Introduction
Acute lung injury (ALI) and acute respiratory
distress syndrome (ARDS) are caused by multiple factors, which
characterize persistent hypoxemia, neutrophil infiltration,
disseminated alveolar damage, acute inflammatory syndrome and
impaired blood gas barrier (1,2).
Numerous studies have focused on the mechanism and therapy of ALI
(3–5); however, there are still no effective
therapeutic methods in the clinic. In previous years, inflammation
has been considered to serve a key role in the development of ALI.
However, specific mechanisms have not yet been fully elucidated.
Inflammatory factors, including nuclear factor-κB (NF-κB), tumor
necrosis factor-α (TNF-α) and interleukin (IL)-1β, could increase
the damage to endothelial and epithelial cells (6,7).
Therefore, it is important to further study the mechanism of
inflammation in ALI.
The inflammasome is a protein complex that includes
the receptor protein apoptosis-associated speck-like protein
containing CARD (ASC) and effector molecules, including
pre-caspase-1 and pro-caspase-1. The inflammasome can promote the
maturation of pro-IL-1β by activating caspase-1. Subsequently,
IL-1β is released to the extracellular environment to participate
in inflammation, injury and other processes (8,9).
Receptor proteins include NOD like receptors (NLRs), including
NLRP1 and NLRP3, and the interferon-inducible p200-proteins,
including absent in melanoma 2. Different receptor proteins can be
activated by different endogenous or exogenous stimuli. The NLRP3
inflammasome is the most studied inflammasome and has been
demonstrated to be activated by numerous factors, including
Listeria, Aeromonas, ATP and insoluble crystals, including
uric acid crystal, silica and asbestos (10,11).
It has been reported that in ALI, pulmonary fibrosis, chronic
obstructive pulmonary disease, asthma and other lung diseases, the
NLRP3 inflammasome serves a key role in inflammation (12). Studies have demonstrated that the
NLRP3 inflammasome can be activated through the toll-like receptor
4 signaling pathway and participates in inflammatory injury in
ventilator-induced lung injury; the ventilator induced lung injury
was markedly alleviated after NLRP3-knockout (13,14).
However, to the best of the authors' knowledge, the mechanism of
NLRP3 inflammasome activation remains unknown in ALI.
Hypoxia-inducible factor-1α (HIF-1α) is a
transcription factor that is widely expressed in the body. Under
normal conditions, HIF-1α can be degraded via the
ubiquitin-proteasome pathway. However, during hypoxia the
degradation of HIF-1α is inhibited; therefore, it accumulates and
is transferred to the nucleus to promote expression of its target
genes (15,16). The lung tissue is in a state of
persistent hypoxia and the expression of HIF-1α is significantly
increased (17–19). It has been demonstrated that HIF-1α
participates in inflammation and promotes the expression of
inflammatory factors, including TNF-α, IL-6 and IL-1β, in ALI
(20,21). In single-stranded RNA
viruses-induced inflammatory reactions, HIF-1α has been reported to
activate the NLRP3 inflammasome, induce the activation of caspase-1
and then convert inactive pro-IL-1β to IL-1β in human THP-1 myeloid
macrophages (22). However,
whether HIF-1α can regulate the activation of the NLRP3
inflammasome and the potential function of HIF-1α in ALI remain
unknown.
Therefore, the aim of the present study was to
investigate whether HIF-1α can regulate the activation of the NLRP3
inflammasome and its potential mechanism in bleomycin (BLM)-induced
ALI.
Materials and methods
Main reagents
BLM was purchased from Selleck Chemicals.
Anti-HIF-1α (1:500; cat. no. BS3514) primary rabbit monoclonal
antibody was obtained from BioWorld Technology, Inc. NF-κB p65
(1:1,000; cat. no. sc-8008), ASC (1:500; cat. no. sc-22514) and
caspase-1 (1:500; cat. no. sc-56036) primary mouse monoclonal
antibodies were purchased from Santa Cruz Biotechnology, Inc.
Anti-NLRP3 (1:1,000; cat. no. ab210491) rabbit monoclonal antibody
and anti-β-actin (1:2,000; cat. no. ab179467) primary mouse
monoclonal antibody were purchased from Abcam. RIPA buffer, BCA
protein assay kit, SDS-PAGE gel preparation kit, 4.0%
paraformaldehyde, Triton X-100, DAPI and fluorescein isothiocyanate
(FITC)-conjugated goat anti-rabbit IgG (1:200; cat. no. A0562) were
obtained from Beyotime Institute of Biotechnology.
Cell culture and transfection
A549 cells and rat type II alveolar cells (RLE-6TN)
were purchased from the American Type Culture Collection. A549
cells were cultured in F-12K medium (Genom Biotech Pvt., Ltd.) with
10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.)
and 1% antibiotics (100 U/ml penicillin and 100 µg/ml
streptomycin). RLE-6TN cells were grown in DMEM/F-12 (HyClone; GE
Healthcare) with 10% FBS and 1% antibiotics (100 U/ml penicillin
and 100 µg/ml streptomycin). Both cell lines were grown at 37°C in
a 5% carbon dioxide incubator. HIF-1α small interfering RNA (siRNA)
and NF-κB siRNA were designed and generated by Shanghai Genepharma
Co., Ltd. The nucleotides sequences of the HIF-1α, NF-κB p65
(abbreviated as NF-κB) and siControl siRNAs are presented in
Table I. Both siRNAs were
transfected into A549 and RLE-6TN cells using Lipofectamine™ 2000
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. After 1 h, A549 cells were treated with
BLM at a concentration of 120 µM and RLE-6TN cells were treated
with BLM at a concentration of 40 µM for 24 h, according to a
previous study (23).
 | Table I.The sequences of HIF-1α and NF-κB
siRNAs. |
Table I.
