Introduction
Hearing loss or hearing impairment refers to a
partial inability to hear sounds in one or both ears, while
deafness is defined by full inability to hear (1). The Global Burden of Disease Study has
demonstrated that hearing loss is the fourth leading cause of
global disability (2). In the
United States, the prevalence of hearing loss doubles every 10
years with an increasingly aging population (3). Risk factors for hearing loss include
genetic factors, aging, a noisy environment, ear trauma or
infection, birth complications, certain medications and toxins
(1). Genetic factors may be
responsible for ~40% of childhood hearing loss, especially in
children born of consanguineous marriages (4–6).
Hundreds of gene mutations have been identified that lead to
hearing loss either as an exclusive clinical feature or in
combination with extra-auditory symptoms as part of a syndrome
(7). Non-syndromic forms of
deafness account for 70% of cases, of which~85% are inherited in an
autosomal recessive manner (8,9).
Owing to recent improvements in research of genetic factors, such
as the identification of gene mutations involved in congenital
hearing loss, genetic counseling has emerged and increased in
availability (1). High-throughput
DNA sequencing technologies, known as next-generation DNA
sequencing or massively parallel sequencing, are used to detect
multiple gene mutations, which results in improved detection and
early treatment of childhood hearing loss (10).
Recessive genetic mutations of cadherin 23 (CDH23)
have been associated with the allelic variants Usher syndrome type
1D (USH1D) and non-syndromic autosomal recessive deafness 12
(DFNB12) (11,12). CDH23 is on chromosome
10q21-q22 and contains 69 exons that encode the CDH23 protein.
CDH23 comprises 3,354 amino acids with 27 cadherin
extracellular (EC) repeats, a single transmembrane domain and a
short unique cytoplasmic domain (12). Each EC domain of the CDH23 protein
contains several cadherin-specific amino acid motifs, such as LDRE,
DXD and DXNDN, and is highly conserved in sequence and spacing,
which is required for cadherin dimerization and calcium-binding
(13). CDH23 mutations
induce disorganization of the stereocilia in the hair cells of the
inner ear in the ‘waltzer’ mouse model of the USH1D (14). Thus, the extracellular domains of
CAD23 are crucial for the correct morphogenesis of hair bundles in
the inner ear neurosensory cells, whereas the cytoplasmic domain of
the CDH23 interacts with other hair bundle proteins, including
myosin VIIA and harmonin (15,16).
However, an increasing number of genetic variants of different
genes have been reported, such as USH1D and DFNB12 (17–20).
Certain gene variants occur with a distinct frequency in different
populations. For example, two variants were screened in patients
with a Swedish background (17),
whereas others were present in Japanese or Korean subjects
(18–20). The aim of the present study was to
elucidate the role of the DFNB12 CDH23 mutation in Chinese
patients with non-syndromic hearing loss. The nature of diverse
CDH23 mutations and their resulting phenotypes in hearing
loss were studied using a comprehensive strategy that included DNA
sequencing and bioinformatics, which provided information regarding
hearing loss in Asian populations.
Materials and methods
Study subjects
The study was approved by the Ethics Committee of
the First Hospital of Jilin University (Changchun, China) and all
participants provided written informed consent prior to being
enrolled in the study. The study enrolled two patients (II:1 and
II:2) with severe sensorineural hearing loss and their parents (I:1
and I:2). Clinical information and peripheral blood samples were
acquired for DNA extraction and DNA sequencing. The patients were
diagnosed using audiologic tests according to the American College
of Medical Genetics and Genomics guidelines (21). Linkage analysis was not carried out
owing to the small number of participants.
DNA extraction and DNA sequencing
Genomic DNA was extracted from the blood samples
using a TIANamp Blood DNA Kit (TianGen Biotech Co., Ltd.) and
quantified using a Nanodrop 2000 spectrophotometer (Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol.
Subsequently, the genomic DNA samples were subjected to DNA
sequencing focusing on nine gene loci: GJB2:35delG, 176del16,
235delC, 299delAT, GJB3:538C>T, SLC26A4:2168A>G,
IVS7-2A>G, mitochondrial 12SrRNA:1494C>T and 1555A>G,
using the LuxScan 10K Microarray Scanner (CapitalBio Corporation).
A customized exome enrichment kit (MyGenostics, Inc.) was designed
to identify genes that induce hearing loss. The prepared samples
were sequenced using Illumina NextSeq 500 (Illumina, Inc.).
