Introduction
Acute lung injury (ALI) is a progressive syndrome,
which is the primary cause of morbidity and mortality in various
types of disease, including acute pulmonary embolism, heart/lung
transplantation, and ventilator and pulmonary thrombosis-induced
lung injury (1–3). An acute inflammatory response and an
increase in pulmonary microvascular permeability are common
pathological changes observed in ALI, which cause a loss of
epithelial barrier function and induce an influx of protein-rich
edema fluid (4). ALI combined with
severe injury or infection in patients induces the activation of
pro-inflammatory signaling pathways and overexpression of
inflammatory mediators, which causes a systemic inflammatory
response that eventually leads to multi-organ failure, severe shock
and death (5). Sepsis syndrome is
a primary cause of ALI and is associated with its development
(6). Inflammation and sepsis
caused by Gram-negative bacteria are primarily due to
lipopolysaccharide (LPS) release from the bacterial outer membrane
(7). LPS-induced ALI is
characterized by lung edema, disruption of endothelial and
epithelial barrier integrity, extensive neutrophil infiltration and
the release of inflammatory mediators (8). A previous study demonstrated that LPS
serves a crucial role in regulating acute damage to the respiratory
epithelium during sepsis (9).
Numerous natural or synthetic chemicals have been extensively
reported to impair the inflammatory responses in sepsis-induced
ALI, including phillyrin, kaempferol and farrerol (5,10,11).
Phillyrin has a protective effect against LPS-induced ALI by
reducing the release of pro-inflammatory cytokines, including tumor
necrosis factor-α (TNF-α), interleukin (IL)-1β and IL-6 (5). However, the transfer of these
findings to clinical treatment remains limited. It is therefore
necessary to determine a novel therapeutic intervention in the
treatment of sepsis-induced ALI.
Ulinastatin, a urinary trypsin inhibitor (UTI), is a
multivalent Kunitz-type serine protease inhibitor found in urine,
plasma and most organs (12). UTI
can reduce the release of various types of inflammatory factor from
neutrophils and inhibit neutrophil elastase activity (13). A previous study reported that UTI
is used to treat numerous severe diseases, such as pancreatitis,
shock and disseminated intravascular coagulation (14–16).
Furthermore, UTI can reduce the inflammatory response in an animal
model of ALI (17). However, the
anti-inflammatory mechanism of UTI in ALI remains unclear. A
previous study revealed that the NF-κB and MAPK signaling pathways
have crucial roles in the development of LPS-induced ALI (10). UTI may therefore attenuate
ALI-induced inflammation responses via the MAPK and NF-κB signaling
pathways.
The present study established a rat model of
LPS-induced ALI and investigated the therapeutic effect of UTI on
rats with ALI. In addition, the molecular mechanism underlying the
anti-inflammatory effects of UTI on ALI were further explored.
Materials and methods
Animals
Adult healthy male Sprague-Dawley (SD) rats (250–300
g) were obtained from the Experimental Animal Center of Zhongshan
Hospital, Fudan University. All animals were housed in a
light-controlled (12:12 light-dark cycle), temperature-controlled
(24±2°C) and humidity-controlled (50%) room with free access to
food and water. Rats underwent an acclimatization period of at
least 1 week prior to experimental manipulation. All experimental
protocols were approved by the Guide for the Care and Use of
Laboratory Animals of the National Institutes of Health (IRB
approval no. 15-000387). Sodium pentobarbital (50 mg/kg
intraperitoneal) was used for anesthesia before each operation, and
all efforts were made to minimize animal suffering. Sacrifice was
performed by intraperitoneal injection of sodium pentobarbital
overdose (200 mg/kg) followed by cervical dislocation, and
mortality was confirmed by observation (18).
Rat model of LPS-induced ALI
Male SD rats were randomly divided into four groups
(n=6 per group) as follows: i) Control group, which received only
normal saline; ii) UTI group, which received 20,000 U/kg UTI; iii)
LPS group, which received 5 mg/kg LPS by intratracheal
instillation; and iv) LPS + UTI group, which received LPS plus
20,000 U/kg UTI (Guangdong Techpool Bio-pharma Co, Ltd.). LPS from
Escherichia coli 055:B5 (5 mg/kg to induce ALI;
Sigma-Aldrich) and normal saline were intratracheally administered
as previously described (19).
Then, 30 min following LPS administration, rats received UTI
(20,000 U/kg) by intraperitoneal injection. Theses doses of drugs
were determined based on preliminary experiments and previous
studies (20–22). Rats were sacrificed with sodium
pentobarbital by intraperitoneal injection (200 mg/kg;
Sigma-Aldrich) 24 h following LPS administration, according to the
Guide for Care and Use of Laboratory Animals. The bronchoalveolar
lavage fluid (BALF) samples were subsequently collected and used
for cell counting. The lower lung tissues were fixed with 4%
paraformaldehyde at 4°C for 24 h. Other parts of the lung tissue
were stored at −80°C until further use.
Histological analysis
Lung tissue samples were fixed in 4%
paraformaldehyde neutral buffer solution for 24 h, dehydrated in a
graded ethanol series (30–100%; v/v), embedded in paraffin, and
sliced into 5-µm thick sections. Following hematoxylin (2 g/l; 10
min at room temperature) and eosin (5 g/l; 30 sec at room
temperature) (H&E) staining, pathological changes in the lung
tissues were observed using light microscopy (magnification, ×200;
BXFM; Olympus Corporation). Histological scoring was performed
blindly according to the following four parameters: i) Presence of
hemorrhage; ii) infiltration; iii) alveolar congestion, aggregation
of neutrophils in the airspace or vessel wall; and iv) thickness of
the alveolar wall/hyaline membrane formation. The severity of
inflammation was graded from 0 to 1 as previously described
(22).
Assessment of Evans blue
extravasation
Evans blue dye (Sigma-Aldrich; Merck KGaA)
extravasation was used to evaluate the pulmonary barrier
permeability. Evans blue dye (20 mg/kg in 1 ml saline) was injected
into the tail vein of rats 30 min before anesthesia in all groups.
Normal saline was immediately injected into the right ventricle
until it effused clear fluid from the left atrium at the end of the
experiment. The right middle lung lobe was collected and dried at
60°C for 72 h. Tissues were sliced and incubated with formamide (3
ml/100 mg; Sigma-Aldrich; Merck KGaA) at room temperature for 24 h,
samples were then centrifuged at 500 × g for 10 min (4°C) for Evans
blue dye extraction. The optical density of the centrifugal
supernatant (Evans blue extravasation) was measured using
spectrophotometry at 620 nm. Evans Blue dye concentration was
determined via a standard curve and was expressed as µg of Evans
Blue dye per 100 mg of lung tissue.
Lung wet/dry ratio determination
The severity of pulmonary edema was evaluated by
assessing the lung wet/dry ratio (W/D ratio). The inferior lobe of
the right lung was removed, rinsed briefly in PBS, blotted and then
weighed to obtain the wet weight. The tissue was then placed in an
incubator at 60°C for 72 h to obtain the dry weight. The W/D ratio
was eventually calculated.