The sequences of HIF-1α and NF-κB
siRNAs.
| Species | Gene | Sense | Antisense |
|---|
| Homo
sapiens | HIF-1α |
5′GCCGAGGAAGAACUAUGAATT3′ |
5′UUCAUAGUUCUUCCUCGGCTT3′ |
|
| NF-κB |
5′GGACAUAUGAGACCUUCAA3′ |
5′UUGAAGGUCUCAUAUGUCC3′ |
|
| siControl |
5′UUCUCCGAACGUGUCACGU3′ |
5′ACGUGACACGUUCGGAGAA3′ |
| Rattus
Norvegicus | HIF-1α |
5′GGGCCGUUCAAUUUAUGAATT3′ |
5′UUCAUAAAUUGAACGGCCCTT3′ |
|
| NF-κB |
5′AAGAAGCACAGAUACCACCAA3′ |
5′UUGGUGGUAUCUGUGCUUCUU3′ |
|
| siControl |
5′UUCUCCGAACGUGUCACGUTT3′ |
5′ACGUGACACGUUCGGAGAATT3′ |
Western blotting
Total proteins were extracted from cells with RIPA
buffer. Concentrations of the total proteins were determined using
a BCA protein assay kit. The total protein samples (4 µg/µl) were
separated via 10% SDS-PAGE g, transferred to PVDF membranes,
blocked with 5% non-fat milk in TBS with 0.1% Tween 20 (TBST) at
37°C for 1.5 h and incubated with relevant primary antibodies
overnight at 4°C. The secondary antibodies were then incubated with
the PVDF membrane for 60 min at room temperature. After the
membranes were washed with TBST, the bands were visualized with an
ECL detection system (Immobilon Western Chemiluminescent HRP
substrate; EMD Millipore), according to the manufacturer's
protocol.
Immunofluorescence
A549 (8×104/well) and RLE-6TN
(1×105/well) cells were seeded in confocal dishes 24 h
prior to treatment. The cells were the treated with BLM (120 µM for
A549 cells and 40 µM for RLE-6TN cells) for 24 h. Subsequently, the
cells were washed with PBS three times, fixed with 4.0%
paraformaldehyde for 10 min at room temperature and permeabilized
with 0.5% Triton X-100 for 10 min at room temperature.
Subsequently, the cells were incubated with primary antibody
(NLRP3, 1:400; Caspase-1, 1:200; ASC, 1:200) overnight at 4°C and
then incubated with FITC-conjugated goat anti-rabbit IgG for 90 min
at room temperature. Nuclei were stained with DAPI for 3 min at
37°C. The cells were observed under a laser confocal microscope
(Leica TCS SP8; Leica Microsystems GmbH).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from A549 and RLE-6TN cells
with RNAiso Plus reagent (Takara Biotechnology, Co., Ltd.) and the
concentration of total RNA in each group was detected with an
ultraviolet spectrophotometer. RT (37°C for 15 min and 85°C for 5
sec, then stored at 4°C until further use) was performed using a
HiScriptII Q RT SuperMix for qPCR (Vazyme), according to the
manufacturer's protocol. qPCR (95°C for 2 min, followed by 40
cycles of 95°C for 10 sec, 60°C for 30 sec and 72°C for 30 sec) was
conducted with a ChamQ™ SYBR qPCR Master mix (Vazyme) using an ABI
ViiA™ 7 system. The specific primers for HIF-1α, NF-κB and β-actin
were designed and generated by BioTNT. The primer sequences of
HIF-1α, NF-κB and β-actin are presented in Table II. All samples were read in
triplicate and β-actin served as a loading control. The
2−∆∆Cq method was used to determine fold changes
(24).
| Table II.The primer sequences of HIF-1α, NF-κB
and β-actin used in reverse transcription-quantitative PCR. |
Table II.
The primer sequences of HIF-1α, NF-κB
and β-actin used in reverse transcription-quantitative PCR.
| Species | Gene | Primer sequence
(Forward primer, 5′-3′; Reverse primer, 5′-3′) |
|---|
| Homo
sapiens | HIF-1α |
GTCTGAGGGGACAGGAGGAT;
CTCCTCAGGTGGCTTGTCAG |
|
| NF-κB |
GAGACATCCTTCCGCAAACT;
TCCTTCCTGCCCATAATCA |
|
| β-actin |
ATGATGATATCGCCGCGCTC;
CCACCATCACGCCCTGG |
| Rattus
Norvegicus | HIF-1α |
AAGTCTAGGGATGCAGCACG;
AGATGGGAGCTCACGTTGTG |
|
| NF-κB |
AGCTCCTGTCCCAGTTCTAGC;
ACTCCTGGGTCTGTGTTGTTG |
|
| β-actin |
TCCTTCCTGGGTATGGAATC;
GCACTGTGTTGGCATAGAGG |
Enzyme linked immunosorbent assay
(ELISA)
The human IL-1β ELISA kit (cat. no. EL10028) and the
rat IL-1β ELISA kit (cat. no. A1010A0301b) were purchased from
Anogen-Yes Biotech Laboratories Ltd., and BioTNT, respectively. The
expression levels of IL-1β in the A549 and RLE-6TN cell culture
supernatants were determined, according to the manufacturer's
protocol. All experiments were performed in triplicate.
Statistical analysis
All data are presented as the mean ± standard error
of mean of at least three experimental repeats. Mean values data
showed a Gaussian distribution. Comparisons between two groups were
performed using a t test. Multigroup comparisons of the means were
carried out using one-way analysis of variance test. The Bonferroni
correction was applied for post hoc analysis. All the statistics
were analyzed GraphPad Prism 7 (GraphPad Software, Inc.). P<0.05
was considered to indicate a statistically significant
difference.