DNA libraries were constructed using the genomic DNA
samples with a Library Preparation kit (MyGenostics, Inc.),
according to the Illumina platform requirements. The enzymatic
fragmentation of genomic DNA samples was end-repaired, and adapters
were added. The length of the prepared libraries was ~350–400 bp,
and they were amplified by PCR and analyzed using an Agilent 2100
Bioanalyzer (Agilent technologies Inc.) by MyGenostics, Inc. The
aligned short reads were compared with the human reference genome
(hg19) (http://hgdownload.cse.ucsc.edu) using the
Burrows-Wheeler Aligner (version 0.7.15; http://bio-bwa.sourceforge.net/). The quality control
assessment and variant calling were processed using the Genome
Analysis Toolkit (GATK; version 3.6; http://www.broadinstitute.org/gatk/) following GATK
best practices (22) and annotated
using ANNOVAR (version 2016-02-01), similarly to a previous study
(23). Potential damaging gene
variants were screened for quality against the 1000 Genomes public
variant databases (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp), the Single
Nucleotide Polymorphism database (http://www.ncbi.nlm.nih.gov/projects/SNP), Sorting
Intolerant from Tolerant (SIFT; http://sift.bii.a-star.edu.sg) and Polyphen-2
(http://genetics.bwh.harvard.edu/pph2). Considering the
deduction of an autosomal recessive pattern of inheritance, only
the variants that were homozygous or compound heterozygous were
selected as candidates.
The variants that met the search criteria were
validated by PCR-based Sanger sequencing using the following
primers: CDH23 exon 9, forward 5′-TACAACGTGCCCCATTCTGC-3′,
reverse 5′-GTCTAGGTTCAGCTATGCCGTT-3′; CDH23 exon 43, forward
5′-TCTTCCGTGGTGGTCCATTT-3′, reverse 5′-AGATGCCTACTGGCTCTCCTT-3′.
The TransStart Taq DNA Polymerase kit (Beijing TransGen Biotech
Co., Ltd.) was used according to the manufacturers protocol and the
thermocycling conditions were as follows: 94°C for 2 min, followed
by 35 cycles of 94°C for 15 sec, 55°C for 30 sec, 68°C for 2 min.
The amplification product was between 200 and 400 bp. The data were
analyzed using Lasergene-SeqMan software (Lasergene 9; DNAStar,