Assessment of lung MPO activity
It has been reported that MPO activity can be used
as a marker of neutrophil activation (23). Thus, MPO activity was examined in
the present study. Lung tissues were collected 24 h following LPS
administration and homogenized in HEPES (pH 8.0) containing 0.5%
cetyltrimethylammonium bromide and underwent three freeze-thaw
cycles. Subsequently, the homogenate was centrifuged (12,000 × g)
at 4°C for 30 min. An ELISA kit (cat. no. ab105136; Abcam) was used
to determine the myeloperoxidase (MPO) activity. Absorbance was
measured at 460 nm on a microplate reader (Model 550; Bio-Rad
Laboratories, Inc.). The concentration of total protein from the
lung tissues was determined using the bicinchoninic acid method.
The MPO activity of the homogenates supernatants was expressed as
units per gram of total protein (U/g).
BALF and cell counting
Following anesthesia with sodium pentobarbital (50
mg/kg intraperitoneal), rats were sacrificed by intraperitoneal
injection of sodium pentobarbital overdose (200 mg/kg) followed by
cervical dislocation and BALF was collected by performing at least
three lavages with 1 ml PBS (pH 7.2) following cannulation of the
upper part of the trachea. The fluid recovery rate was >90%.
BALF samples were kept on ice and BALF was centrifuged (700 × g) at
4°C for 5 min. The supernatants were stored at −80°C for analysis
of cytokine concentrations. Subsequently, 50 µl PBS was used to
resuspend the cell pellet and BALF cells were stained with 0.4%
trypan blue dye (Invitrogen; Thermo Fisher Scientific, Inc.) at
room temperature for 3 min and total cells were counted under a
light microscope. BALF cells were fixed using 4% paraformaldehyde
at 4°C for 24 h on slides and stained with 0.5% H&E for 30 sec
at room temperature for differential random counting (24). Subsequently, total cells,
neutrophils and leukocytes were counted in a double-blind manner
using a hemocytometer, according to morphology (25).
Cytokine measurement
Following BALF centrifugation for 4 min at 3,000 ×
g, BALF supernatant was collected and stored at −80°C prior to
measuring cytokines. The following ELISA assay kits were used:
TNF-α (cat. no. BMS607-3FIVE; Thermo Fisher Scientific, Inc.), IL-6
(cat. no. RAB0308; Sigma-Aldrich; Merch KGaA), IL-1β (cat. no.
RAB0274; Sigma-Aldrich; Merck KGaA) and interferon-γ (IFN-γ; cat.
no. MIF00; R&D Systems, Inc.), according to the manufacturer's
instructions.
Extraction of lung nuclear
proteins
Lung tissues were collected from each group and
immediately stored in liquid nitrogen until homogenization. Frozen
lung tissue (50 mg) was homogenized using a Precellys 24 bead-based
tissue homogenizer in 0.5 ml ice-cold buffer A [1.5 mM
MgCl2, 10 mM HEPES (pH 7.9), 10 mM KCl, 0.5 mM
dithiothreitol (DTT), 1% Nonidet P-40, 0.1 mM Na2EDTA,
0.5 mM phenylmethylsulfonyl fluoride (PMSF), 125 µg/ml aprotinin,
25 µg/ml pepstatin A and 0.5 µg/ml leupeptin]. Cell debris were
removed by centrifugation at 700 × g for 30 sec. Supernatant was
then collected and placed on ice for 5 min, and centrifuged for 10
min at 5,000 × g. Cytoplasmic proteins were collected in the
supernatant and the pellet was resuspended in cold buffer B (0.5
ml) [1.5 mM MgCl2, 20 mM HEPES (pH 7.9), 0.42 mM NaCl,
0.5 mM DTT, 20% glycerol, 0.5 mM PMSF, 125 µg/ml aprotinin, 25
µg/ml pepstatin A and 0.5 µg/ml leupeptin], and kept on ice for 30
min. The nuclear fraction from the supernatant was obtained by
centrifugation at 12,000 × g for 2 min, the pellet was collected
and stored at-70°C.
Western blotting
The lower lobe of the right lung was collected and
frozen at −80°C. Total proteins from lung tissues were isolated
using RIPA buffer (Sigma Aldrich; Merch KGaA) and their
concentrations were measured using bicinchoninic acid assay.
Proteins (20 µg) were separated on 12% SDS-PAGE, and transferred
onto polyvinylidene difluoride membranes (EMD Millipore). Following
blocking with 5% non-fat milk at 4°C overnight, membranes were
incubated at 4°C overnight with primary antibodies against ERK1/2
(cat. no. 353097; 1:1,000), p-ERK1/2 (cat. no. 8146; 1:1,000), JNK
(cat. no. 2855; 1:1,000), phosphorylated (p-)JNK (cat. no. 2055;
1:1,000), p38 MAPK (cat. no. 16005; 1:1,000), p-p38 MAPK (cat. no.
9102; 1:1,000), p-NF-κB (Ser536)-specific p65 (cat. no. 3031;
1:1,000), IκBα (cat. no. 3025; 1:1,000), p-(Ser32/Ser36)-specific
IκBα (cat. no. 9246; 1:1,000), which were all obtained from Cell
Signaling Technology, Inc. β-actin (cat. no. 2832; 1:1,000;
Sigma-Aldrich; Merck KGaA), incubated overnight at 4°C, was used as
an internal control. Membranes were then incubated with horseradish
peroxidase-conjugated secondary antibody (cat. no. ab205719;
1:2,000; Abcam) for 1 h at room temperature. The protein bands were
measured using the ECLPlus Detection System (GE Healthcare) and
analyzed using AlphaImager software version 2000 (ProteinSimple).
Experiments were repeated at least three times, and the values
obtained for the relative intensity were used for statistical
analysis.