Results
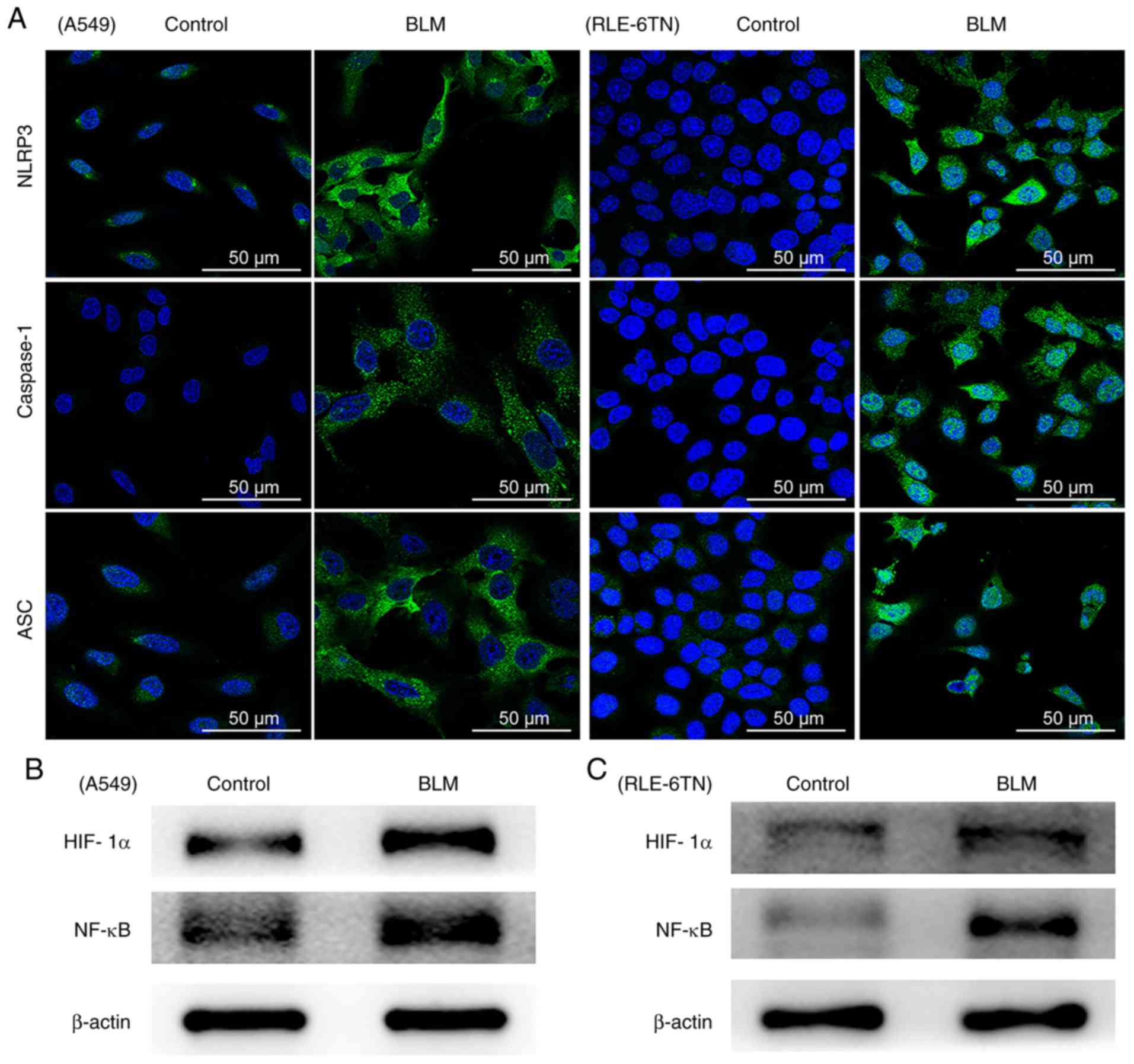
NLRP3 inflammasome is activated after
BLM-treatment
Activation of the NLRP3 inflammasome and the protein
expression of HIF-1α and NF-κB in A549 and RLE-6TN cells were
evaluated following treatment with BLM 24 h. The levels of proteins
associated with the NLRP3 inflammasome, including NLRP3, ASC and
caspase-1, were analyzed by immunofluorescence. In the BLM-treated
groups, the expression levels of NLRP3, ASC and caspase-1 markedly
increased in A549 and RLE-6TN cells (Fig. 1A). In addition, the results
demonstrated that HIF-1α and NF-κB expression markedly increased in
both cell lines (Fig. 1B and C).
These data confirmed that the NLRP3 inflammasome is activated in
BLM-treated alveolar epithelial cells.
HIF-1α regulates BLM-induced
activation of the NLRP3 inflammasome and the expression of
NF-κB
Next, the present study investigated the role of
HIF-1α in BLM-induced activation of the NLRP3 inflammasome by
transfecting A549 and RLE-6TN cells with HIF-1α siRNA. The
expression of HIF-1α significantly increased when both cell lines
were treated with BLM for 24 h (P<0.05). Furthermore, HIF-1α
siRNA significantly reduced the expression of HIF-1α mRNA in the
siHIF-1α group and the siHIF-1α + BLM group (P<0.05; Fig. 2A and B). The protein level of
HIF-1α was also inhibited in both cell lines following transfection
with HIF-1α siRNA. The levels of proteins associated with the NLRP3
inflammasome, including NLRP3, ASC and caspase-1, and NF-κB were
notably decreased in the siHIF-1α + BLM group compared with the BLM
group (Fig. 2C and D). The
immunofluorescence results also demonstrated that the expression of
NLRP3, ASC and caspase-1 deceased after transfection with HIF-1α
siRNA (Fig. 2E). These data
indicate that HIF-1α could regulate the activation of the NLRP3
inflammasome in BLM-induced ALI.
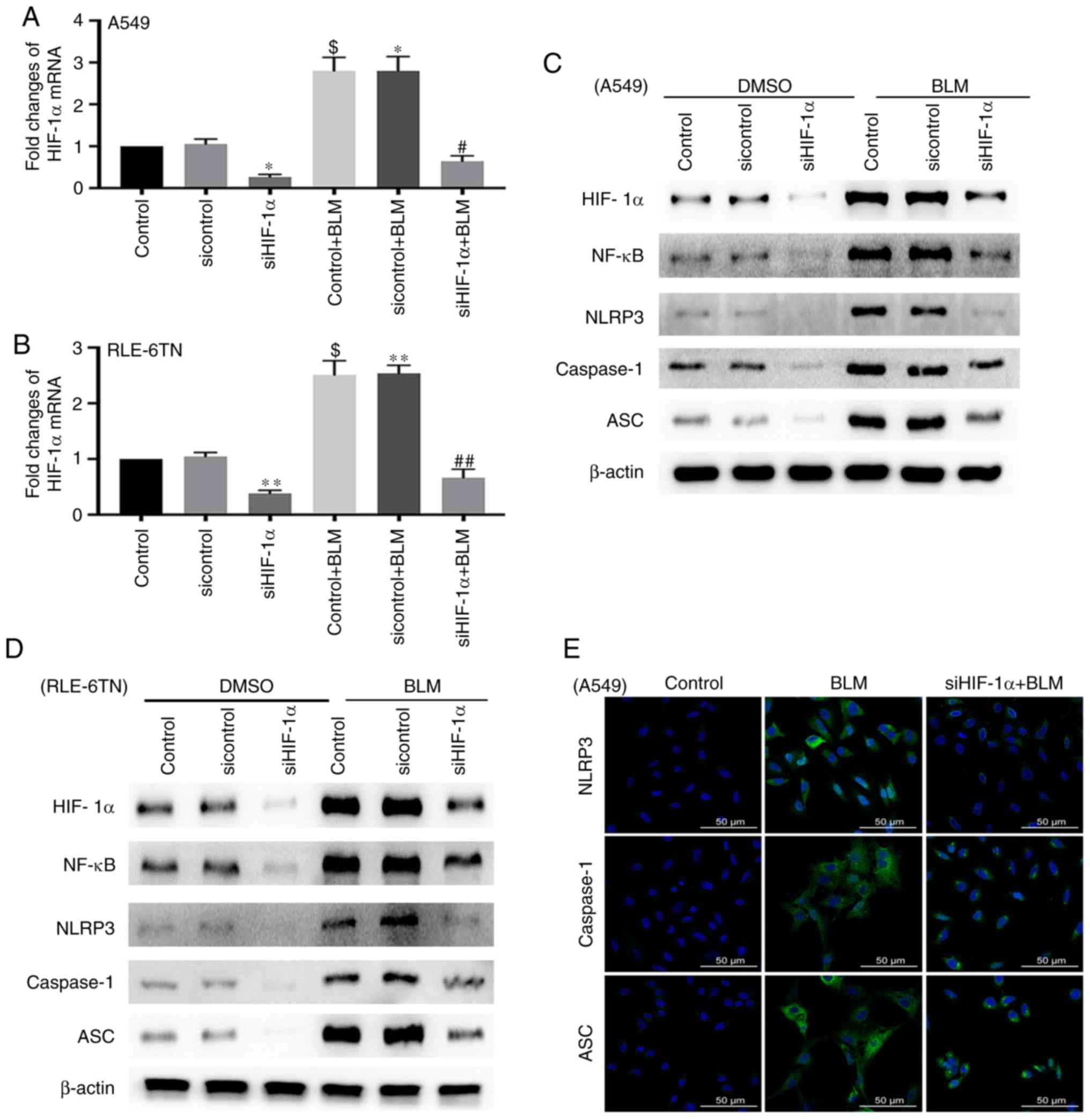 | Figure 2.Expression of NF-κB and NLRP3
inflammasome-associated proteins after silencing HIF-1α. (A)
RT-qPCR for detecting the mRNA expression level of HIF-1α in A549
cells after transfection with HIF-1α siRNA for 48 h and treatment
with BLM for 24 h. β-actin served as a loading control. These data
are presented as the mean ± standard error of the mean. *P<0.05
[siHIF-1α vs. sicontrol; P=0.0206 (Bonferroni correction, n=2);
sicontrol + BLM vs. sicontrol; P=0.0234 (Bonferroni correction,
n=2), $P<0.05 (control + BLM vs. control; P=0.0158