Inc.) and the DNA sequences were compared with the sequence of
CDH23 (NM_022124.5) and corresponding CDH23 protein
sequences (NP_071407.4). In total, three sequences were compared to
establish a consensus.
In silico analysis
Pathogenicity of the missense variants of
CDH23 was predicted by using SIFT and Polyphen-2. The
pathogenicity was determined using the following criteria: i) The
variants were considered as ‘likely damaging or damaging’ by either
SIFT or Polyphen-2; ii) the variants occurred at the residues that
were highly conserved among various species; iii) the variants
occurred in affected subjects in homozygous or compound
heterozygous phenotypes.
A 3D molecular structure model of the extracellular
domain was generated using I-TASSER (http://zhanglab.ccmb.med.umich.edu/I-TASSER) and
compared with the confidence score and TM-score using the predicted
models. The confidence score is used to estimate the quality of the
predicted models using I-TASSER (https://zhanglab.ccmb.med.umich.edu/I-TASSER/) It is
calculated based on the significance of threading template
alignments and the convergence parameters of the structure assembly
simulations. TM-scores are a recently proposed scale for measuring
the structural similarity between two structures (24). The DeepView/Swiss-PdbViewer
(http://spdbv.vital-it.ch) was used for structural
analysis and visualization.
Results
Patient clinicopathological
features
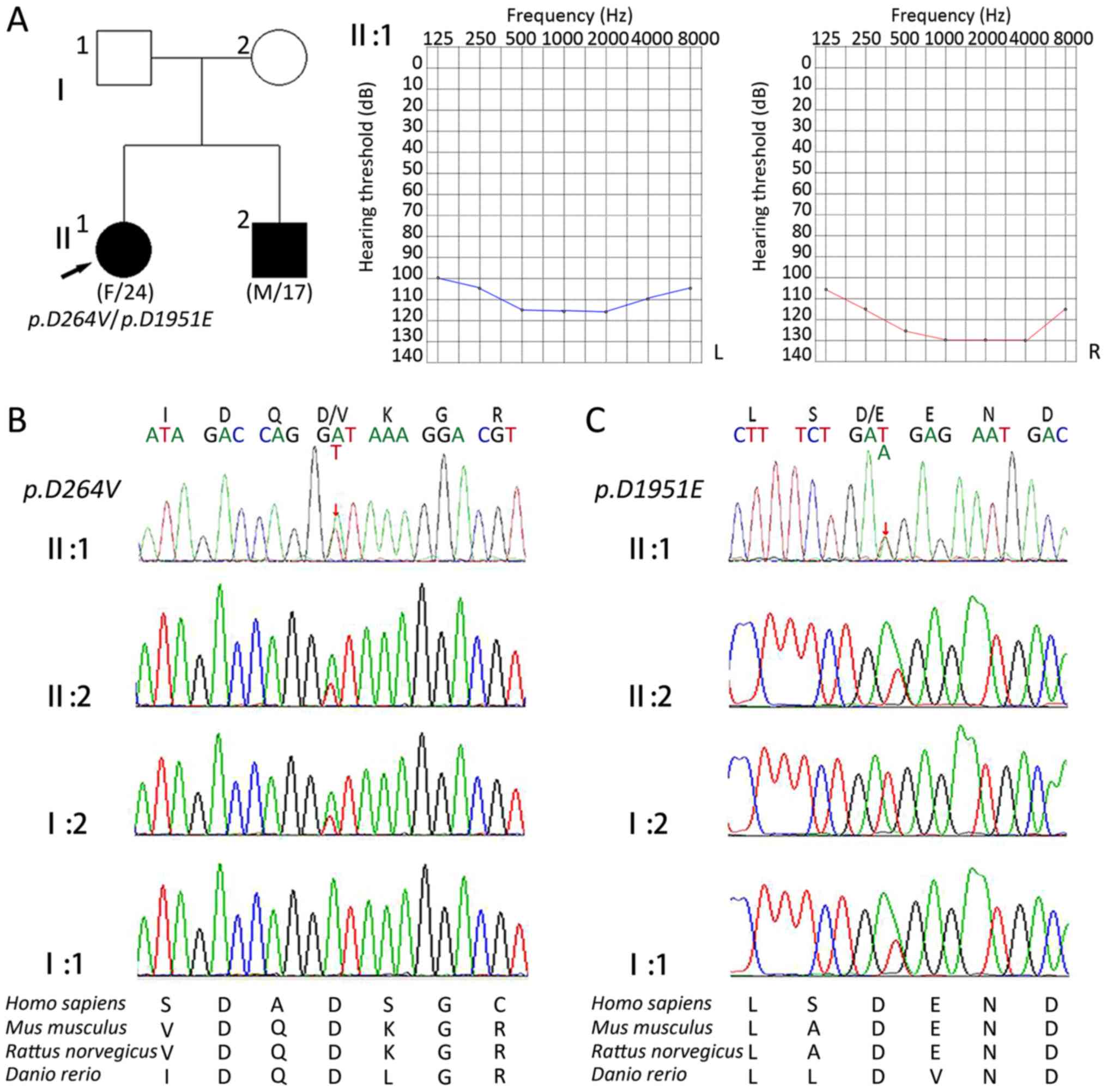
The proband was a 24-year-old female (II:1) that had
bilateral hearing loss, and the sibling of the proband was a
17-year-old male (II:2) with severe deafness. Pure tone audiometry
revealed profound sensorineural hearing loss at all frequencies.
The patients' parents (I:1 and I:2) had normal hearing, and all
other family members exhibited no impairment in movement,
vestibular or visual functions.
Genetic analysis
Nine gene loci (GJB2:35delG, 176del16, 235delC,
299delAT, GJB3:538C>T, SLC26A4:2168A>G, IVS7-2A>G,
mitochondrial 12SrRNA:1494C>T and 1555A>G) were analyzed and
none was detected in either of the siblings or in the mother.
However, mitochondrial 12SrRNA 1555A>G was detected in the