Statistical analysis
All statistical analysis was conducted using SPSS
14.0 software (SPSS, Inc.). Data are presented as the mean ±
standard deviation. Statistical analyses were evaluated by
Student's t-test (comparison between two groups) or one way ANOVA
to make multiple-group comparisons, followed by the post hoc
Tukey's test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Effects of UTI treatment on rats with
LPS-induced ALI
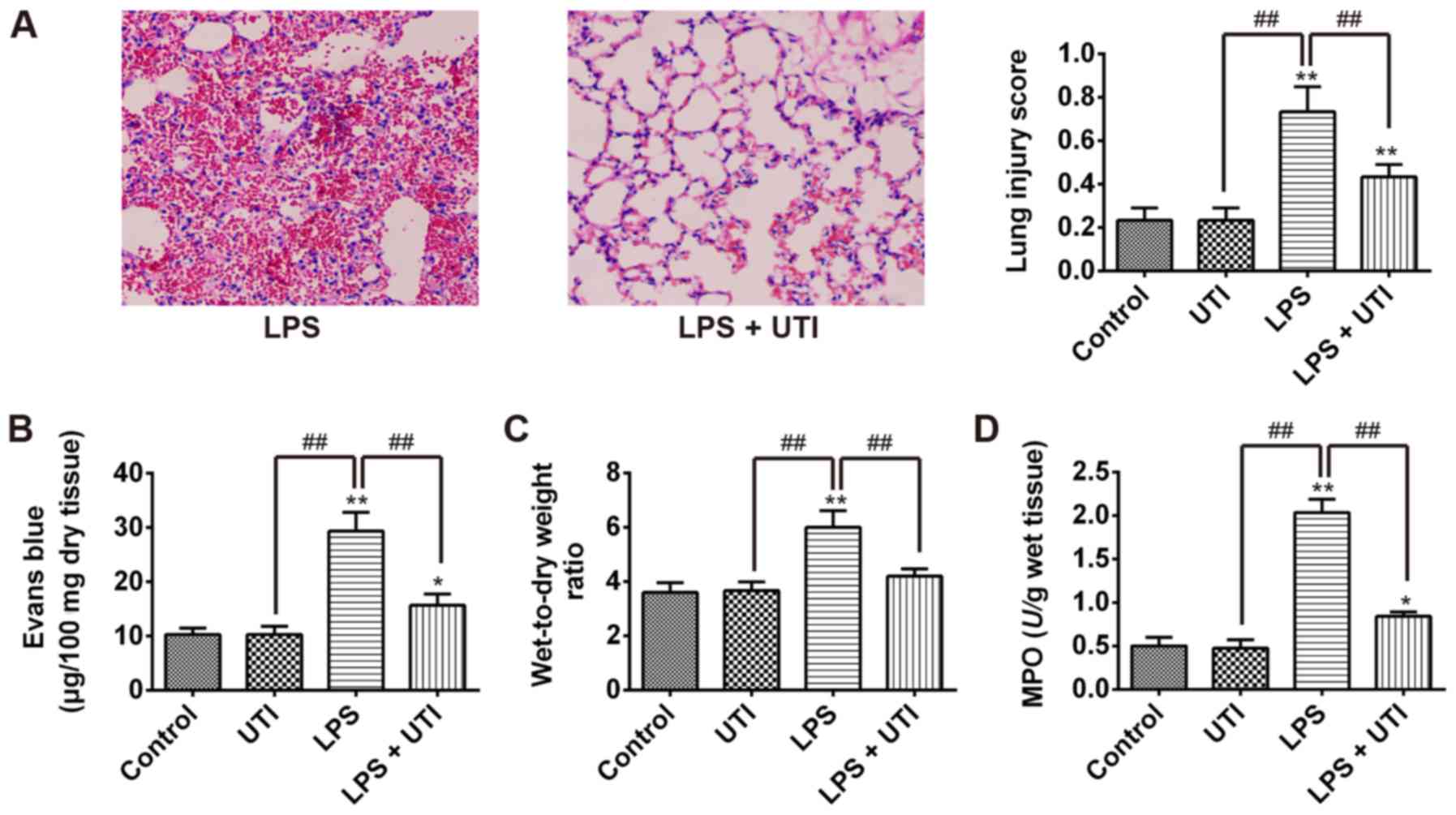
To evaluate the potential of UTI to treat ALI, the
rat model of LPS-induced ALI treated with 20,000 U/kg UTI was
established. The results demonstrated that LPS induced lung
inflammatory responses, including thickening of the alveolar walls
and notable interstitial infiltration of inflammatory cells;
however, UTI treatment distinctly reversed these phenomena
(Fig. 1A). Furthermore, an Evans
blue dye assay was performed to evaluate the severity of lung
vascular leakage. As presented in Fig.
1B, Evans blue dye extravasation from the lung vasculature was
significantly increased in the LPS group compared with the control
group (P<0.01). However, treatment with UTI significantly
reduced Evans blue dye extravasation compared with the LPS group
(P<0.01; Fig. 1B). To further
evaluate the effect of UTI on LPS-induced lung edema, pulmonary
edema was assessed by calculating the lung W/D ratio. Rats who
received LPS by intratracheal instillation exhibited a higher W/D
ratio compared with the controls. However, UTI administration
significantly decreased the W/D ratio in the LPS + UTI group
compared with the LPS group (P<0.01; Fig. 1C). Furthermore, MPO activity is
considered a key indicator of neutrophil migration into the lung
(26). Therefore, the MPO activity
was used to assess the accumulation of activated neutrophils in the
lung tissues. The results revealed that MPO activity in the LPS
group was significantly higher compared with the control group;
however, MPO activity in LPS + UTI group was significantly lower
than in the LPS group (P<0.01; Fig.
1D). In addition, the lung W/D ratio and MPO activity exhibited
no differences in the lung tissues between the control and UTI
groups. These data indicated that UTI administration may attenuate
LPS-induced ALI in rats.
UTI reduces inflammatory cytokine
expression in rats with LPS-induced ALI
Upregulation of inflammatory cytokines is an
important feature of ALI. Therefore, the levels of TNF-α, IL-6,
IL-1β and IFN-γ were determined in the BALF by ELISA. The TNF-α,
IL-6, IL-1β and IFN-γ levels in the BALF were significantly
increased in the LPS group compared with the control group
(P<0.01; Fig. 2). However, UTI
treatment significantly decreased the TNF-α, IL-6, IL-1β and IFN-γ
levels compared with the LPS group (P<0.01; Fig. 2). These results suggested that UTI
may reduce TNF-α, IL-6, IL-1β and IFN-γ expression in rats with
