Introduction
Retinitis pigmentosa (RP) is caused by a progressive
degeneration of photoreceptor cells that initially primarily affect
the rods whereas the function of the cones is compromised as the
disease progresses (1). Due to its
heterogeneity and diversity of inheritance patterns, the clinical
presentation of RP is variable. Some RP patients lose vision in
their childhood, whereas other patients retain normal vision until
mid-adulthood. The clinical symptoms of RP include night blindness
and gradual loss of the peripheral visual field caused by rod
photoreceptor lost. Later with the loss of cone photoreceptors, the
central vision is impaired and patients may ultimately suffer from
legal blindness ultimately (1).
The worldwide prevalence of RP is approximately 1 in 4,000, or more
than 1 million individuals (2).
RP is clinically and genetically heterogeneous. It
can be inherited as an autosomal dominant (30–40%), autosomal
recessive (50–60%), or X-linked (5–15%) trait (3). Some patients who have no known family
history are classified as simplex RP. Autosomal recessive RP (arRP)
is the most common form of RP worldwide. To date, more than 80
genes (https://sph.uth.edu/retnet/) have
been implicated in syndromic and non-syndromic forms of RP.
However, these identified genes account for no more than 60% of RP
cases, and there has been limited success in screening known genes
for RP using conventional Sanger sequencing. Recently, exome
sequencing has been successfully used to identify genes associated
with Mendelian disorders (4,5).
Coupled with DNA capture technology, the next-generation sequencing
(NGS) platform enables rapid and cost-effective parallel sequencing
of a large panel of disease genes. In some recent studies, NGS has
provided a promising alternative for the molecular diagnosis and
gene identification of RP (6–10).
In the present study, the identification of
disease-causing mutations of RP was presented by using exome
sequencing in two Chinese families with arRP. This report concerns
the clinical features and genetic findings of the two arRP
families.
Patients and methods
Study subjects
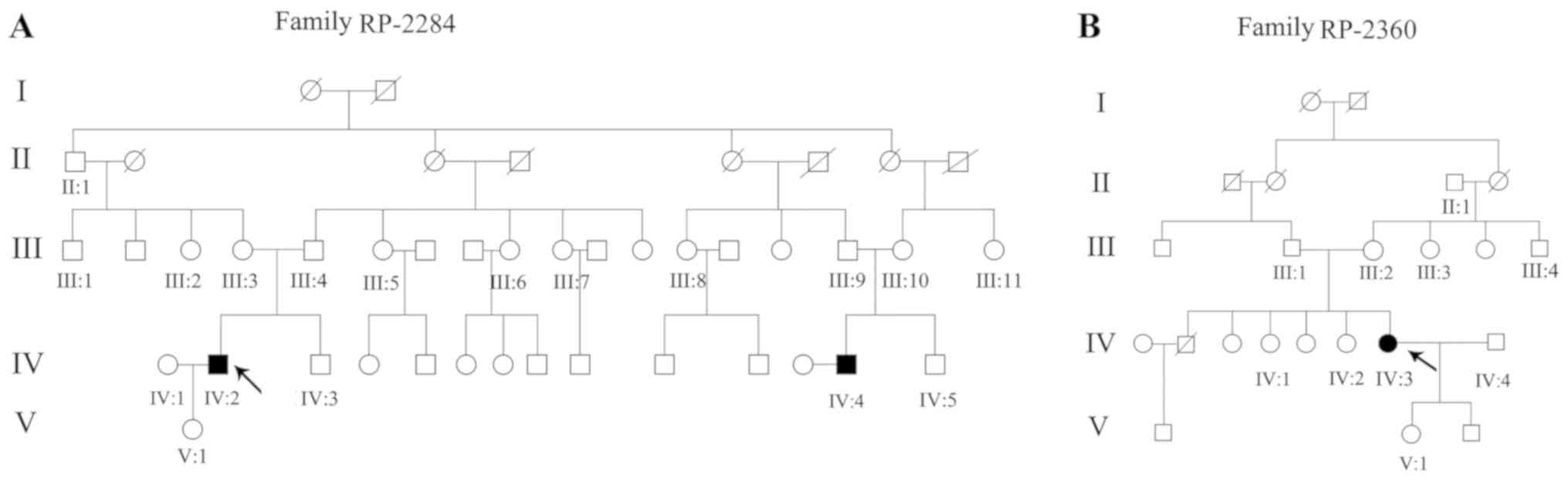
Two arRP pedigrees with Chinese origin were
recruited from Sichuan Provincial People's Hospital, including a
four-generation family with two consanguineous arRP patients and 16
unaffected members (RP-2284; Fig.
1A), and a four-generation family with one consanguineous RP
patient and nine unaffected members (RP-2360; Fig. 1B). In addition, 1,260 unrelated
healthy individuals of Chinese Han origin were also recruited. This
study was approved by the Institutional Review Board of the Sichuan
Provincial People's Hospital, University of Electronic Science and
Technology of China. Informed written consent was obtained from
each individual prior to participation in this study. All
procedures were carried out in accordance with the tenets of the
Declaration of Helsinki.