father. Following exclusion of variants of these four genes by
Sanger sequencing, the target region capture sequence was used to
resolve the genetic causes. Compound heterozygous variants in
CDH23 from the proband were identified following DNA
sequencing as one of the two shifts caused by a transition
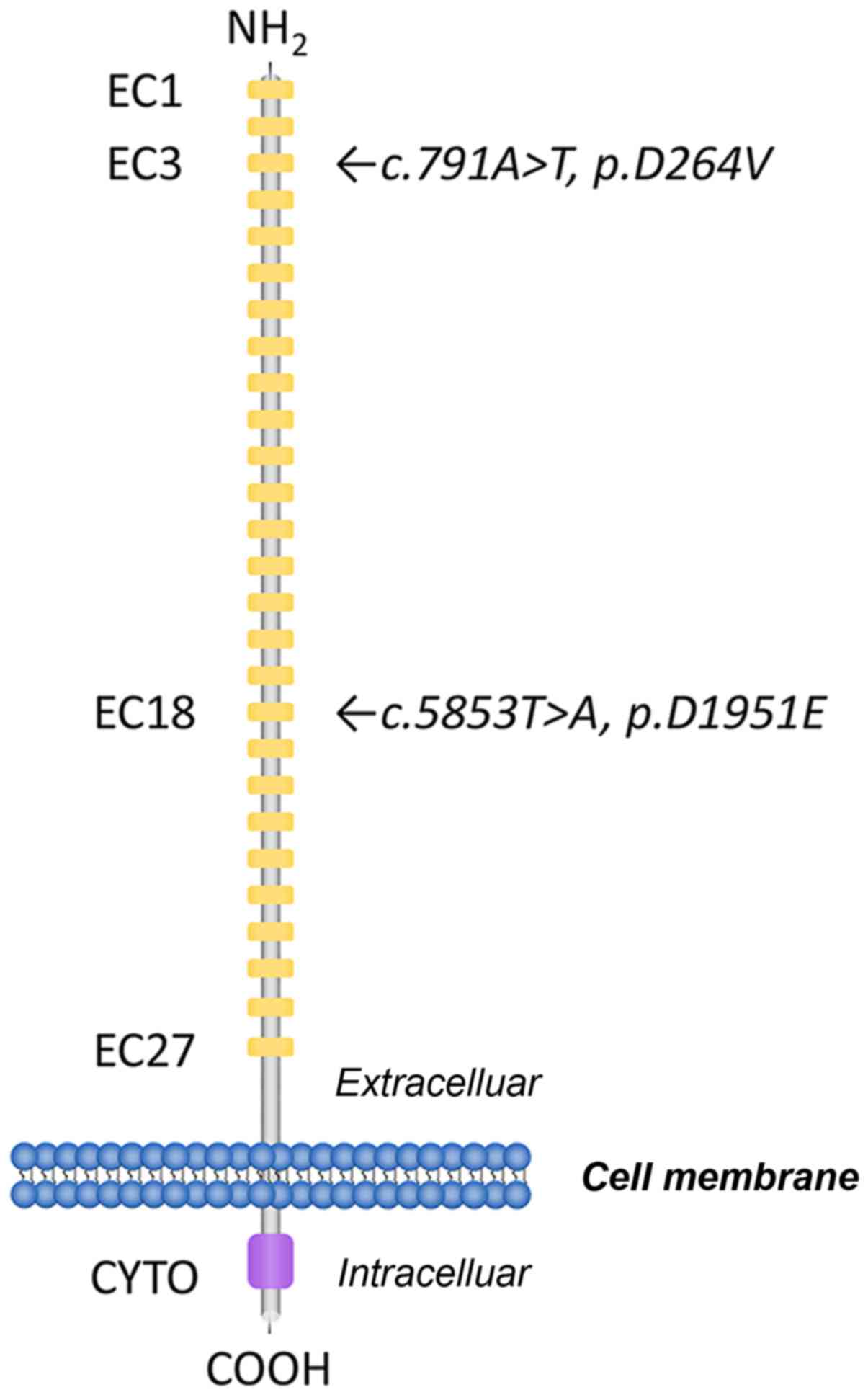
CDH23:c.791A>T in exon 9 (Fig. 1). This transition resulted in the
substitution of a valine with an aspartic acid at position 264
(p.D264V). The other variant identified was
CDH23:c.5853T>A in exon 43. This resulted in the
substitution of an aspartic acid for a glutamic acid residue at
position 1951 (p.D1951E; Fig.
1).
The associated regions of CDH23 were
subsequently sequenced in other family members (Fig. 2) and it was revealed that
p.D264V also occurred in the patients' mother.
p.D1951E was considered to be a paternal gene, and multiple
sequence alignment of the two affected EC domains revealed that the
amino acids residues were well-conserved among various species,
including Homo sapiens, Mus musculus, Rattus norvegicus and
Danio rerio (Fig. 2C).
In silico analysis
Two novel mutations of CDH23
(c.791A>T and c.5853T>A) were identified in the
coding region, both of which were missense variants. The data were
subsequently scored on the compatibility with the 1000 Genome
sequence database, the Single Nucleotide Polymorphism database,
SIFT and PolyPhen2 score (Table I)
and according to the scores, the two gene variants were damaged,
which was indicated by the comparison with SIFT and Polyphen-2.
 | Table I.Pathogenicity prediction of cadherin
23 variants associated with hearing loss using in silico
analysis. |
Table I.
Pathogenicity prediction of cadherin
23 variants associated with hearing loss using in silico
analysis.
| Nucleotide
change | Amino acid
change | Exon | Domain | dbSNP | 1000 Genomes | Polyphen-2
scorea | SIFT
scoreb |
|---|
|
c.791A>T | p.D264V | 9 | 3 | – | N/A | 0.998 | 0.00 |
|
c.5853T>A |
p.D1951E | 43 | 18 | – | N/A | 0.997 | 0.00 |
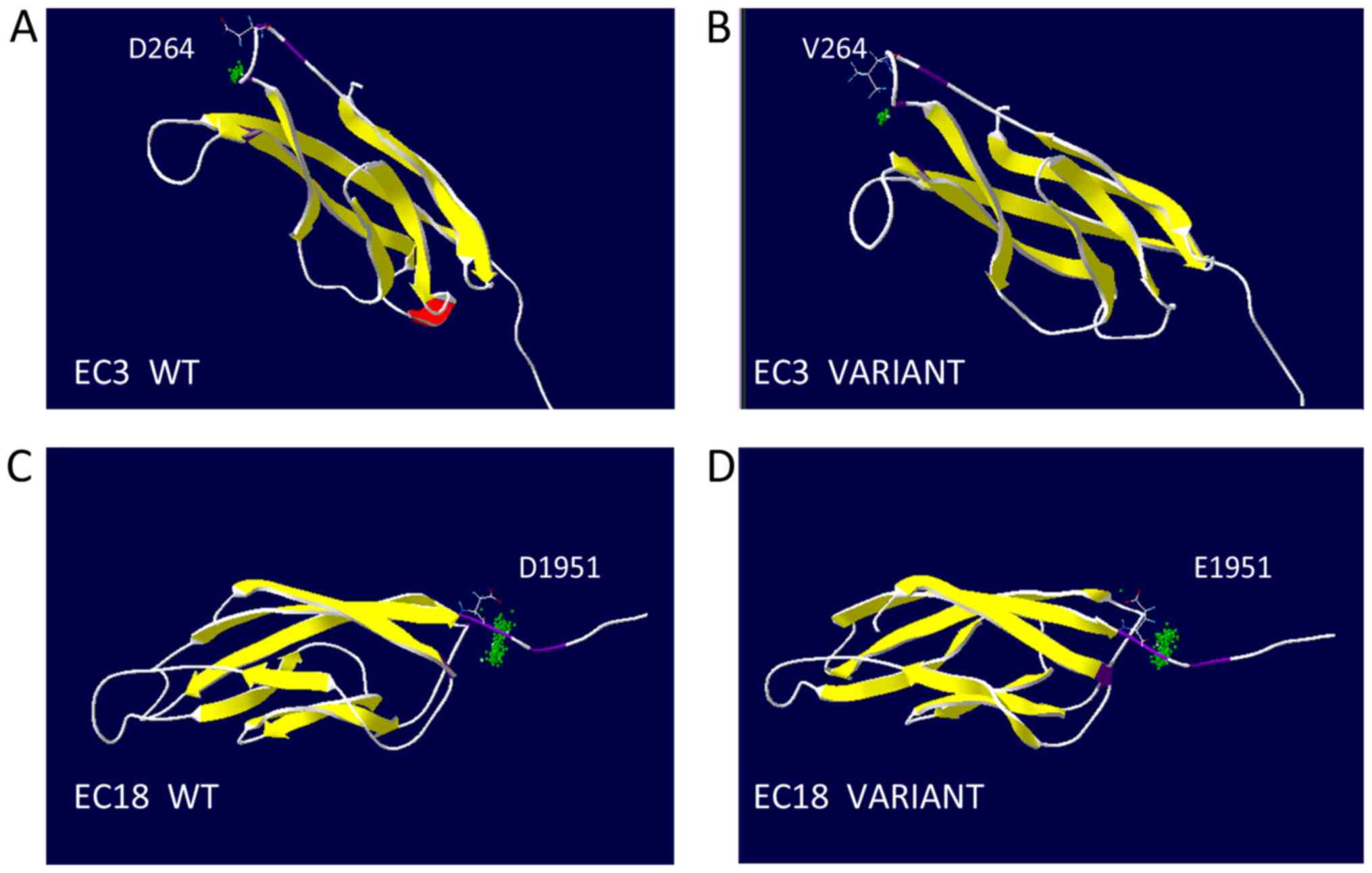
3D models of these variants were created and
revealed that affected EC domains (containing aspartic acid