LPS-induced ALI.
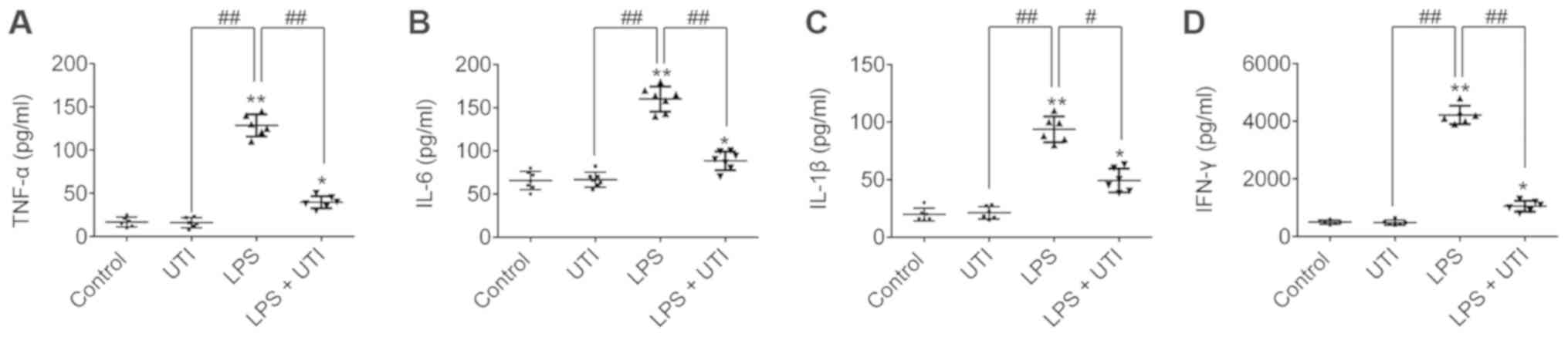 | Figure 2.UTI reduces inflammatory cytokine
expression in rat LPS-induced ALI. Rats were sacrificed at 24 h
following LPS administration, and BALF was collected. The (A)
TNF-α, (B) IL-6, (C) IL-1β and (D) IFN-γ levels in BALF were
measured by ELISA. Data are presented as the mean ± standard
deviation (n=6 rats/group). *P<0.05, **P<0.01 vs. control;
##P<0.01. ALI, acute lung injury; BALF,
bronchoalveolar lavage fluid; IFN, interferon; IL, interleukin;
LPS, lipopolysaccharide; MPO, myeloperoxidase; UTI, urinary trypsin
inhibitor; TNF, tumor necrosis factor. |
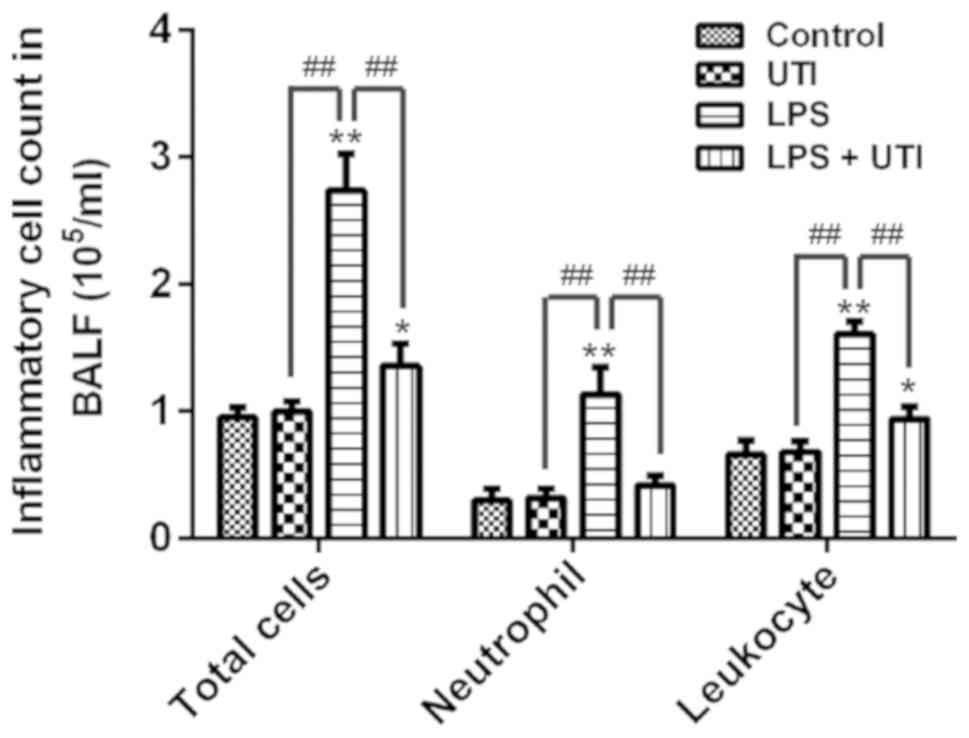
UTI reduces cell counts in BALF
Neutrophils are the main inflammatory cells found in
ALI, and serve important roles in the development of ALI (25). To further investigate the
anti-inflammatory effects of UTI, the total cells, neutrophils and
leukocytes were measured in BALF. The results demonstrated that
total cells, neutrophils and leukocytes were significantly
increased in BALF following LPS administration compared with the
control group (P<0.01; Fig. 3).
However, UTI treatment significantly decreased the total cells,
neutrophils and leukocytes in BALF from the LPS + UTI group. These
data indicated that UTI may reduce total cells, neutrophils and
leukocytes in the lungs of rats with LPS-induced ALI.
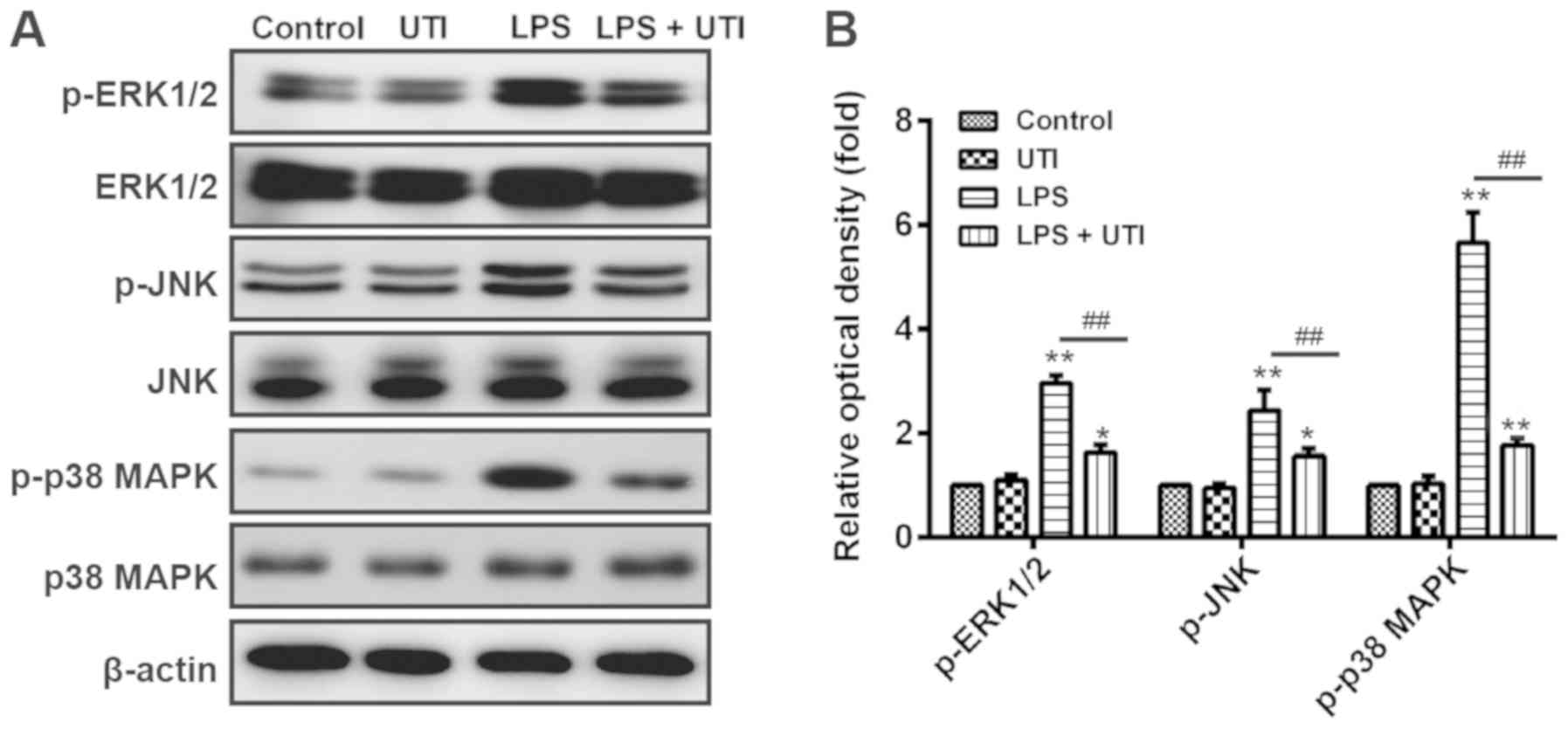
UTI inhibits the MAPK signaling
pathway in rats with LPS-induced ALI
MAPK signaling pathways serve crucial roles in
LPS-induced ALI pathogenesis. Notably, it was reported that LPS
stimulation can result in the phosphorylation of MAPKs, including
in the ERK, JNK and p38 MAPK pathways (27). A previous study revealed that UTI
inhibits LPS-induced TNF-α and subsequent IL-1β and IL-6 secretion
by macrophages, via suppression of MAPK signaling pathways,
including JNK, ERK1/2 and p38 in vitro (28). UTI may therefore have an
anti-inflammatory effect in rats with LPS-induced ALI through
repression of MAPK signaling pathways. To further investigate this
mechanism, the rats from the present study were intraperitoneally
injected with UTI following LPS administration, and western blot
analyses were conducted to detect the expression of ERK1/2, JNK and
p38 MAPK in lung tissues. As presented in Fig. 4, LPS administration significantly
increased the expression of p-ERK1/2, p-JNK and p-p38 MAPK in the
LPS group compared with the control group (P<0.01). Conversely,
p-ERK1/2, p-JNK and p-p38 MAPK levels were significantly decreased
in the lung tissues of rats with LPS-induced ALI that were treated
with UTI. However, there was no significant difference between the
UTI group and the control group. These results indicated that UTI
may exert an anti-inflammatory effect on LPS-induced ALI by
suppressing the MAPK signaling pathways.