Clinical diagnosis
The clinical information of the two families is
listed in Table I. Each family
member underwent a complete ophthalmic examination, including
best-corrected visual acuity (BCVA), slit-lamp biomicroscopy,
fundus photography if possible, visual field testing (Octopus;
Interzeag AG) and electroretinography (ERG). ERGs were performed
using a multifocal ERG recorder (GT-2008V–IV; Chongqing Kanghua
Ruiming S&T Co., Ltd) and corneal contact lens electrodes. The
ERG protocol complied with the standards of the International
Society for Clinical Electrophysiology of Vision. Diagnosis of RP
in the three patients was based on poor night vision at an early
age, followed by progressive loss of peripheral visual fields, and
extinguished ERG responses, along with abnormal fundus changes
(2). In contrast, all the
unaffected family members, and the unrelated control individuals
had no signs of retinal diseases.
 | Table I.Genotypes and phenotypes of the two
RP family members. |
Table I.
Genotypes and phenotypes of the two
RP family members.
| A, Family
RP-2284 |
|---|
|
|---|
| Family member | Age
(years)/sex | Phenotype | Age at onset
(years) | Visual acuity
(OD/OS) | Fundus
appearance | Mutation | Mutation type |
|---|
| V:1 | 6/F | Normal | – | – | – | C.1997 T>A | Het |
| IV:1 | 30/F | Normal | – | 1.0/1.0 | Normal | No mutation | – |
| IV:2 | 31/M | RP | 24 | Light
perception | RVA and DBSP | C.1997 T>A |
Hom |
| IV:3 | 28/M | Normal | – | 1.0/1.0 | Normal | C.1997 T>A | Het |
| IV:4 | 23/M | RP | 22 | Counting
fingers | RVA, DBSP and
MD | C.1997 T>A |
Hom |
| IV:5 | 26/M | Normal |
| 1.0/1.0 | Normal | No mutation | – |
| III:1 | 50/M | Normal | – | – | – | C.1997 T>A | Het |
| III:2 | 52/F | Normal | – | 1.0/0.9 | Normal | No mutation | – |
| III:3 | 54/F | Normal | – | 1.0/1.0 | Normal | C.1997 T>A | Het |
| III:4 | 55/M | Normal | – | 1.0/0.8 | Normal | C.1997 T>A | Het |
| III:5 | 53/F | Normal | – | – | – | C.1997 T>A | Het |
| III:6 | 50/F | Normal | – | 0.8/0.8 | Normal | No mutation | – |
| III:7 | 48/F | Normal | – | – | – | C.1997 T>A | Het |
| III:8 | 49/F | Normal | – | 0.8/0.8 | Normal | No mutation | – |
| III:9 | 51/M | Normal | – | 0.9/0.8 | Normal | C.1997 T>A | Het |
| III:10 | 48/F | Normal | – | 0.8/1.0 | Normal | C.1997 T>A | Het |
| III:11 | 46/F | Normal | – | – | – | No mutation | – |
| II:1 | 78/M | Cataract | – | – | – | C.1997 T>A | Het |
|
| B, Family
RP-2360 |
|
| Family
member | Age
(years)/sex |
Phenotype | Age at onset
(years) | Visual acuity
(OD/OS) | Fundus
appearance |
Mutation | Mutation
type |
|
| V:1 | 18/F | Normal | – | – | Normal | C.2426 A>C | Het |
| IV:1 | 41/F | Normal | – | 1.0/1.0 | Normal | C.2426 A>C | Het |
| IV:2 | 45/M | Normal | – | 1.0/0.9 | Normal | No mutation | – |
| IV:3 | 43/F | RP | 23 | Counting
fingers | OS: RVA and
DBSP | C.2426 A>C | Hom |
| IV:4 | 46/F | Normal | – | – | – | No mutation | – |
| III:1 | 70/M | Normal | – | 0.8/0.7 | Normal | C.2426 A>C | Het |
| III:2 | 69/F | Normal | – | 0.7/0.8 | Normal | C.2426 A>C | Het |
| III:3 | 67/F | Normal | – | – | – | C.2426 A>C | Het |
| III:4 | 66/F | Normal | – | 0.8/0.8 | Normal | No mutation | – |
| II:1 | 92/M | Cataract | – | – | – | No mutation | – |
DNA extraction
Genomic DNA were extracted from the peripheral blood
of all family members (RP-2284 and RP-2360) and 1,260 controls,
using a TIANamp Blood DNA kit (cat. no. DP348) according to the
manufacturer's protocol (Tiangen Biotech Co., Ltd.).