residues) contained the highly conserved calcium-binding motifs,
EC3 and EC18, in the location of the linking cadherin repeats
(Fig. 3). The substitution at
residue 264 was the second aspartic acid residue of the DXD
calcium-binding motif in EC3. The substitution at residue 1,951 was
the first aspartic acid residue of DXNDN calcium-binding motif in
EC18. Modeling analysis of the mutated CDH23 used in the present
study revealed that these CDH23 variants were related to DXD and
DXNDN motifs. Consistently, DNFB12 mutations were more likely to be
involved in the highly conserved regions, such as LDRE, DXNDN and
DXD, compared with USH1D (Fig.
4).
Discussion
Previous studies have indicated that CDH23
expression is vital for the bundle of hair cells in the cochlea and
serves an important role in the establishment and maintenance of
the proper organization of the stereocilia (25,26).
CDH23 forms a functional network with USH1C, USH1G,
CDH23 and myosin VIIA, and participates in
mechanotransduction in auditory processing; therefore, CDH23
variants may contribute to hearing loss. The present study
identified two novel missense mutations of CDH23,
c.791A>T and c.5853T>A, in the coding region.
The gene mutation spectrum can be distinguished
across different ethnic origins. For example, GJB2:c.35delG
occurs more frequently in people of Caucasian origin, whereas
GJB2:c.167delT is prevalent in Ashkenazi Jews;
GJB2:p.R143W was found in an African farmer, and
GJB2:c.235delC is commonly reported in East Asian countries
(27,28). The frequency of the gene mutations
involved in hearing loss is due to the founder effect (1). In the present study, nine gene loci
(GJB2:35delG, 176del16, 235delC, 299delAT, GJB3:538C>T,
SLC26A4:2168A>G, IVS7-2A>G, mitochondrial
12SrRNA:1494C>T and 1555A>G) were assessed, but
none of these mutations was identified in either affected sibling.
However, using the target region captured DNA sequencing
technology, two novel pathological CDH23 variants were
identified, which may be possible pathological variants and
missense CDH23 mutations; this was in agreement with
previous studies (11,17,18).