(ANOVA), #P<0.05 (siHIF-1α + BLM vs. sicontrol + BLM;
P=0.0232 (Bonferroni correction, n=2)]. (B) RT-qPCR for detecting
the mRNA expression level of HIF-1α in RLE-6TN cells after
transfection with HIF-1α siRNA for 48 h and treatment with BLM for
24 h. β-actin served as a loading control. These data are presented
as the mean ± standard error of the mean. $P<0.05
[control + BLM vs. control, P=0.0141 (ANOVA)]. **P<0.01
(siHIF-1α vs. sicontrol, P=0.0046 (Bonferroni correction, n=2);
sicontrol + BLM vs. sicontrol, P=0.0038 (Bonferroni correction,
n=2), ##P<0.01 (siHIF-1α + BLM vs. sicontrol + BLM,
P=0.0046 (Bonferroni correction, n=2). Western blotting for
detecting the expression levels of HIF-1α, NF-κB and NLRP3
inflammasome-associated proteins in (C) A549 and (D) RLE-6TN cell
lines after transfection with HIF-1α siRNA and treatment with BLM.
β-actin served as a loading control. (E) Immunofluorescence for
evaluating changes in the expression levels of proteins associated
with the NLRP3 inflammasome (NLRP3, ASC and caspase-1) in A549
cells after inhibition of HIF-1α. Scale bar, 50 µm. NLRP3, NOD-like
receptor 3; HIF-1α, hypoxia-inducible factor-1α; BLM, bleomycin;
ASC, apoptosis-associated speck-like protein containing CARD;
RT-qPCR, reverse transcription-quantitative PCR; siRNA, small
interfering RNA; NF-κB, nuclear factor-κB; ANOVA, analysis of
variance. |
BLM-induced activation of the NLRP3
inflammasome is modulated by NF-κB
Subsequently, the current study aimed to confirm the
role of NF-κB in BLM-induced activation of the NLRP3 inflammasome
by transfecting A549 and RLE-6TN cells with NF-κB siRNA. The level
of NF-κB significantly increased when both cell lines were treated
with BLM for 24 h (P<0.01). In addition, NF-κB siRNA
significantly decreased the level of NF-κB mRNA in the siNF-κB
group and the siNF-κB + BLM group (P<0.05; Fig. 3A and B). The protein level of NF-κB
was also inhibited in both cell lines after transfection with NF-κB
siRNA. Proteins associated with the NLRP3 inflammasome, including
NLRP3, ASC and caspase-1, decreased in the siNF-κB + BLM group
(Fig. 3C and D). The
immunofluorescence results also demonstrated that the expression
levels of NLRP3, ASC and caspase-1 deceased following transfection
with NF-κB siRNA (Fig. 3E). These
data indicate that NF-κB may also participate in modulating
activation of the NLRP3 inflammasome in BLM-induced ALI.