Exome sequencing and variant
detection
Exome sequencing was performed on patients IV:2 and
IV:4 in RP-2284 and patient IV:3 in RP-2360 by Genergy
Biotechnology, Co., Ltd. The sequenced samples were prepared
according to the Illumina protocols of the SureSelect Target
Enrichment System Capture Process (Illumina, Inc.) and exome
sequencing analysis was performed as described previously (11). Briefly, the reads were mapped
against UCSC hg19 (http://genome.ucsc.edu/) by BWA (http://bio-bwa.sourceforge.net/). The SNPs and
Indels were detected by SAMTOOLS (http://samtools.sourceforge.net/). The detected
variants were annotated and filtered based on public and in-house
databases, under the following conditions: i) variants within
intergenic, intronic, and UTR regions and synonymous mutations were
excluded from downstream analysis; ii) variants in dbSNP138
(http://www.ncbi.nlm.nih.gov/projects/SNP/), 1000
Genome project (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp), Exome
Aggregation Consortium, YH Database (http://yh.genomics.org.cn/), the HapMap Project
(ftp://ftp.ncbi.nlm.nih.gov/hapmap)
and our in-house database generated from 2,010 samples of exome
sequencing, were excluded; iii) Polyphen2 (http://genetics.bwh.harvard.edu/pph2/) was used to
predict the possible damaging impacts of each mutation.
Mutation validation
The CRB1 mutations identified by exome
sequencing were further confirmed in all members of the two
families and in 1,260 controls by direct Sanger sequencing, using
the PCR primers (CRB1-c.1997T>A-F,
5′-AGTGGCATTTCGTGGAGGTA-3′ and CRB1-c.1997T>A-R:
5′-GCTGTTTCTGCTCTGCTCTG-3′; CRB1-c.2426A>C-F:
5′-TTTGTCCGAACGCTTCAACC-3′ and CRB1-c.2426A>C-R:
5′-TCTTGCTTGTCAGGTAGGCC-3′) designed by Primer 3.0 (http://primer3.ut.ee/) and synthesized by Invitrogen
(Thermo Fisher Scientific, Inc.). Direct sequencing was performed
in an ABI 3130XL genetic analyzer according to ABI BigDye
sequencing protocols (Thermo Fisher Scientific, Inc.).
Results
Patients and clinical information
Two Chinese consanguineous arRP families (RP-2284
and RP-2360, Fig. 1) were
recruited in this study. Two subjects (IV:2 and IV:4) were
diagnosed with RP in family RP-2284, which exhibited a pattern of
recessive inheritance. They presented typical clinical symptoms,
including night blindness rapidly progressing to severe visual
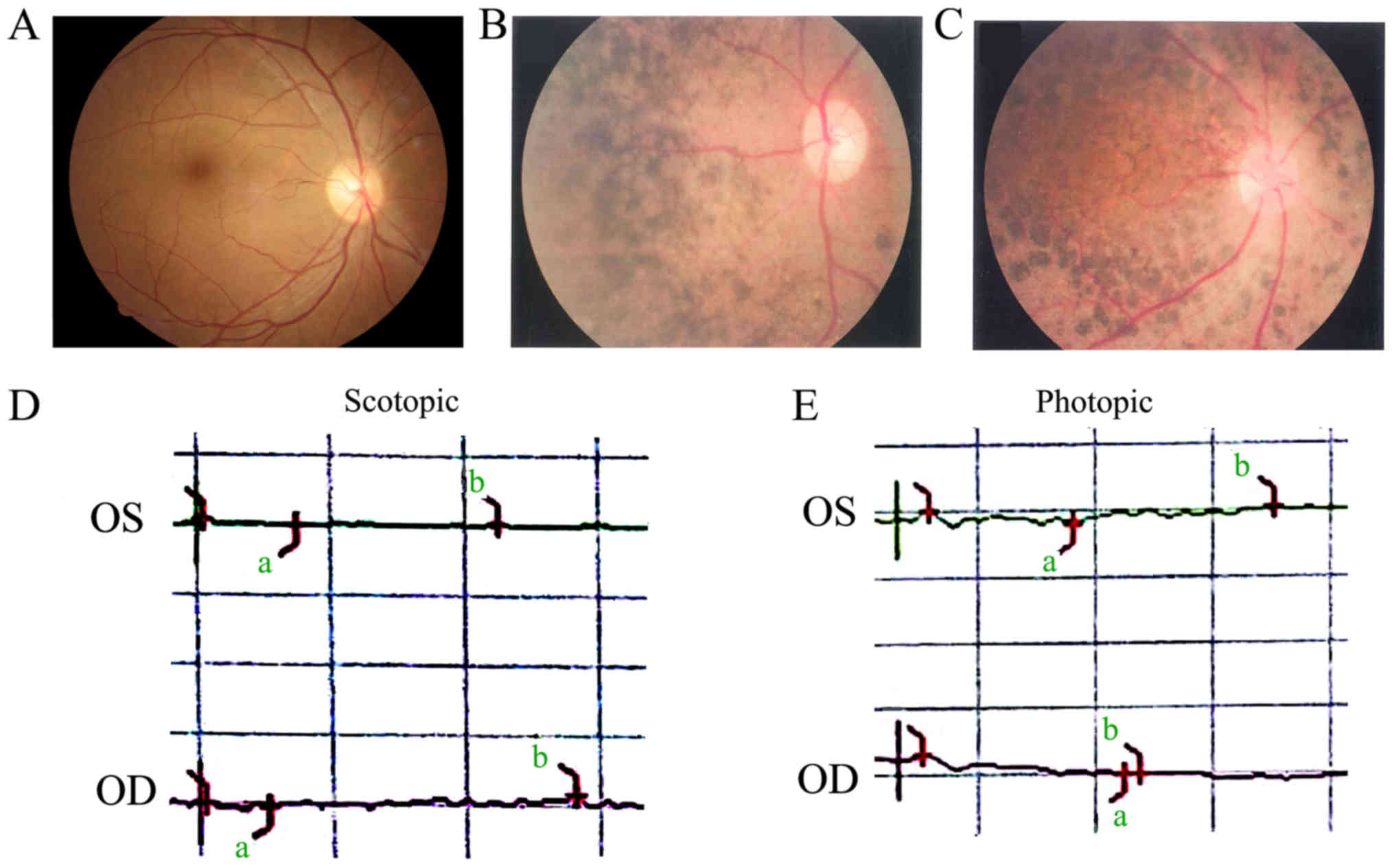
disability. The fundus examination of the proband (IV:2) showed
pallor in the optic nerve head, retinal artery attenuation, and
mid-peripheral defuse and dense bone-spicule pigmentation (Fig. 2A and B). Another affected member
(IV:4) had similar clinical manifestations to the proband and
presented with macular degeneration (Fig. 2C). Family RP-2360 included one RP
patient (IV:3, the proband), who had experienced night blindness
since 23 years of age and progressive bilateral visual loss
(Table I). She presented with
early-onset and markedly decreased visual acuity (OD: 20/400, OS:
20/400) in both eyes (Table I).