A previous study demonstrated nonsense, frame shift or splice
mutations of CDH23 in patients with USH1D (17). Aspartic acid is a medium-sized
acidic residue, whereas, valine is hydrophobic. Substitution of the
aspartic acid residues with valine strongly reduced the number of
oxygen atoms binding to calcium, indicating that these variants may
have an effect on CDH23 structure. Thus, this valine
substitution may be pathological in hearing loss. These motifs have
been suggested to be essential for a calcium-binding ability, which
may influence linearization, rigidification and dimerization of
cadherin (29,30).
Calcium ions are usually enclosed by several oxygen
atoms (31), and the absence of
these atoms normally weakens calcium-binding capacity (32). Substitution of the aspartic acid
residues with a valine impaired the interaction of the CDH23
protein either with itself or with other protein, owing to the loss
of oxygen atoms present in aspartic acid. The c.791A>T
mutation involved an amino acid change from an aspartic acid to a
hydrophobic valine (p.D264V), which altered the function of
the CDH23 protein. In addition, the results of Polyphen-2
and SIFT predicted the impact of these changes and indicated that
they may pathogenically affect protein structure or function.
Calcium ions serve a crucial role in the process of adaptation of
mechanical transduction, frequency tuning, neurotransmitter release
and efferent synaptic signaling in the auditory and vestibular
systems (33). A previous study
has also demonstrated that the CDH23 protein associates with
myosin-1c to form a protein complex that is a component of the
mechanotransduction apparatus (34). This suggested that CDH23 may
participate in the activity of mechanically gated ion channels in
hair cells. Missense variants of genes, including CDH23, may
be a common cause of disease, depending on the mutation site within
the protein. In the present study, two novel CDH23 missense
mutations were identified, and the results revealed that there may
be an association between gene mutation and disease pathogenicity.
In addition, in silico analysis predicted pathogenic
variants of a given gene (35),
which was used to demonstrate that the two CDH23 variants
may contribute to the hearing impairment of the two patients.
Previous studies have revealed that CDH23 variants are
associated with hearing loss in Chinese patients. For example, a
homozygous c.5985C>A (p.Y1995X) variant has been
linked to USH1D (36). Patients
from the Chinese Jiangxi province were heterozygous with
p.E1006K and p.D1663V in CDH23 (37). Regarding the CDH23 spectrum
in the Japanese, the frequency was 3.7% (including the heterozygous
phenotype) in the hearing loss population and 5.7% (including the
heterozygous phenotype) among the recessive inherited cases
(19). Two out of 13 (15%) Korean
families with non-syndromic hearing loss were affected by
CDH23 mutations in an autosomal recessive pattern and three
of 93 patients exhibited a heterozygous phenotype (20). Consequently, some CDH23
variants frequently occurred in certain ethnic origins (19,20)
and, based on these data, it was speculated that CDH23 variants may
be an important cause of hearing loss in Asian populations
(Table II). Although the precise
pathogenesis and molecular mechanism of hearing have not been
defined, the data from the present study and the literature
indicate that DFNB12 CDH23 variants may impact the Chinese
population with non-syndromic hearing loss.
| Table II.Frequency of cadherin 23 variants in
hearing loss. |
Table II.
Frequency of cadherin 23 variants in
hearing loss.
|
|
|
|
| Number of
patients |
|
|
|---|
|
|
|
|
|
|
|
|
|---|
| Author, year | Country | Nucleotide
change | Amino acid
change | -/- | Compound +/- | +/- | No. patients | (Refs.) |
|---|
| Chai et al,
2015 | China |
c.5985C>A |
p.Y1995X | 1 | N/A | N/A | 945 | (36) |
| Lu et al,
2014 | China |
c.3016G>A |
p.E1006K | N/A | 2 | N/A | 9 | (37) |
|
|
|
c.4988A>T |
p.D1663V |
|
|
|
|
|
| Miyagawa et
al, 2012 | Japan |
c.719C>T | p.P240L | N/A | 2 | 1 | 1,396 | (19) |
|
|
|
c.902G>A | p.R301Q | N/A | 3 | N/A |
|
|
|
|
|
c.2866G>A | p.E956K | N/A | 1 | 2 |
|
|
|
|
|
c.4103C>T |
p.T1368M | N/A | 1 | N/A |
|
|
|
|
|
c.4249C>T |
p.R1417W | 1 | N/A | 2 |
|
|
|
|
|
c.4877A>C |
p.D1626A | N/A | 1 | N/A |
|
|
|
|
|
c.5147A>C |
p.Q1716P | N/A | 3 | N/A |
|
|
|
|
|
c.6085C>T |
p.R2029W | 2 | 2 | 6 |
|
|
|
|
|
c.6861T>G |
p.N2287K | N/A | 2 | N/A |
|
|
|
|
|
c.7312G>A |
p.E2438K | N/A | 1 | N/A |
|
|
| Woo et al,
2014 | Korea |
c.719C>T | p.P240L | N/A | 3 | N/A | 16 | (20) |
|
|
|
c.1025A>G | p.N342S | N/A | 2 | N/A |
|
|
|
|
|
c.4783G>A | p.1595K | N/A | 1 | N/A |
|
|
| Present study | China |
c.791A>T | p.D264V | N/A | 2 | N/A | 2 |
|
|
|
|
c.5853T>A |
p.D1951E | N/A | 2 | N/A |
|
|
In conclusion, the present study identified two
novel CDH23 mutations in one Chinese family with autosomal
recessive non-syndromic hearing loss using the target region
captured DNA sequencing. However, the exact frequency requires
further investigation as only data from two generations of this
family were obtained. Future studies with a larger cohort of
patient samples are needed to confirm the present results, which
may be used for early detection of hearing loss. In addition,
target region captured DNA sequencing technology may be effective
for detecting causative gene variants in patients with no mutations
in GJB2, GJB3, SLC26A4 and mitochondrial 12SrRNA.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
All authors made substantial contributions to
conception and design, or acquisition of data, or analysis and
interpretation of data. TX, HL and SY performed the experiments. TX
drafted the article. PW and WZ critically revised the article. ZW
reviewed the submitted version of manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
the First Hospital of Jilin University (Changchun, China) and all
participants provided written informed consent prior to being
enrolled in the study.