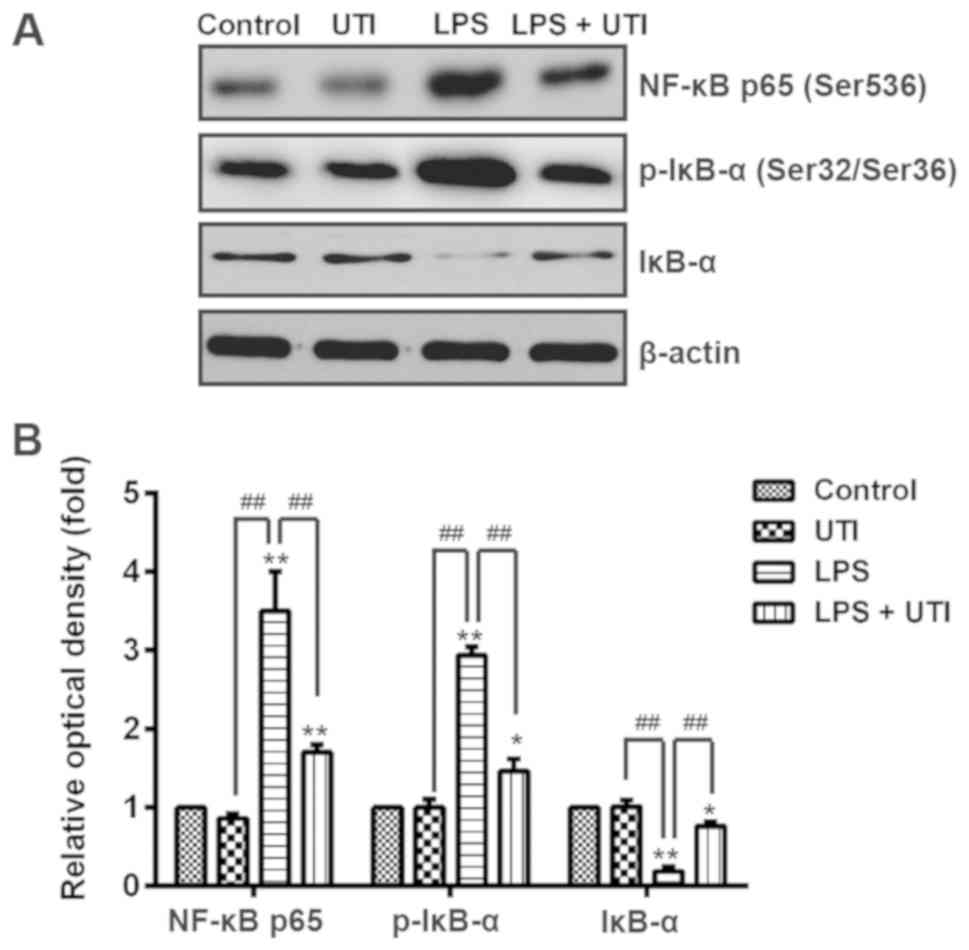
UTI inhibits the NF-κB signaling
pathway in rats with LPS-induced ALI
NF-κB p65 has a crucial role in the inflammatory
response (10). To further explore
the anti-inflammatory mechanism of UTI in LPS-induced ALI, the
effect of UTI on NF-κB activation in lung tissues was evaluated by
western blotting. The results demonstrated that NF-κB expression in
the nuclear extracts of the LPS group was significantly higher
compared with that in the control group; however, UTI treatment
significantly decreased the NF-κB p65 protein level in nuclear
extracts from the LPS + UTI group compared with the LPS group
(P<0.01; Fig. 5). Conversely,
LPS significantly inhibited IκB-α expression in cytosolic extracts
compared with the control group, whereas UTI treatment
significantly induced IκB-α expression in the LPS + UTI group
compared with the LPS group (P<0.01; Fig. 5). Furthermore, the results
demonstrated that LPS significantly upregulated p-IκB-α compared
with the control group, whereas UTI treatment inhibited p-IκB-α
expression in the LPS + UTI group compared with the LPS group
(P<0.01; Fig. 5). These results
suggested that UTI may suppress the NF-κB signaling pathway by
blocking IκB-α degradation in the lungs of rats with LPS-induced
ALI.
Discussion
The present study investigated the protective
effects of UTI in rats with LPS-induced ALI and explored the
molecular mechanism of the anti-inflammatory effects of UTI in ALI.
The results demonstrated that UTI treatment reduced pathological
changes, and decreased lung vascular leakage, pulmonary edema and
MPO activity in lung tissues from rats with LPS-induced ALI.
Conversely, UTI reduced the levels of inflammatory cytokine
expression, neutrophils and leukocytes in BALF from rats with
LPS-induced ALI. In addition, the results revealed that the
anti-inflammatory effect of UTI on LPS-induced ALI may be mediated
by NF-κB and MAPK signaling pathway suppression.
LPS, which is a main component of the outer membrane
of Gram-negative bacteria, induces the upregulation of numerous
inflammatory cytokines that are considered principal components in
sepsis-induced ALI (29). A model
of LPS-induced ALI was therefore established by infusing LPS (5
mg/kg) into the rat trachea. The results revealed significant lung
injury following LPS administration, particularly histopathological
changes and increases in vascular leakage, the W/D ratio and MPO
activity, which was consistent with previous studies (29,30).
Conversely, UTI treatment reversed the histopathological changes,
and decreased vascular permeability, the W/D ratio and MPO activity
in rats with LPS-induced ALI. These results suggested that UTI
treatment may attenuate LPS-induced ALI in rats.
ALI is closely associated with various inflammatory
mediators. Numerous studies have reported that TNF-α, IL-6 and
IL-1β are the most important inflammatory mediators in the early
development of inflammation (31–34).
A previous study demonstrated that a complex network of
inflammatory cytokines serves a crucial role in amplifying,
mediating and perpetuating the lung injury process (35,36).
Many studies have also revealed that levels of the inflammatory
cytokines TNF-α, IL-6, IL-1β and IFN-γ are increased in rats with
LPS-induced ALI (22,37,38).