The fundus image in the right eye was blurred due to dense
posterior subcapsular cataract. Fundus examination of her left eye
showed similar features to those of patient IV:2 in RP-2284 (data
not shown). None of the three patients exhibited glaucoma features,
and their ERG examination showed no recordable response under
either scotopic or photopic condition, indicating significant loss
of function in the both rods and cones (Fig. 2D and E).
Exome sequencing and data
analysis
Exome sequencing was performed on two patients (IV:2
and IV:4) in the RP-2284 family and on one patient (IV:3) in the
RP-2360 family with a mean read depth of target regions of 52.3×.
There were 18,793 SNPs in the coding regions (8,763 non-synonymous
SNPs, 9,836 synonymous SNPs, and 873 other types of SNPs) and 387
coding indels identified. To identify the disease-causing
mutations, the researchers paid close attention to the functional
SNPs/indels in homozygous or compound heterozygous status including
frameshift coding-region insertion or deletions and non-synonymous
variants which were more likely to be pathogenic. Detected variants
were compared with HapMap Project, dbSNP138, 1000 Genomes Project,
Exome Aggregation Consortium, YH Database, and our in-house
database. The rare homozygous variants shared by the two affected
patients in RP-2284 and RP-2360 families are listed in Table SI. Using the autosomal recessive
mode, two homozygous mutations, c.1997 T>A and c.2426 A>C,
were identified in the CRB1 gene (NM_201253.2) in the
RP-2284 and RP-2360 families, respectively.
Mutation validation and analysis
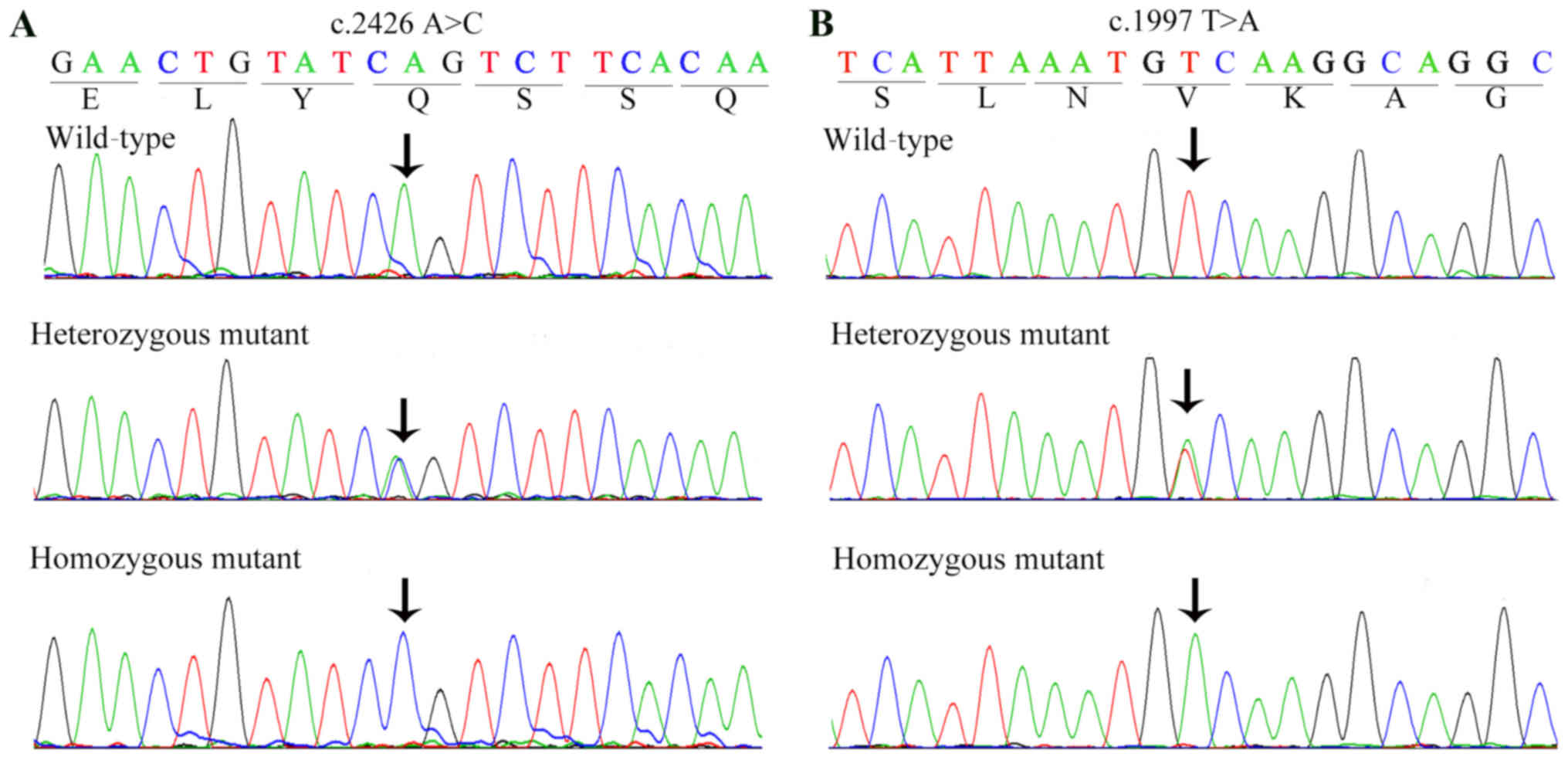
In the RP-2284 family, the presence of homozygous
mutation c.1997 T>A in two patients (IV:2 and IV:4) was
confirmed by Sanger sequencing (Fig.