Patient consent for publication
Written informed consent was obtained from the
parent of the patient for the publication of this case report.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Shearer AE, Hildebrand MS and Smith RJH:
Hereditary hearing loss and deafness overview. Adam MP, Ardinger
HH, Pagon RA, Wallace SE, Bean LJH, Stephens K and Amemiya A:
GeneReviews® [Internet]. Seattle (WA). University of
Washington; Seattle: 1993-2019
|
|
2
|
GB D, 2015 Disease, Injury Incidence and
Prevalence Collaborators: Global, regional, and national incidence,
prevalence, and years lived with disability for 310 diseases and
injuries, 1990–2015: A systematic analysis for the global burden of
disease study 2015. Lancet. 388:1545–1602. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cunningham LL and Tucci DL: Hearing loss
in adults. N Engl J Med. 377:2465–2473. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Olusanya BO, Neumann KJ and Saunders JE:
The global burden of disabling hearing impairment: A call to
action. Bull World Health Organ. 92:367–373. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Smith RJH, Bale JF Jr and White KR:
Sensorineural hearing loss in children. Lancet. 365:879–890. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Paludetti G, Conti G, Di Nardo W, De Corso
E, Rolesi R, Picciotti PM and Fetoni AR: Infant hearing loss: From
diagnosis to therapy official report of XXI conference of italian
society of pediatric otorhinolaryngology. Acta Otorhinolaryngol
Ital. 32:347–370. 2012.PubMed/NCBI
|
|
7
|
Friedman TB and Griffith AJ: Human
nonsyndromic sensorineural deafness. Annu Rev Genomics Hum Genet.
4:341–402. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hilgert N, Smith RJ and Van Camp G:
Function and expression pattern of nonsyndromic deafness genes.
Curr Mol Med. 9:546–564. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Morton NE: Genetic epidemiology of hearing
impairment. Ann N Y Acad Sci. 630:16–31. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Funamura JL: Evaluation and management of
nonsyndromic congenital hearing loss. Curr Opin Otolaryngol Head
Neck Surg. 25:385–389. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bork JM, Peters LM, Riazuddin S, Bernstein
SL, Ahmed ZM, Ness SL, Polomeno R, Ramesh A, Schloss M,
Srisailpathy CR, et al: Usher syndrome 1D and nonsyndromic
autosomal recessive deafness DFNB12 are caused by allelic mutations
of the novel cadherin-like gene CDH23. Am J Hum Genet. 68:26–37.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bolz H, von Brederlow B, Ramirez A, Bryda
EC, Kutsche K, Nothwang HG, Seeliger M, del C-Salcedó Cabrera M,
Vila MC, Molina OP, et al: Mutation of CDH23, encoding a new member
of the cadherin gene family, causes Usher syndrome type 1D. Nat
Genet. 27:108–112. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rowlands TM, Symonds JM, Farookhi R and
Blaschuk OW: Cadherins: Crucial regulators of structure and
function in reproductive tissues. Rev Reprod. 5:53–61. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Di Palma F, Holme RH, Bryda EC,
Belyantseva IA, Pellegrino R, Kachar B, Steel KP and Noben-Trauth
K: Mutations in Cdh23, encoding a new type of cadherin, cause
stereocilia disorganization in waltzer, the mouse model for Usher
syndrome type 1D. Nat Genet. 27:103–107. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Boëda B, El Amraoui A, Bahloul A, Goodyear
R, Daviet L, Blanchard S, Perfettini I, Fath KR, Shorte S, Reiners
J, et al: Myosin VIIa, harmonin and cadherin 23, three usher I gene
products that cooperate to shape the sensory hair cell bundle. EMBO
J. 21:6689–6699. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Siemens J, Kazmierczak P, Reynolds A,
Sticker M, Littlewood-Evans A and Müller U: The usher syndrome
proteins cadherin 23 and harmonin form a complex by means of
PDZ-domain interactions. Proc Natl Acad Sci USA. 99:14946–14951.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Astuto LM, Bork JM, Weston MD, Askew JW,
Fields RR, Orten DJ, Ohliger SJ, Riazuddin S, Morell RJ, Khan S, et
al: CDH23 mutation and phenotype heterogeneity: A profile of 107
diverse families with usher syndrome and nonsyndromic deafness. Am
J Hum Genet. 71:262–275. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wagatsuma M, Kitoh R, Suzuki H, Fukuoka H,
Takumi Y and Usami S: Distribution and frequencies of CDH23
mutations in Japanese patients with non-syndromic hearing loss.