UTI has been identified as an innate anti-inflammatory regulator.
Clinically, UTI is used for the treatment of acute circulatory
disorders, pancreatitis, and for the regulation of hemodynamic
stability during surgical stress (22). Furthermore, UTI acted as an
anti-inflammatory agent in an infant piglet model by inhibiting
inflammatory markers, including IL-10, TNF-α and neuron-specific
enolase (39). Consistent with
previous reports, the results from the present study also
demonstrated that the TNF-α, IL-6, IL-1β and IFN-γ levels were
significantly increased in the BALF from rats with LPS-induced ALI.
Conversely, UTI treatment significantly reduced the levels of these
inflammatory mediators compared with the LPS group. These results
suggested that UTI may have a protective effect on LPS-induced ALI
by inhibiting the secretion of the inflammatory cytokines TNF-α,
IL-6, IL-1β and IFN-γ.
Neutrophils arrive quickly at an infection site and
are considered the first line of defense against invading
microorganisms (40). Neutrophils
are predominant inflammatory cells in ALI and have key roles in the
development of most cases of ALI (41). Multiple inflammatory mediators are
able to activate neutrophils and macrophages during LPS-induced
inflammation, including TNF-α and IL-8 (5,42).
Neutrophils are subsequently attracted to the inflammation site by
chemotactic factors (43,44), and invade the inflamed endothelial
tissue via adhesion molecules, including L-selectin on neutrophils
and P-selectin and E-selectin on endothelial cells (45–47).
In the present study, total cells, neutrophils and leukocytes were
significantly increased in BALF from rats with LPS-induced ALI;
however, UTI treatment reduced the amount of these cells in BALF.
These data indicated that the anti-inflammatory effect of UTI on
LPS-induced ALI may be due to neutrophil and leukocyte
repression.
NF-κB, which is a DNA binding protein, can modulate
the transcriptional regulation of numerous genes. A number of
cellular stimuli, including LPS, TNF-α, reactive oxygen species and
ultraviolet light, are able to active NF-κB (48). The activated NF-κB pathway acts as
a trigger that initiates an inflammatory cascade, which results in
the upregulation of many pro-inflammatory cytokines (49). In addition, NF-κB regulates the
expression of these cytokines through binding with the consensus
sequence of their enhancer/promoter regions (50–52).
The main form of NF-κB is a heterodimer (NF-κB p50 and NF-κB p65),
which is localized in the cytoplasm and binds to the inhibitory
proteins of the IκB family (28).
NF-kB pathway activation mainly depends on the IκB kinase (IKKγ)
complex, which consists of a regulatory subunit (IKK) and catalytic
subunits (IKKα and IKKβ), and on IκBα as its downstream substrate.
In unstimulated cells, the heterodimer NF-κB p50 and NF-κB p65
binds to IκBα. Phosphorylated IKKα and IKKβ promote the
phosphorylation of IκBα following stimulation. Subsequently, the
ubiquitin proteasome pathway degrades phosphorylated IκBα, which
causes NF-κB p50/NF-κB p65 dimer phosphorylation and translocation,
eventually leading to gene transcription (53,54).
A previous study demonstrated that NF-κB is activated via MAPKs,
including SAPK/JNK, p38 MAPK and ERK1/2. MAPKs are considered to be
evolutionarily conserved and respond to stress by activating signal
transduction during a phosphorylation cascade from cytoplasmic to
nuclear targets (55,56).
To explore the underlying mechanisms of the
inhibitory effect of UTI on cytokine production, the influence of
UTI on the NF-κB and MAPK signaling pathways was evaluated in the
present study. Increasing evidence indicates that the expression of
pro-inflammatory mediators is tightly modulated at the
transcriptional level via the MAPK (ERK, JNK and p38) and NF-κB
signaling pathways (57–59). A previous study demonstrated that
activation of JNK and p38 results in sepsis-induced organ injury,
whereas JNK and p38 repression improves survival in septic mice
(60). The results from the
present study demonstrated that LPS administration resulted in
p-ERK1/2, p-JNK and p-p38 MAPK upregulation in lung tissues.
However, UTI treatment significantly downregulated the
phosphorylation of ERK1/2, JNK and p38 MAPK in lung tissues from
rats with LPS-induced ALI. These data indicated that UTI may have
anti-inflammatory properties in LPS-induced ALI via downregulation
of the MAPK signaling pathways. NF-κB is considered a key factor in
the inflammatory response and is involved in the development of ALI
(61). Activated NF-κB is able to
promote the expression of numerous inflammatory cytokines,
including TNF-α, IL-1 and IL-6, which causes inflammation (60). It has been suggested that NF-κB
activation could be regulated by the phosphorylation of ERK, JNK
and p38 MAPK (62). In particular,
the p38 MAPK pathway has an essential role in the expression of
multiple pro-inflammatory cytokines via activation of NF-κB
(63). The anti-inflammatory
effect of UTI was therefore investigated, notably on the NF-κB
signaling pathway in rats with LPS-induced ALI. The results
demonstrated that UTI treatment may suppress the NF-κB signaling
pathway by reducing the level of NF-κB p65 and blocking IκB-α
degradation in rats with LPS-induced ALI.
In conclusion, the results from the present study
suggested that UTI may exert anti-inflammatory effects in
LPS-induced ALI by alleviating pathological changes, decreasing
lung vascular leakage and pulmonary edema, and reducing lung
inflammatory responses. In addition, UTI affected the
phosphorylation of ERK1/2, JNK, p38 MAPK and NF-κB p65, which
suggested that the inhibitory effect of UTI on pro-inflammatory
cytokine release may be mediated by the MAPK and NF-κB signaling
pathways. These findings indicated that UTI may be used as a
potential therapeutic agent in the treatment of ALI.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Independent
Research Projects of Fudan University (grant no. 20520133491) and
the Talent Training Program-Excellent Youth Program of Zhongshan
Hospital affiliated to Fudan University (grant no.
2015ZSYXQN29).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MJ, HH, SC, YiL, YuL, SP, YZ, LX and DZ performed
the experiments, contributed to data analysis and wrote the
manuscript. MJ, HH, SC and YiL analyzed the data. ZL designed the
study, contributed to data analysis and experimental materials. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by Zhongshan
Hospital, Fudan University Ethics Committees.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ambrosio G and Tritto I: Reperfusion
injury: Experimental evidence and clinical implications. Am Heart
J. 138:S69–S75. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
de Perrot M, Liu M, Waddell TK and
Keshavjee S: Ischemia-reperfusion-induced lung injury. Am J Respir
Crit Care Med. 167:490–511. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yellon DM and Hausenloy DJ: Myocardial
Reperfusion Injury. N Engl J Med. 357:1121–1135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vadász I, Morty RE, Kohstall MG,
Olschewski A, Grimminger F, Seeger W and Ghofrani HA: Oleic acid
inhibits alveolar fluid reabsorption: A role in acute respiratory
distress syndrome? Am J Respir Crit Care Med. 171:469–479. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhong WT, Wu YC, Xie XX, Zhou X, Wei MM,
Soromou LW, Ci XX and Wang DC: Phillyrin attenuates LPS-induced
pulmonary inflammation via suppression of MAPK and NF-κB activation
in acute lung injury mice. Fitoterapia. 90:132–139. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nagase T, Uozumi N, Ishii S, Kume K, Izumi
T, Ouchi Y and Shimizu T: Acute lung injury by sepsis and acid
aspiration: A key role for cytosolic phospholipase A2. 1:42–46.