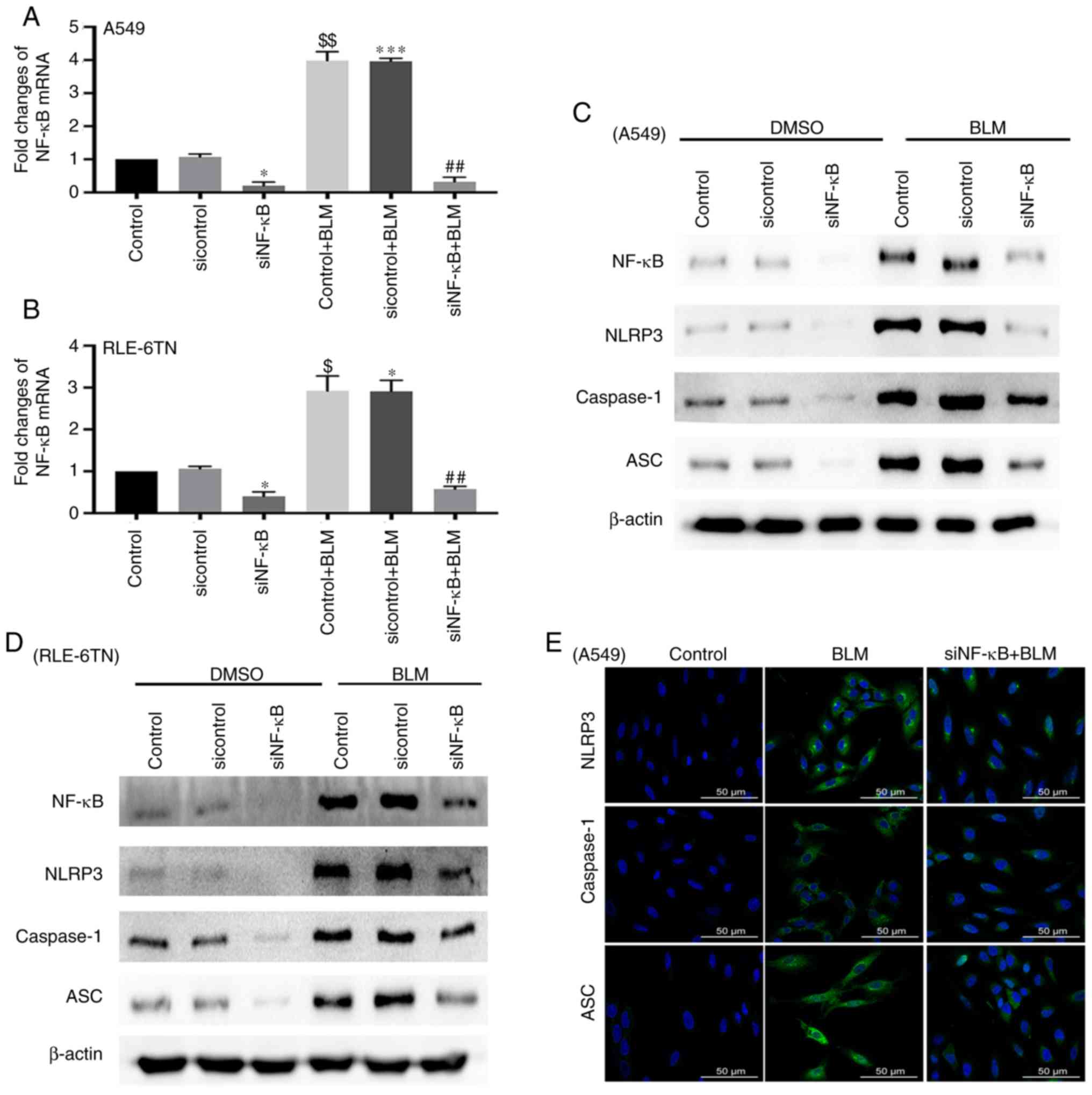 | Figure 3.Expression of NLRP3
inflammasome-associated proteins after silencing NF-κB. (A) RT-qPCR
for detecting the mRNA expression level of NF-κB in A549 cells
after transfection with NF-κB siRNA for 48 h and treatment with BLM
for 24 h. β-actin served as a loading control. These data are
presented as the mean ± standard error of the mean. *P<0.05
[siNF-κB vs. sicontrol; P=0.0246 (Bonferroni correction, n=2)],
***P<0.001 [sicontrol + BLM vs. sicontrol; P=0.0008 (Bonferroni
correction, n=2)], $$P<0.01 [control + BLM vs.
control; P=0.0041 (ANOVA)], ##P<0.01 [siNF-κB + BLM
vs. sicontrol + BLM; P=0.002 (Bonferroni correction, n=2)]. (B)
RT-qPCR for detecting the mRNA expression level of NF-κB in RLE-6TN
cells after transfection with NF-κB siRNA for 48 h and treatment
with BLM for 24 h. β-actin served as a loading control. These data
are presented as the mean ± standard error of the mean. *P<0.05
[siNF-κB vs. sicontrol; P=0.023 (Bonferroni correction, n=2);
sicontrol + BLM vs. sicontrol; P=0.021 (Bonferroni correction,
n=2)], $P<0.05 [control + BLM vs. control; P=0.0168
(ANOVA)], ##P<0.01 [siNF-κB + BLM vs. sicontrol +
BLM; P=0.0046 (Bonferroni correction, n=2)]. Western blotting for
detecting the expression levels of NF-κB and NLRP3
inflammasome-associated proteins in (C) A549 and (D) RLE-6TN cell
lines after transfection with NF-κB siRNA and treatment with BLM.
β-actin served as a loading control. (E) Immunofluorescence for
evaluating changes in the expression levels of proteins associated
with the NLRP3 inflammasome (NLRP3, ASC and caspase-1) in A549
cells after inhibition of NF-κB. Scale bar, 50 µm. NLRP3, NOD-like
receptor 3; BLM, bleomycin; ASC, apoptosis-associated speck-like
protein containing CARD; RT-qPCR, reverse
transcription-quantitative PCR; siRNA, small interfering RNA;
siRNA, small interfering RNA; NF-κB, nuclear factor-κB. |
IL-1β level is regulated by HIF-1α and
NF-κB
It has been reported that IL-1β expression
significantly increases after activation of the NLRP3 inflammasome
(25,26). Therefore, the present study
detected the levels of IL-1β in the cellular culture supernatants
of A549 following inhibition of HIF-1α or NF-κB. The results
demonstrated that IL-1β expression significantly increased in the
BLM-treatment group (P<0.05) and significantly decreased in the
siHIF-1α + BLM group and the siNF-κB + BLM group (P<0.05;
Fig. 4A and B). The similar
results were observed in RLE-6TN cells (Fig. 4C and D). This result indicates that
BLM-induced IL-1β expression may also be regulated by HIF-1α and
NF-κB.