Clin Genet. 72:339–344. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Miyagawa M, Nishio SY and Usami S:
Prevalence and clinical features of hearing loss patients with
CDH23 mutations: A large cohort study. PLoS One. 7:e403662012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Woo HM, Park HJ, Park MH, Kim BY, Shin JW,
Yoo WG and Koo SK: Identification of CDH23 mutations in Korean
families with hearing loss by whole-exome sequencing. BMC Med
Genet. 15:462014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Alford RL, Arnos KS, Fox M, Lin JW, Palmer
CG, Pandya A, Rehm HL, Robin NH, Scott DA, Yoshinaga-Itano C, et
al: American college of medical genetics and genomics guideline for
the clinical evaluation and etiologic diagnosis of hearing loss.
Genet Med. 16:347–355. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, et
al: The Genome Analysis Toolkit: a MapReduce framework for
analyzing next-generation DNA sequencing data. Genome research.
20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Huang XF, Xiang P, Chen J, Xing DJ, Huang
N, Min Q, Gu F, Tong Y, Pang CP, Qu J and Jin ZB: Targeted exome
sequencing identified novel USH2A mutations in Usher syndrome
families. PloS One. 8:e638322013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang Y and Skolnick J: Scoring function
for automated assessment of protein structure template quality.
Proteins. 57:702–710. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Siemens J, Lillo C, Dumont RA, Reynolds A,
Williams DS, Gillespie PG and Müller U: Cadherin 23 is a component
of the tip link in hair-cell stereocilia. Nature. 428:950–955.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Müller U: Cadherins and
mechanotransduction by hair cells. Curr Opin Cell Biol. 20:557–566.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tsukada K, Nishio S and Usami S: Deafness
Gene Study Consortium. A large cohort study of GJB2 mutations in
Japanese hearing loss patients. Clin Genet. 78:464–470. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Van Laer L, Coucke P, Mueller RF,
Caethoven G, Flothmann K, Prasad SD, Chamberlin GP, Houseman M,
Taylor GR, Van de Heyning CM, et al: A common founder for the
35delG GJB2 gene mutation in connexin 26 hearing impairment. J Med
Genet. 38:515–518. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nagar B, Overduin M, Ikura M and Rini JM:
Structural basis of calcium-induced E-cadherin rigidification and
dimerization. Nature. 380:360–364. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Angst BD, Marcozzi C and Magee AI: The
cadherin superfamily: Diversity in form and function. J Cell Sci.
114:629–641. 2001.PubMed/NCBI
|
|
31
|
Strynadka NC and James MN: Crystal
structures of the helix-loop-helix calcium-binding proteins. Annu
Rev Biochem. 58:951–998. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chakrabarti P: Systematics in the
interaction of metal ions with the main-chain carbonyl group in
protein structures. Biochemistry. 29:651–658. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yamoah EN, Lumpkin EA, Dumont RA, Smith
PJ, Hudspeth AJ and Gillespie PG: Plasma membrane Ca2+-ATPase
extrudes Ca2+ from hair cell stereocilia. J Neurosci. 18:610–624.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Holt JR, Gillespie SK, Provance DW, Shah
K, Shokat KM, Corey DP, Mercer JA and Gillespie PG: A
chemical-genetic strategy implicates myosin-1c in adaptation by
hair cells. Cell. 108:371–381. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the american college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chai Y, Chen D, Sun L, Li L, Chen Y, Pang
X, Zhang L, Wu H and Yang T: The homozygous p.V37I variant of GJB2
is associated with diverse hearing phenotypes. Clin Genet.
87:350–355. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lu Y, Zhou X, Jin Z, Cheng J, Shen W, Ji
F, Liu L, Zhang X, Zhang M, Cao Y, et al: Resolving the genetic
heterogeneity of prelingual hearing loss within one family:
Performance comparison and application of two targeted next
generation sequencing approaches. J Hum Genet. 59:599–607. 2014.
View Article : Google Scholar : PubMed/NCBI
|