3B). The parents and other unaffected siblings of these two
patients were either unaffected heterozygous carriers of c.1997
T>A, or had a normal CRB1 sequence without any mutation
(Table I). In the RP-2360 family,
direct Sanger sequencing confirmed the presence of homozygous
mutation c.2426 A>C (Fig. 3A)
in the proband (IV:3), whereas all other unaffected family members
were heterozygotes or normal. The two homozygous mutations
described above were absent in the 1,260 Han Chinese healthy
controls. These results reveal a complete co-segregation of the two
CRB1 mutations with RP within the respective families. The
genotypes of the members of families RP-2284 and RP-2360 are shown
in Table I.
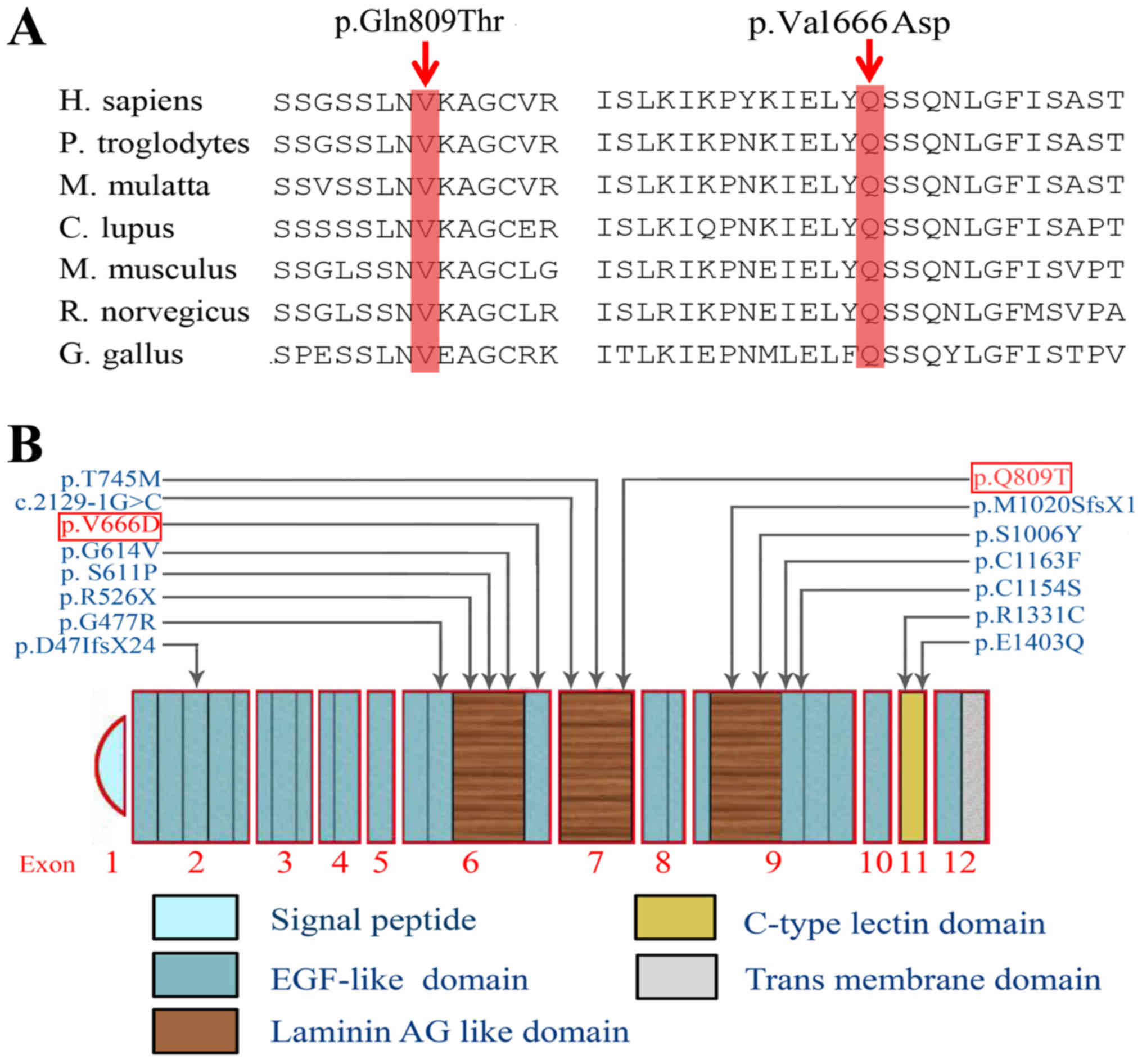
The c.1997 T>A (p.V666D) homozygous mutation is
expected to change valine (hydrophobic non-polar) to aspartic acid
(hydrophilic positively charged) at codon 666 in exon 6. The
substituted amino acid is predicted to alter the hydrophobicity and
chargeability of the CRB1 protein. The c.2426 A>C (p.Q809T)
mutation introduced a substitution of glutamine for threonine at
codon 809 in exon 7 (Fig. 4B).
Comparative amino acid sequence alignment of the CRB1 proteins
across human and other species revealed that the two mutations
occurred at highly conserved positions (Fig. 4A). These changes were predicted to
be damaging to CRB1 using Polyohen-2.
Discussion
A previous study demonstrated that the CRB1
gene was responsible for retinitis pigmentosa (RP) at the RP12
locus (12) and more than 150
disease-associated variants in CRB1 have been reported
(13). Most of the mutations occur
in exon 7 (27%) and exon 9 (41%), which encode the second and the
third laminin AG-like domains, respectively (13). In the present study, two homozygous
