathways.
Cardiovasc Diabetol. 14:182015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ha M and Kim VN: Regulation of microRNA
biogenesis. Nat Rev Mol Cell Biol. 15:509–524. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Natarajan R and Nadler JL: Lipid
inflammatory mediators in diabetic vascular disease. Arterioscler
Thromb Vasc Biol. 24:1542–1548. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu H, Kong L, Zhou S, Cui W, Xu F, Luo M,
Li X, Tan Y and Miao L: The role of microRNAs in diabetic
nephropathy. J Diabetes Res. 2014:9201342014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Alexander MR and Owens GK: Epigenetic
control of smooth muscle cell differentiation and phenotypic
switching in vascular development and disease. Annu Rev Physiol.
74:13–40. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Maegdefessel L, Rayner KJ and Leeper NJ:
MicroRNA regulation of vascular smooth muscle function and
phenotype: Early career committee contribution. Arterioscler Thromb
Vasc Biol. 35:2–6. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Davis-Dusenbery BN, Chan MC, Reno KE,
Weisman AS, Layne MD, Lagna G and Hata A: Down-regulation of
Kruppel-like factor-4 (KLF4) by microRNA-143/145 is critical for
modulation of vascular smooth muscle cell phenotype by transforming
growth factor-beta and bone morphogenetic protein 4. J Biol Chem.
286:28097–28110. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang RN, Zheng B, Li LM, Zhang J, Zhang
XH and Wen JK: Tongxinluo inhibits vascular inflammation and
neointimal hyperplasia through blockade of the positive feedback
loop between miR-155 and TNF-alpha. Am J Physiol Heart Circ
Physiol. 307:H552–H562. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sun SG, Zheng B, Han M, Fang XM, Li HX,
Miao SB, Su M, Han Y, Shi HJ and Wen JK: miR-146a and Krüppel-like
factor 4 form a feedback loop to participate in vascular smooth
muscle cell proliferation. EMBO Rep. 12:56–62. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Reddy MA, Das S, Zhuo C, Jin W, Wang M,
Lanting L and Natarajan R: Regulation of vascular smooth muscle
cell dysfunction under diabetic conditions by miR-504. Arterioscler
Thromb Vasc Biol. 36:864–873. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang J, Chen L, Ding J, Fan Z, Li S, Wu H,
Zhang J, Yang C, Wang H, Zeng P and Yang J: MicroRNA-24 inhibits
high glucose-induced vascular smooth muscle cell proliferation and
migration by targeting HMGB1. Gene. 586:268–273. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou J, Li YS, Nguyen P, Wang KC, Weiss A,
Kuo YC, Chiu JJ, Shyy JY and Chien S: Regulation of vascular smooth
muscle cell turnover by endothelial cell-secreted microRNA-126:
Role of shear stress. Circ Res. 113:40–51. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nudelman AS, DiRocco DP, Lambert TJ,
Garelick MG, Le J, Nathanson NM and Storm DR: Neuronal activity
rapidly induces transcription of the CREB-regulated microRNA-132,
in vivo. Hippocampus. 20:492–498. 2010.PubMed/NCBI
|
|
25
|
Zhang S, Liang Z, Sun W and Pei L:
Repeated propofol anesthesia induced downregulation of hippocampal
miR-132 and learning and memory impairment of rats. Brain Res.
1670:156–164. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tian H, Hou L, Xiong YM, Huang JX, Zhang
WH, Pan YY and Song XR: miR-132 targeting E2F5 suppresses cell
proliferation, invasion, migration in ovarian cancer cells. Am J
Transl Res. 8:1492–1501. 2016.PubMed/NCBI
|
|
27
|
Xu B, Zhang Y, Du XF, Li J, Zi HX, Bu JW,
Yan Y, Han H and Du JL: Neurons secrete miR-132-containing exosomes
to regulate brain vascular integrity. Cell Res. 27:882–897. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Qin B, Liu J, Liu S, Li B and Ren J:
MiR-20b targets AKT3 and modulates vascular endothelial growth
factor-mediated changes in diabetic retinopathy. Acta Biochim
Biophys Sin (Shanghai). 48:732–740. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang J, Chen L, Ding J, Rong H, Dong W and
Li X: High mobility group box-1 induces migration of vascular
smooth muscle cells via TLR4-dependent PI3K/Akt pathway activation.
Mol Biol Rep. 39:3361–3367. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yuan Y, Shen Y, Xue L and Fan H: miR-140
suppresses tumor growth and metastasis of non-small cell lung
cancer by targeting insulin-like growth factor 1 receptor. PLoS
One. 8:e736042013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zheng F, Liao YJ, Cai MY, Liu YH, Liu TH,
Chen SP, Bian XW, Guan XY, Lin MC, Zeng YX, et al: The putative
tumour suppressor microRNA-124 modulates hepatocellular carcinoma
cell aggressiveness by repressing ROCK2 and EZH2. Gut. 61:278–289.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Torella D, Iaconetti C, Tarallo R, Marino
F, Giurato G, Veneziano C, Aquila I, Scalise M, Mancuso T,
Cianflone E, et al: MicroRNA regulation of the hyper-proliferative
phenotype of vascular smooth muscle cells in diabetes mellitus.