2000.PubMed/NCBI
|
|
7
|
Ware LB and Matthay MA: The acute
respiratory distress syndrome. N Engl J Med. 342:1334–1349. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Suda K, Tsuruta M, Eom J, Or C, Mui T, Jaw
JE, Li Y, Bai N, Kim J, Man J, et al: Acute lung injury induces
cardiovascular dysfunction: Effects of IL-6 and
budesonide/formoterol. Am J Respir Cell Mol Biol. 45:510–516. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rojas M, Woods CR, Mora AL, Xu J and
Brigham KL: Endotoxin-induced lung injury in mice: Structural,
functional, and biochemical responses. Am J Physiol Lung Cell Mol
Physiol. 288:L333–L341. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen X, Yang X, Liu T, Guan M, Feng X,
Dong W, Chu X, Liu J, Tian X, Ci X, et al: Kaempferol regulates
MAPKs and NF-κB signaling pathways to attenuate LPS-induced acute
lung injury in mice. Int Immunopharmacol. 14:209–216. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ci X, Chu X, Wei M, Yang X, Cai Q and Deng
X: Different effects of farrerol on an OVA-induced allergic asthma
and lps-induced acute lung injury. PLoS One. 7:e346342012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pugia Michael J and Lott John A:
Pathophysiology and diagnostic value of urinary trypsin inhibitors.
Clin Chem Lab Med. 43:1–16. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gando S and Tedo I: Increased neutrophil
elastase release in patients with cardiopulmonary arrest: Role of
elastase inhibitor. Intensive Care Med. 21:636–640. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Inoue K and Takano H: Urinary trypsin
inhibitor as a therapeutic option for endotoxin-related
inflammatory disorders. Expert Opin Investig Drugs. 19:513–520.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Maciejewski R, Burdan F, Burski K, Madej
B, Ziemiakowicz R, Dabrowski A and Wallner G: Selected biochemical
parameters and ultrastructural picture of pancreas due to
Ulinastatin treatment of experimental acute pancreatitis. Exp
Toxicol Pathol. 56:305–311. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Park KH, Lee KH, Kim H and Hwang SO: The
anti-inflammatory effects of ulinastatin in trauma patients with
hemorrhagic shock. J Korean Med Sci. 25:128–134. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rui M, Duan Y, Zhang XH, Wang HL and Wang
DP: Urinary trypsin inhibitor attenuates seawater-induced acute
lung injury by influencing the activities of nuclear factor-ĸB and
its related inflammatory mediators. Respiration. 83:335–343. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hickman DL and Johnson SW: Evaluation of
the aesthetics of physical methods of euthanasia of anesthetized
rats. J Am Assoc Lab Anim Sci. 50:695–701. 2011.PubMed/NCBI
|
|
19
|
Shen W, Gan J, Xu S, Jiang G and Wu H:
Penehyclidine hydrochloride attenuates LPS-induced acute lung
injury involvement of NF-kappaB pathway. Pharmacol Res. 60:296–302.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cao YZ, Tu YY, Chen X, Wang BL, Zhong YX
and Liu MH: Protective effect of Ulinastatin against murine models
of sepsis: Inhibition of TNF-α and IL-6 and augmentation of IL-10
and IL-13. Exp Toxicol Pathol. 64:543–547. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gao C, Li R and Wang S: Ulinastatin
protects pulmonary tissues from lipopolysaccharide-induced injury
as an immunomodulator. J Trauma Acute Care Surg. 72:169–176.
2012.PubMed/NCBI
|
|
22
|
Luo Y, Che W and Zhao M: Ulinastatin
post-treatment attenuates lipopolysaccharide-induced acute lung
injury in rats and human alveolar epithelial cells. Int J Mol Med.
39:297–306. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Feng G, Sun B, Liu H, Liu QH, Zhao L and
Wang TL: EphA2 antagonism alleviates LPS-induced acute lung injury
via Nrf2/HO-1, TLR4/MyD88 and RhoA/ROCK pathways. Int
Immunopharmacol. 72:176–185. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Narasaraju T, Yang E, Samy RP, Ng HH, Poh
WP, Liew AA, Phoon MC, van Rooijen N and Chow VT: Excessive
neutrophils and neutrophil extracellular traps contribute to acute
lung injury of influenza pneumonitis. Am J Pathol. 179:199–210.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Grommes J and Soehnlein O: Contribution of
neutrophils to acute lung injury. Mol Med. 17:293–307. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kremserova S, Perecko T, Soucek K, Klinke
A, Baldus S, Eiserich JP and Kubala L: Lung Neutrophilia in
myeloperoxidase deficient mice during the course of acute pulmonary
inflammation. Oxid Med Cell Longev. 2016:52190562016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee HS, Kang P, Kim KY and Seol GH:
Foeniculum vulgare Mill. Protects against
lipopolysaccharide-induced acute lung injury in mice through
ERK-dependent NF-κB activation. Korean J Physiol Pharmacol.
19:183–189. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu YJ, Ling Q, Zhou XH, Wang Y, Xie HY, Yu
JR and Zheng SS: Urinary trypsin inhibitor attenuates hepatic
ischemia-reperfusion injury by reducing nuclear factor-kappa B
activation. Hepatobiliary Pancreat Dis Int. 8:53–58.
2009.PubMed/NCBI
|
|
29
|
Li W, Qiu X, Jiang H, Zhi Y, Fu J and Liu
J: Ulinastatin inhibits the inflammation of LPS-induced acute lung
injury in mice via regulation of AMPK/NF-κB pathway. Int
Immunopharmacol. 29:560–567. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xie K, Yu Y, Huang Y, Zheng L, Li J, Chen
H, Han H, Hou L, Gong G and Wang G: Molecular hydrogen ameliorates
lipopolysaccharide-induced acute lung injury in mice through
reducing inflammation and apoptosis. Shock. 37:548–555.
2012.PubMed/NCBI
|
|
31
|
Christofidou-Solomidou M, Kennel S,
Scherpereel A, Wiewrodt R, Solomides CC, Pietra GG, Murciano JC,
Shah SA, Ischiropoulos H, Albelda SM and Muzykantov VR: Vascular
immunotargeting of glucose oxidase to the endothelial antigens
induces distinct forms of oxidant acute lung injury: Targeting to
thrombomodulin, but not to PECAM-1, causes pulmonary thrombosis and
neutrophil transmigration. Am J Pathol. 160:1155–1169. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Meduri GU, Kohler G, Headley S, Tolley E,
Stentz F and Postlethwaite A: Inflammatory Cytokines in the BAL of
Patients With ARDS: Persistent elevation over time predicts poor
outcome. Chest. 108:1303–1314. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ito K, Mizutani A, Kira S, Mori M, Iwasaka
H and Noguchi T: Effect of Ulinastatin, a human urinary trypsin
inhibitor, on the oleic acid-induced acute lung injury in rats via
the inhibition of activated leukocytes. Injury. 36:387–394. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ware LB: Pathophysiology of acute lung
injury and the acute respiratory distress syndrome. Semin Respir
Crit Care Med. 27:337–349. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bhatia M and Moochhala S: Role of
inflammatory mediators in the pathophysiology of acute respiratory
distress syndrome. J Pathol. 202:145–156. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Goodman RB, Pugin J, Lee JS and Matthay
MA: Cytokine-mediated inflammation in acute lung injury. Cytokine
Growth Factor Rev. 14:523–535. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rao R, Nagarkatti P and Nagarkatti M:
Staphylococcal eterotoxin B-induced microRNA-155 targets SOCS1 to
promote acute inflammatory lung injury. J Immunol. 82:2971–2979.