mutations c.1997 T>A and c.2426 A>C in CRB1 causing
autosomal recessive retinitis pigmentosa (arRP) in two Chinese
families were identified using exome sequencing. This result
expanded the CRB1 mutation spectrum causing RP, which may
contribute to improvement of the molecular prognosis of RP.
Studies have reported that approximately 194
mutations in CRB1 have been found to be related to the
pathogenesis of arRP in diverse populations, including 189
mutations in the extracellular region (14–17).
CRB1 is a human homolog of Drosophila crumbs, a well
conserved gene with homologue across multiple phyla (18). It is located at 1q31.3 and consists
of 12 exons spanning 210 kb of genomic DNA (12). This gene encodes two different
proteins of 1,376 and 1,406 amino acids, and both protein are
composed of three parts: 19 epidermal growth factor (EGF)-like
domains, three laminin A globular (AG)-like domains and a signal
peptide sequence. Beyond that, the longer isoform also contains
additional transmembrane and cytoplasmatic domains (19). CRB1 is a transmembrane protein and
is mainly expressed in the inner segments of mammalian
photoreceptors as well as in the brain (12,18).
It plays an essential role in the development of the retina
(20,21). CRB1 is involved in photoreceptor
morphogenesis in the retina (22).
Mutations in CRB1 may restrain retinal development and result in a
loss of photoreceptor signaling (23). In the mouse and human retina, the
protein Crb1 or CRB1 is concentrated in the vicinity of the outer
limiting membrane and is expressed in photoreceptor inner segments,
in Muller cell and the epithelial cells (24). The Crb1 mutant in mouse
showed a developmental defect of retina, including a disrupted
outer limiting membrane and folded retina (25).