Diabetes. 67:2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Choe N, Kwon JS, Kim JR, Eom GH, Kim Y,
Nam KI, Ahn Y, Kee HJ and Kook H: The microRNA miR-132 targets
Lrrfip1 to block vascular smooth muscle cell proliferation and
neointimal hyperplasia. Atherosclerosis. 229:348–355. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ucar A, Gupta SK, Fiedler J, Erikci E,
Kardasinski M, Batkai S, Dangwal S, Kumarswamy R, Bang C, Holzmann
A, et al: The miRNA-212/132 family regulates both cardiac
hypertrophy and cardiomyocyte autophagy. Nat Commun. 3:10782012.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rawal S, Munasinghe PE, Shindikar A,
Paulin J, Cameron V, Manning P, Williams MJ, Jones GT, Bunton R,
Galvin I and Katare R: Down-regulation of proangiogenic
microRNA-126 and microRNA-132 are early modulators of diabetic
cardiac microangiopathy. Cardiovasc Res. 113:90–101. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Katare R, Riu F, Mitchell K, Gubernator M,
Campagnolo P, Cui Y, Fortunato O, Avolio E, Cesselli D, Beltrami
AP, et al: Transplantation of human pericyte progenitor cells
improves the repair of infarcted heart through activation of an
angiogenic program involving micro-RNA-132. Circ Res. 109:894–906.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bijkerk R, de Bruin RG, van Solingen C,
van Gils JM, Duijs JM, van der Veer EP, Rabelink TJ, Humphreys BD
and van Zonneveld AJ: Silencing of microRNA-132 reduces renal
fibrosis by selectively inhibiting myofibroblast proliferation.
Kidney Int. 89:1268–1280. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jin W, Reddy MA, Chen Z, Putta S, Lanting
L, Kato M, Park JT, Chandra M, Wang C, Tangirala RK and Natarajan
R: Small RNA sequencing reveals microRNAs that modulate angiotensin
II effects in vascular smooth muscle cells. J Biol Chem.
287:15672–15683. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yerneni KK, Bai W, Khan BV, Medford RM and
Natarajan R: Hyperglycemia-induced activation of nuclear
transcription factor kappaB in vascular smooth muscle cells.
Diabetes. 48:855–864. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen M, Zhang Y, Li W and Yang J:
MicroRNA-145 alleviates high glucose-induced proliferation and
migration of vascular smooth muscle cells through targeting ROCK1.
Biomed Pharmacother. 99:81–86. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
He M, Xue ZM, Li J and Zhou BQ:
Breviscapine inhibits high glucose-induced proliferation and
migration of cultured vascular smooth muscle cells of rats via
suppressing the ERK1/2 MAPK signaling pathway. Acta Pharmacol Sin.
33:606–614. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Anand S, Majeti BK, Acevedo LM, Murphy EA,
Mukthavaram R, Scheppke L, Huang M, Shields DJ, Lindquist JN,
Lapinski PE, et al: MicroRNA-132-mediated loss of p120RasGAP
activates the endothelium to facilitate pathological angiogenesis.
Nat Med. 16:909–914. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu GF, Zhang SH, Li XF, Cao LY, Fu ZZ and
Yu SN: Overexpression of microRNA-132 enhances the radiosensitivity
of cervical cancer cells by down-regulating Bmi-1. Oncotarget.
8:80757–80769. 2017.PubMed/NCBI
|
|
45
|
Chen HZ, Tsai SY and Leone G: Emerging
roles of E2Fs in cancer: An exit from cell cycle control. Nat Rev
Cancer. 9:785–797. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Dimova DK and Dyson NJ: The E2F
transcriptional network: Old acquaintances with new faces.
Oncogene. 24:2810–2826. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zheng Y, Zhu C, Ma L, Shao P, Qin C, Li P,
Cao Q, Ju X, Cheng G, Zhu Q, et al: miRNA-154-5p inhibits
proliferation, migration and invasion by targeting E2F5 in prostate
cancer cell lines. Urol Int. 98:102–110. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cai C, Huo Q, Wang X, Chen B and Yang Q:
SNHG16 contributes to breast cancer cell migration by competitively
binding miR-98 with E2F5. Biochem Biophys Res Commun. 485:272–278.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chen S, Ding Y, Tao W, Zhang W, Liang T
and Liu C: Naringenin inhibits TNF-alpha induced VSMC proliferation
and migration via induction of HO-1. Food Chem Toxicol.
50:3025–3031. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Portaluppi F, Smolensky MH and Touitou Y:
Ethics and methods for biological rhythm research on animals and
human beings. Chronobiol Int. 27:1911–1929. 2010. View Article : Google Scholar : PubMed/NCBI
|