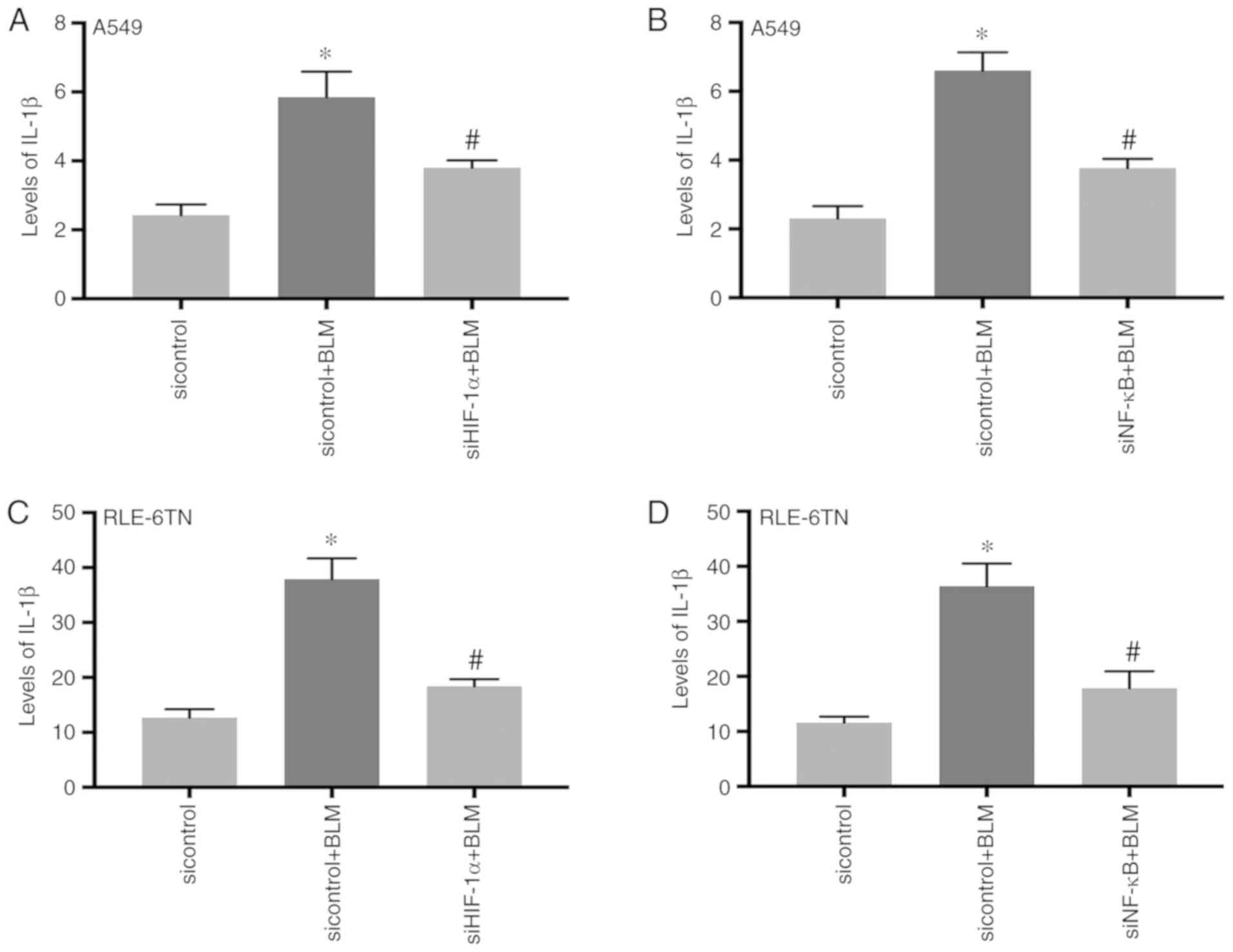 | Figure 4.Levels of IL-1β in cellular culture
supernatants after silencing HIF-1α or NF-κB. (A) The cellular
culture supernatants of A549 cells were collected after
transfection with HIF-1α siRNA. The change of IL-1β in the cellular
culture supernatants was detected by ELISA. *P<0.05 [sicontrol +
BLM vs. sicontrol; P=0.0354 (Bonferroni correction, n=2)],
#P<0.05 [siHIF-1α + BLM vs. sicontrol + BLM; P=0.0403
(Bonferroni correction, n=2)]. (B) The cellular culture
supernatants of A549 cells were collected after transfection with
NF-κB siRNA. The change of IL-1β in the cellular culture
supernatants was detected by ELISA. *P<0.05 [sicontrol + BLM vs.
sicontrol; P=0.022 (Bonferroni correction, n=2)],
#P<0.05 [siNF-κB + BLM vs. sicontrol + BLM; P=0.0432
(Bonferroni correction, n=2)]. (C) The cellular culture
supernatants of RLE-6TN cells were collected after transfection
with HIF-1α siRNA. The change of IL-1β in the cellular culture
supernatants was detected by ELISA. *P<0.05 [siHIF-1α + BLM vs.
sicontrol + BLM; P=0.0256 (Bonferroni correction, n=2)],
#P<0.05 [siHIF-1α + BLM vs. sicontrol + BLM; P=0.0406
(Bonferroni correction, n=2)]. (D) The cellular culture
supernatants of RLE-6TN cells were collected after transfection
with NF-κB siRNA. The change of IL-1β in the cellular culture
supernatants was detected by ELISA. *P<0.05 [sicontrol + BLM vs.
sicontrol; P=0.029 (Bonferroni correction, n=2)],
#P<0.05 [siNF-κB + BLM vs. sicontrol + BLM; P=0.0366
(Bonferroni correction, n=2)]. SiRNA, small interfering RNA; BLM,
bleomycin; HIF-1α, hypoxia-inducible factor-1α; IL,
interleukin. |
Discussion
ALI and ARDS, which can be caused by multiple
factors, are common clinical syndromes. It has been confirmed that
over-regulation of the inflammatory response is one of the most
important factors leading to the occurrence and development of ALI
(27,28). IL-1β is the main pro-inflammatory
cytokine that may be a potential molecular biomarker for predicting
morbidity and mortality (4). The
present study identified that HIF-1α may regulate activation of the
NLRP3 inflammasome via NF-κB and could promote the expression of
IL-1β in BLM-induced ALI.
The NLRP3 inflammasome is currently the most studied
and most widely activated inflammasome. It can be activated by
bacteria, fungus, virus and damage-associated molecular patterns,
including uric acid crystals and silicon dioxide (29–33).
It has been reported that the NLRP3 inflammasome is activated in
transfusion-associated acute lung injury, ventilator-induced lung
injury, asthma, chronic obstructive pulmonary disease and other
pulmonary diseases. The NLRP3 inflammasome has been demonstrated to
increase the release of IL-1β to participate in inflammation and
the immune reaction (12). Tian
et al (23) demonstrated
that the NLRP3 inflammasome could also regulate the
epithelial-mesenchymal transition in BLM-induced pulmonary
fibrosis. The present study identified that the expression of
proteins associated with the NLRP3 inflammasome, including NLRP3,
ASC and caspase-1, were significantly increased in the
BLM-treatment group. Therefore, it was confirmed that the NLRP3
inflammasome could be activated in BLM-induced ALI. However, the
mechanism of the activation of the NLRP3 inflammasome requires
further investigation.