The present study involved two Han Chinese arRP
families. An analysis of the exome sequencing data revealed that
only the two homozygous mutations c.1997 T>A (p.V666D) and
c.2426 A>C (p.Q809T) in CRB1 were found to be
co-segregated with arRP in the families. The two mutations have
never been reported in the ExAC database (http://exac.broadinstitute.org/) or the human gene
mutation database (http://www.hgmd.org/). The clinical findings
associated with retinal dystrophies caused by CRB1 mutations
include congenital blindness, LCA, to early-onset rod-cone
dystrophy and arRP. Mutations in CRB1 account for 10–15% of
all patients with LCA and as many as 6.5% of all patients with
arRP. In this study, the age of onset of patients in the two RP
families was approximately 22–24 years of age. The homozygous
mutation c.2426 A>C found in family RP-2360 is located at exon 7
of the CRB1 gene, encoding the second laminin AG-like
domain. Apart from the proband (IV:3) who carried the homozygous
mutation c.2426 A>C, other family members carried either
heterozygous mutation or no mutation. The proband's parents had a
consanguineous marriage, and they were each heterozygous for c.2426
A>C, which was also found in members V:1, IV:1 and III:3
(Table I). As previously reported,
laminin AG-like domain is predicted to affect interactions among
proteins, calcium binding, and protein folding (26). This mutation introduces a
substitution of glutanine to threonine, which may dramatically
affect the function of the second laminin AG-like domain of the
CRB1 protein (PolyPhen2 scores ~0.824). There were many disease
causative mutations reported in the laminin AG-like domain, which
indicates that this domain plays an important role in CRB1 function
(24). The mutation c.1997 T>A
identified in family RP-2284, was previously reported by Wang et
al as present in RP patents in compound heterozygote (27), which further confirmed its
pathogenic role in RP. In this pedigree with a consanguineous
marriage, the proband's parents (III:3 and III:4), daughter (V:1),
aunts (III:5 and III:7), uncle (III:1) and brother (IV:3), as well
as another patient's parents (III:9 and III:10), were found to
carry the heterozygous mutation (Table
I). The change could convert valine to aspartic acid at codon
666 (p.V666 D). This substitution was predicted to be damaging by
Polyohen-2 and to alter the hydrophobicity and chargeability of the
CRB1 protein. Nevertheless, how these mutations exactly affect the
function of CRB1 protein remains unclear. Further functional
research is essential to elucidate the underlying mechanism
governing how the two homozygous mutations result in RP.
In conclusion, the present study revealed two
homozygous mutations c.1997 T>A (p.V 666D) and c.2426 A>C
(p.Q809T) in the CRB1 gene in two consanguineous Chinese
families with arRP via exome sequencing. These data expand the
CRB1 mutation spectrum causing RP and may contribute to an
improvement in the molecular diagnosis for retinal dystrophies.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
Natural Science Foundation of China (81670853, 81700841, 81670893,
81570882 and 81670877), CAS ‘Light of West China’ Program (to BG),
Science and Technology Department of Sichuan Province (2019JDJQ0031
and 2014FZ0124), Foundation for Technology & Science &
Technology Bureau of Chengdu (2015-HM02-00094-SF and
2018-YF05-00348-SN).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
BG, CQ, JL, QW and JY conceived the experiment. XG,
JW and YS performed the experiments. YS, FL, JY, LJ, CQ and HZ
recruited the patients. BG, FL, LC, QW, LJ, CQ and HZ analyzed the
data. XG and JL drafted the manuscript. JY and HZ critically
revised the manuscript for important intellectual content. All
authors read and approved the manuscript and agreed to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
This study was approved by the Institutional Review
Board of the Sichuan Provincial People's Hospital, University of
Electronic Science and Technology of China. Informed written
consent was obtained from each individual prior to participation in
this study. All procedures were carried out in accordance with the
tenets of the Declaration of Helsinki.
Patient consent for publication
Not applicable.
Competing interests
The authors delare that they have no competing
interests.
References
|
1
|
Bird AC: Retinal photoreceptor dystrophies
LI. Edward Jackson Memorial Lecture. Am J Ophthalmol. 119:543–562.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hartong DT, Berson EL and Dryja TP:
Retinitis pigmentosa. Lancet. 368:1795–1809. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rivolta C, Sharon D, DeAngelis MM and
Dryja TP: Retinitis pigmentosa and allied diseases: Numerous
diseases, genes, and inheritance patterns. Hum Mol Genet.
11:1219–1227. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ng SB, Turner EH, Robertson PD, Flygare
SD, Bigham AW, Lee C, Shaffer T, Wong M, Bhattacharjee A, Eichler
EE, et al: Targeted capture and massively parallel sequencing of 12
human exomes. Nature. 461:272–276. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ng SB, Buckingham KJ, Lee C, Bigham AW,
Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, et
al: Exome sequencing identifies the cause of a mendelian disorder.
Nat Genet. 42:30–35. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Neveling K, Collin RW, Gilissen C, van
Huet RA, Visser L, Kwint MP, Gijsen SJ, Zonneveld MN, Wieskamp N,
de Ligt J, et al: Next-generation genetic testing for retinitis
pigmentosa. Hum Mutat. 33:963–972. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
O'Sullivan J, Mullaney BG, Bhaskar SS,
Dickerson JE, Hall G, O'Grady A, Webster A, Ramsden SC and Black
GC: A paradigm shift in the delivery of services for diagnosis of
inherited retinal disease. J Med Genet. 49:322–326. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Katagiri S, Akahori M, Sergeev Y,
Yoshitake K, Ikeo K, Furuno M, Hayashi T, Kondo M, Ueno S, Tsunoda
K, et al: Whole exome analysis identifies frequent CNGA1 mutations
in Japanese population with autosomal recessive retinitis
pigmentosa. PLoS One. 9:e1087212014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu J, Chen L, Tam OS, Huang XF, Pang CP
and Jin ZB: Whole exome sequencing reveals genetic predisposition
in a large family with retinitis pigmentosa. Biomed Res Int.