2014.
|
|
38
|
Zeng Z, Gong H, Li Y, Jie K, Ding C, Shao
Q, Liu F, Zhan Y, Nie C, Zhu W and Qian K: Upregulation of miR-146a
contributes to the suppression of inflammatory responses in
LPS-induced acute lung injury. Exp Lung Res. 39:275–282. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang X, Xue Q, Yan F, Li L, Liu J, Li S
and Hu S: Ulinastatin as a neuroprotective and anti-inflammatory
agent in infant piglets model undergoing surgery on hypothermic
low-flow cardiopulmonary bypass. Paediatr Anae. 23:209–216. 2013.
View Article : Google Scholar
|
|
40
|
Zhu T, Wang DX, Zhang W, Liao XQ, Guan X,
Bo H, Sun JY, Huang NW, He J, Zhang YK, et al: Andrographolide
protects against LPS-induced acute lung injury by inactivation of
NF-κB. PLoS One. 8:e564072013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cepkova M and Matthay MA: Pharmacotherapy
of acute lung injury and the acute respiratory distress syndrome. J
Intensive Care Med. 21:119–143. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tianzhu Z and Shumin W: Esculin inhibits
the inflammation of LPS-induced acute lung injury in mice via
regulation of TLR/NF-κB pathways. Inflammation. 38:1529–1536. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Reddy RC and Standiford TJ: Effects of
sepsis on neutrophil chemotaxis. Curr Opin Hematol. 17:18–24. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lavoie-Lamoureux A, Moran K, Beauchamp G,
Mauel S, Steinbach F, Lefebvre-Lavoie J, Martin JG and Lavoie JP:
IL-4 activates equine neutrophils and induces a mixed inflammatory
cytokine expression profile with enhanced neutrophil chemotactic
mediator release ex vivo. Am J Physiol Lung Cell Mol Physiol.
299:L472–L482. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Whitley MZ, Thanos D, Read MA, Maniatis T
and Collins T: A striking similarity in the organization of the
E-selectin and beta interferon gene promoters. Mol Cell Biol.
14:6464–6475. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shu HB, Agranoff AB, Nabel EG, Leung K,
Duckett CS, Neish AS, Collins T and Nabel GJ: Differential
regulation of vascular cell adhesion molecule 1 gene expression by
specific NF-kappa B subunits in endothelial and epithelial cells.
Mol Cell Biol. 13:6283–6289. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Akira S, Uematsu S and Takeuchi O:
Pathogen recognition and innate immunity. Cell. 124:783–801. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
DiDonato J, Mercurio F, Rosette C, Wu-Li
J, Suyang H, Ghosh S and Karin M: Mapping of the inducible IkappaB
phosphorylation sites that signal its ubiquitination and
degradation. Mol Cell Biol. 16:1295–1304. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Park GY and Christman JW: Nuclear factor
kappa B is a promising therapeutic target in inflammatory lung
disease. Curr Drug Targets. 7:661–668. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Barnes PJ and Karin M: Nuclear
factor-kappaB: A pivotal transcription factor in chronic
inflammatory diseases. N Engl J Med. 336:1066–1071. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Filgueiras LR Jr, Martins JO, Serezani CH,
Capelozzi VL, Montes MB and Jancar S: Sepsis-induced acute lung
injury (ALI) is milder in diabetic rats and correlates with
impaired NFκB activation. PLoS One. 7:e449872012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Huang X, Tang J, Cai H, Pan Y, He Y, Dai
C, Chen A, Yu X, Chen M, Zou L and Wang L: Anti-inflammatory
effects of monoammonium glycyrrhizinate on
lipopolysaccharide-induced acute lung injury in mice through
regulating nuclear factor-kappa B signaling pathway. Evid Based
Complement Alternat Med. 2015:2724742015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Xia YF, Ye BQ, Li YD, Wang JG, He XJ, Lin
X, Yao X, Ma D, Slungaard A, Hebbel RP, et al: Andrographolide
attenuates inflammation by inhibition of NF-kappa B activation
through covalent modification of reduced cysteine 62 of p50. J
Immunol. 173:4207–4217. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Li Q and Verma IM: NF-kappaB regulation in
the immune system. Nat Rev Immunol. 2:725–734. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Guha M and Mackman N: LPS induction of
gene expression in human monocytes. Cell Signal. 13:85–94. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhang Y, Chen G, Zhong S, Zheng F, Gao F,
Chen Y, Huang Z, Cai W, Li W, Liu X, et al: N-n-butyl haloperidol
iodide ameliorates cardiomyocytes hypoxia/reoxygenation injury by
extracellular calcium-dependent and-independent mechanisms. Oxid
Med Cell Longev. 2013:9123102013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Schuh K and Pahl A: Inhibition of the MAP
kinase ERK protects from lipopolysaccharide-induced lung injury.
Biochem Pharmacol. 77:1827–1834. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Jian MY, Alexeyev MF, Wolkowicz PE,
Zmijewski JW and Creighton JR: Metformin-stimulated AMPK-α1
promotes microvascular repair in acute lung injury. Am J Physiol
Lung Cell Mol Physiol. 305:L844–L855. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Liu S, Feng G, Wang GL and Liu GJ: p38MAPK
inhibition attenuates LPS-induced acute lung injury involvement of
NF-kappaB pathway. Eur J Pharmacol. 584:159–165. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Liu SF and Malik AB: NF-kappa B activation
as a pathological mechanism of septic shock and inflammation. Am J
Physiol Lung Cell Mol Physiol. 290:L622–L645. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Everhart MB, Han W, Sherrill TP, Arutiunov
M, Polosukhin VV, Burke JR, Sadikot RT, Christman JW, Yull FE and
Blackwell TS: Duration and intensity of NF-kappaB activity
determine the severity of endotoxin-induced acute lung injury. J
Immunol. 176:4995–5005. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zhao S, Zhang L, Lian G, Wang X, Zhang H,
Yao X, Yang J and Wu C: Sildenafil attenuates LPS-induced
pro-inflammatory responses through down-regulation of intracellular
ROS-related MAPK/NF-κB signaling pathways in N9 microglia. Int
Immunopharmacol. 11:468–474. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Cho SY, Park SJ, Kwon MJ, Jeong TS, Bok
SH, Choi WY, Jeong WI, Ryu SY, Do SH, Lee CS, et al: Quercetin
suppresses proinflammatory cytokines production through MAP kinases
and NF-kappaB pathway in lipopolysaccharide-stimulated macrophage.
Mol Cell Biochem. 243:153–160. 2003. View Article : Google Scholar : PubMed/NCBI
|