It is understood that the body is in a state of
hypoxia when ALI occurs. Numerous studies have confirmed that
HIF-1α is an important regulatory factor under hypoxic conditions
(34–36). In addition, a recent study
demonstrated that HIF-1α could also increase under normoxic
conditions (37). HIF-1α could
promote the expression of inflammatory cytokines, including TNF-α,
IL-6 and IL-1β, in septic lymph treated A549 cells and human
pulmonary microvascular endothelial cells (18). Ouyang et al (38) reported that HIF-1α sustains
inflammasome activity via the cAMP/PKA/CREB/HIF-1α pathway in
adenosine-stimulated murine peritoneal macrophages. However,
whether HIF-1α can regulate the activation of the NLRP3
inflammasome in ALI has not been reported. The current study
identified that HIF-1α was increased in BLM-treated A549 and
RLE-6TN cells. In addition, activation of the NLRP3 inflammasome
was inhibited when the expression of HIF-1α was silenced in
BLM-induced ALI. These results indicate that BLM-induced activation
of the NLRP3 inflammasome could be regulated by HIF-1α.
NF-κB is a transcription factor that serves an
important role in regulating the transcription of multiple
inflammatory factors and cytokines. A number of studies have
reported that NF-κB can increase in response to multiple factors
induced in ALI (39,40). In a lipopolysaccharide-treated
alveolar macrophages cell line, myeloid differentiation protein 2
could regulate the activation of the NLRP3 inflammasome and IL-1β
expression via the MyD88/NF-κB pathway (41). Under hypoxic conditions, NF-κB is
activated by HIF-1α to participate in regulation of inflammation,
cell death and angiogenesis (42).
In the present study, activation of the NLRP3 inflammasome was
inhibited when HIF-1α and NF-κB were silenced in BLM-treated
alveolar epithelial cells. Additionally, the levels of IL-1β were
markedly decreased in the cellular culture supernatants after
inhibition of HIF-1α and NF-κB. Thus, these data indicate that
HIF-1α may modulate the activation of the NLRP3 inflammasome and
the secretion of IL-1β via NF-κB signaling.
In conclusion, it was confirmed that the NLRP3
inflammasome is activated in BLM-induced ALI. Furthermore, HIF-1α
was demonstrated to modulate activation of the NLRP3 inflammasome
through NF-κB and subsequently promote the expression of IL-1β. The
present results promoted understanding regarding the mechanism of
ALI and may provide new ideas in identifying therapeutic targets of
ALI. However, the current study is limited to clinical guidance as
it was primarily performed in vitro. In the future, further
research will be performed in in vivo experiments and
clinical research.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
JH and YL conceived and designed the study, and
analyzed and interpreted the results. JH performed the experiments
and wrote the manuscript. JX and LH conducted the experiments. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ALI
|
acute lung injury
|
|
ARDS
|
acute respiratory distress
syndrome
|
|
ASC
|
apoptosis-associated speck-like
protein containing CARD
|
|
BLM
|
bleomycin
|
|
HIF-1α
|
hypoxia-inducible factor-1 α
|
|
IL-1β
|
interleukin-1β
|
|
NF-κB
|
nuclear factor-κB
|
|
NLRP3
|
NOD-like receptor 3
|
|
TNF-α
|
tumor necrosis factor-α
|
References
|
1
|
ARDS Definition Task Force, Ranieri VM,
Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E,
Camporota L and Slutsky AS: Acute respiratory distress syndrome:
The Berlin Definition. JAMA. 307:2526–2533. 2012.PubMed/NCBI
|
|
2
|
Matthay MA and Zemans RL: The acute
respiratory distress syndrome: Pathogenesis and treatment. Annu Rev
Pathol. 6:147–163. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Spragg RG, Bernard GR, Checkley W, Curtis
JR, Gajic O, Guyatt G, Hall J, Israel E, Jain M, Needham DM, et al:
Beyond mortality: Future clinical research in acute lung injury. Am
J Respir Crit Care Med. 181:1121–1127. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Butt Y, Kurdowska A and Allen TC: Acute
lung injury: A clinical and molecular review. Arch Pathol Lab Med.
140:345–350. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Qu L, Chen C, Chen Y, Li Y, Tang F, Huang
H, He W, Zhang R and Shen L: High-Mobility group box 1 (HMGB1) and
autophagy in acute lung injury (ALI): A review. Med Sci Monit.
25:1828–1837. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bouwmeester T, Bauch A, Ruffner H, Angrand
PO, Bergamini G, Croughton K, Cruciat C, Eberhard D, Gagneur J,
Ghidelli S, et al: A physical and functional map of the human
TNF-alpha/NF-kappa B signal transduction pathway. Nat Cell Biol.
6:97–105. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bhatia M and Moochhala S: Role of
inflammatory mediators in the pathophysiology of acute respiratory
distress syndrome. J Pathol. 202:145–156. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schroder K and Tschopp J: The
inflammasomes. Cell. 140:821–832. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Strowig T, Henao-Mejia J, Elinav E and
Flavell R: Inflammasomes in health and disease. Nature.
481:278–286. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bauernfeind F, Ablasser A, Bartok E, Kim
S, Schmid-Burgk J, Cavlar T and Hornung V: Inflammasomes: Current
understanding and open questions. Cell Mol Life Sci. 68:765–783.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Eisenbarth SC and Flavell RA: Innate
instruction of adaptive immunity revisited: The inflammasome. EMBO
Mol Med. 1:92–98. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hosseinian N, Cho Y, Lockey RF and
Kolliputi N: The role of the NLRP3 inflammasome in pulmonary
diseases. Ther Adv Respir Dis. 9:188–197. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kuipers MT, Aslami H, Janczy JR, van der
Sluijs KF, Vlaar AP, Wolthuis EK, Choi G, Roelofs JJ, Flavell RA,
Sutterwala FS, et al: Ventilator-induced lung injury is mediated by
the NLRP3 inflammasome. Anesthesiology. 116:1104–1115. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dai H, Pan L, Lin F, Ge W, Li W and He S:
Mechanical ventilation modulates Toll-like receptors 2, 4, and 9 on
alveolar macrophages in a ventilator-induced lung injury model. J
Thorac Dis. 7:616–624. 2015.PubMed/NCBI
|
|
15
|
Vriend J and Reiter RJ: Melatonin and the
von Hippel- Lindau/HIF-1 oxygen sensing mechanism: A review.