2014:3024872014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Villanueva A, Willer JR, Bryois J,
Dermitzakis ET, Katsanis N and Davis EE: Whole exome sequencing of
a dominant retinitis pigmentosa family identifies a novel deletion
in PRPF31. Invest Ophthalmol Vis Sci. 55:2121–2129. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou Y, Shuai P, Li X, Wang J, Yang Y, Hao
F, Lin H, Zhang D and Gong B: Association of SOD2 polymorphisms
with primary open angle glaucoma in a Chinese population.
Ophthalmic Genet. 36:43–49. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
den Hollander AI, ten Brink JB, de Kok YJ,
van Soest S, van den Born LI, van Driel MA, van de Pol DJ, Payne
AM, Bhattacharya SS, Kellner U, et al: Mutations in a human
homologue of Drosophila crumbs cause retinitis pigmentosa
(RP12). Nat Genet. 23:217–221. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Slavotinek AM: The family of crumbs genes
and human disease. Mol Syndromol. 7:274–281. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cordovez JA, Traboulsi EI, Capasso JE,
Sadagopan KA, Ganesh A, Rychwalski PJ, Neely KA, Brodie SE and
Levin AV: Retinal dystrophy with intraretinal cystoid spaces
associated with mutations in the crumbs homologue (CRB1) gene.
Ophthalmic Genet. 36:257–264. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Booij JC, Florijn RJ, ten Brink JB, Loves
W, Meire F, van Schooneveld MJ, de Jong PT and Bergen AA:
Identification of mutations in the AIPL1, CRB1, GUCY2D, RPE65, and
RPGRIP1 genes in patients with juvenile retinitis pigmentosa. J Med
Genet. 42:e672005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Beryozkin A, Zelinger L, Bandah-Rozenfeld
D, Harel A, Strom TA, Merin S, Chowers I, Banin E and Sharon D:
Mutations in CRB1 are a relatively common cause of autosomal
recessive early-onset retinal degeneration in the Israeli and
Palestinian populations. Invest Ophthalmol Vis Sci. 54:2068–2075.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen Y, Lin Y, Vithana EN, Jia L, Zuo X,
Wong TY, Chen LJ, Zhu X, Tam PO, Gong B, et al: Common variants
near ABCA1 and in PMM2 are associated with primary open-angle
glaucoma. Nat Genet. 46:1115–1119. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Richard M, Roepman R, Aartsen WM, van
Rossum AG, den Hollander AI, Knust E, Wijnholds J and Cremers FP:
Towards understanding CRUMBS function in retinal dystrophies. Hum
Mol Genet. 15:R235–R243. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
den Hollander AI, Johnson K, de Kok YJ,
Klebes A, Brunner HG, Knust E and Cremers FP: CRB1 has a
cytoplasmic domain that is functionally conserved between human and
Drosophila. Hum Mol Genet. 10:2767–2773. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tepass U: Crumbs, a component of the
apical membrane, is required for zonula adherens formation in
primary epithelia of Drosophila. Dev Biol. 177:217–225.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pellikka M, Tanentzapf G, Pinto M, Smith
C, McGlade CJ, Ready DF and Tepass U: Crumbs, the Drosophila
homologue of human CRB1/RP12, is essential for photoreceptor
morphogenesis. Nature. 416:143–149. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Beheshtian M, Saee Rad S, Babanejad M,
Mohseni M, Hashemi H, Eshghabadi A, Hajizadeh F, Akbari MR, Kahrizi
K, Riazi Esfahani M and Najmabadi H: Impact of whole exome
sequencing among Iranian patients with autosomal recessive
retinitis pigmentosa. Arch Iran Med. 18:776–785. 2015.PubMed/NCBI
|
|
23
|
Thompson DA, Janecke AR, Lange J, Feathers
KL, Hübner CA, McHenry CL, Stockton DW, Rammesmayer G, Lupski JR,
Antinolo G, et al: Retinal degeneration associated with RDH12
mutations results from decreased 11-cis retinal synthesis due to
disruption of the visual cycle. Hum Mol Genet. 14:3865–3875. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bujakowska K, Audo I, Mohand-Saïd S,
Lancelot ME, Antonio A, Germain A, Léveillard T, Letexier M,
Saraiva JP, Lonjou C, et al: CRB1 mutations in inherited retinal
dystrophies. Hum Mutat. 33:306–315. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mehalow AK, Kameya S, Smith RS, Hawes NL,
Denegre JM, Young JA, Bechtold L, Haider NB, Tepass U, Heckenlively
JR, et al: CRB1 is essential for external limiting membrane
integrity and photoreceptor morphogenesis in the mammalian retina.
Hum Mol Genet. 12:2179–2189. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhou Y, Tao S, Chen H, Huang L, Zhu X, Li
Y, Wang Z, Lin H, Hao F, Yang Z, et al: Exome sequencing analysis
identifies compound heterozygous mutation in ABCA4 in a Chinese
family with Stargardt disease. PLoS One. 9:e919622014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang H, Wang X, Zou X, Xu S, Li H, Soens
ZT, Wang K, Li Y, Dong F, Chen R and Sui R: Comprehensive molecular
diagnosis of a large Chinese Leber congenital Amaurosis cohort.
Invest Ophthalmol Vis Sci. 56:3642–3655. 2015. View Article : Google Scholar : PubMed/NCBI
|