Biochim Biophys Acta. 1865:176–183. 2016.PubMed/NCBI
|
|
16
|
Rhim T, Lee DY and Lee M: Hypoxia as a
target for tissue specific gene therapy. J Control Release.
172:484–494. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tang M, Tian Y, Li D, Lv J, Li Q, Kuang C,
Hu P, Wang Y, Wang J, Su K and Wei L: TNF-α mediated increase of
HIF-1α inhibits VASP expression, which reduces alveolar-capillary
barrier function during acute lung injury (ALI). PLoS One.
9:e1029672014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jiang H, Hu R, Sun L, Chai D, Cao Z and Li
Q: Critical role of toll-like receptor 4 in hypoxia-inducible
factor 1α activation during trauma/hemorrhagic shocky induced acute
lung injury after lymph infusion in mice. Shock. 42:271–278. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Suresh MV, Ramakrishnan SK, Thomas B,
Machado-Aranda D, Bi Y, Talarico N, Anderson E, Yatrik SM and
Raghavendran K: Activation of hypoxia-inducible factor-1α in type 2
alveolar epithelial cell is a major driver of acute inflammation
following lung contusion. Crit Care Med. 42:e642–e653. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sun HD, Liu YJ, Chen J, Chen MY, Ouyang B
and Guan XD: The pivotal role of HIF-1α in lung inflammatory injury
induced by septic mesenteric lymph. Biomed Pharmacother.
91:476–484. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vohwinkel CU, Hoegl S and Eltzschig HK:
Hypoxia signaling during acute lung injury. J Appl Physiol (1985).
119:1157–1163. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nicholas SA, Bubnov VV, Yasinska IM and
Sumbayev VV: Involvement of xanthine oxidase and hypoxia-inducible
factor 1 in Toll-like receptor 7/8-mediated activation of caspase 1
and interleukin-1β. Cell Mol Life Sci. 68:151–158. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tian R, Zhu Y, Yao J, Meng X, Wang J, Xie
H and Wang R: NLRP3 participates in the regulation of EMT in
bleomycin-induced pulmonary fibrosis. Exp Cell Res. 357:328–334.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Grebe A, Hoss F and Latz E: NLRP3
Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ Res.
122:1722–1740. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sarvestani ST and McAuley JL: The role of
the NLRP3 inflammasome in regulation of antiviral responses to
influenza A virus infection. Antiviral Res. 148:32–42. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sciuto AM, Clapp DL, Hess ZA and Moran TS:
The temporal profile of cytokines in the bronchoalveolar lavage
fluid in mice exposed to the industrial gas phosgene. Inhal
Toxicol. 15:687–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang P, Ye XL, Liu R, Chen HL, Liang X, Li
WL, Zhang XD, Qin XJ, Bai H, Zhang W, et al: Mechanism of acute
lung injury due to phosgene exposition and its protection by cafeic
acid phenethyl ester in the rat. Exp Toxicol Pathol. 65:311–318.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Toma C, Higa N, Koizumi Y, Nakasone N,
Ogura Y, McCoy AJ, Franchi L, Uematsu S, Sagara J, Taniguchi S, et
al: Pathogenic Vibrio activate NLRP3 inflammasome via cytotoxins
and TLR/nucleotide-binding oligomerization domain-mediated NF-kappa
B signaling. J Immunol. 184:5287–5297. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gross O, Poeck H, Bscheider M, Dostert C,
Hannesschläger N, Endres S, Hartmann G, Tardivel A, Schweighoffer
E, Tybulewicz V, et al: Syk kinase signalling couples to the Nlrp3
inflammasome for anti-fungal host defence. Nature. 459:433–436.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Thomas PG, Dash P, Aldridge JR Jr,
Ellebedy AH, Reynolds C, Funk AJ, Martin WJ, Lamkanfi M, Webby RJ,
Boyd KL, et al: The intracellular sensor NLRP3 mediates key innate
and healing responses to influenza A virus via the regulation of
caspase-1. Immunity. 30:566–575. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Martinon F, Petrilli V, Mayor A, Tardivel
A and Tschopp J: Gout-associated uric acid crystals activate the
NALP3 inflammasome. Nature. 440:237–241. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hornung V, Bauernfeind F, Halle A, Samstad
EO, Kono H, Rock KL, Fitzgerald KA and Latz E: Silica crystals and
aluminum salts activate the NALP3 inflammasome through phagosomal
destabilization. Nat Immunol. 9:847–856. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Semenza GL: Hypoxia-inducible factors in
physiology and medicine. Cell. 148:399–408. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Palazon A, Goldrath AW, Nizet V and
Johnson RS: HIF transcription factors, inflammation, and immunity.
Immunity. 41:518–528. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rohwer N, Zasada C, Kempa S and Cramer T:
The growing complexity of HIF-1α's role in tumorigenesis: DNA
repair and beyond. Oncogene. 32:3569–3576. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Koyasu S, Kobayashi M, Goto Y, Hiraoka M
and Harada H: Regulatory mechanisms of hypoxia-inducible factor 1
activity: Two decades of knowledge. Cancer Sci. 109:560–571. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ouyang X, Ghani A, Malik A, Wilder T,
Colegio OR, Flavell RA, Cronstein BN and Mehal WZ: Adenosine is
required for sustained inflammasome activation via the
A2A receptor and the HIF-1α pathway. Nat Commun.
4:29092013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li YC, Yeh CH, Yang ML and Kuan YH:
Luteolin suppresses inflammatory mediator expression by blocking
the Akt/NFκB pathway in acute lung injury induced by
lipopolysaccharide in mice. Evid Based Complement Alternat Med.
2012:3836082012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Niu X, Hu H, Li W, Li Y, Huang H, Mu Q,
Yao H and Li H: Protective effect of total alkaloids on
lipopolysaccharide-induced acute lung injury. J Surg Res.
189:126–134. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Luo M, Hu L, Li D, Wang Y, He Y, Zhu L and
Ren W: MD-2 regulates LPS-induced NLRP3 inflammasome activation and
IL-1beta secretion by a MyD88/NF-κB-dependent pathway in alveolar
macrophages cell line. Mol Immunol. 90:1–10. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
D'Ignazio L and Rocha S: Hypoxia Induced
NF-κB. Cells. 5(pii): E102016. View Article : Google Scholar : PubMed/NCBI
|