Introduction
Esophageal squamous cell carcinoma (ESCC) is one of
the most common types of malignancies in China (1). Significant advances in the diagnosis
of ESCC have been made; however, the outcome of ESCC remains poor
(1). Dysregulations in metabolism
are one of the hallmarks of cancer cells (2,3). An
improved understanding of the molecular mechanisms underlying the
dysregulated metabolism in cancer cells may aid developments in the
treatment of ESCC.
The mevalonate (MVA) pathway is responsible for the
de novo synthesis of cholesterol (4,5). The
3-hydroxy 3-methylglutaryl (HMG)-coenzyme A reductase (HMGCR) is
the rate-limiting enzyme of cholesterol synthesis and is regulated
via a negative feedback mechanism mediated by sterols and
non-sterol metabolites derived from MVA (6,7).
Statins inhibit the activity of HMGCR, which is regulated by sterol
regulatory element-binding protein 2 (SREBP2) (8,9). A
lack of intracellular sterols leads to the translocation of SREBP2
from the endoplasmic reticulum to the Golgi; SREBP2 is cleaved by
proteases in the Golgi membrane (10,11).
The N-terminal of SREBP2 serves as a transcription factor and
enters the nucleus to activate the transcription of enzymes
involved in cholesterol biosynthesis (10,11).
In our previous study, it was demonstrated that the
expression of enzymes involved in the MVA pathway was upregulated
in ESCC clinical tissues, and treatment with statin inhibited the
growth and tumorigenesis of ESCC cells (12). Additionally, it was demonstrated
that HMGCR promoted the growth, migration and colony formation of
ESCC cells (13); however, whether
SREBP2 associates with other factors to regulate HMGCR expression
in ESCC cells remains unknown.
In the present study, the expression profile and
biological functions of SREBP2 in ESCC cells were investigated and
an SREBP2-binding protein was identified. In addition, the
underlying molecular mechanisms of SREBP2 in ESCC were
examined.
Materials and methods
Cell culture and clinical samples
A normal human esophageal cell line (Het-1A) was
obtained from the American Type Culture Collection (Manassas, VA,
USA). 293T, Eca109 and KYSE150 cells were obtained from the cell
bank (the Chinese Academy of Science, Shanghai, China). The Caes17
and KYSE180 cells were gifts from Professor Xu (Shantou Medical
University, Shantou, China) (14).
Cells were cultured in Dulbecco's modified Eagle's medium (DMEM;
Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (Thermo Fisher Scientific,
Inc.), penicillin (100 U/ml) and streptomycin (100 µg/ml). Cell
cultures were maintained in a humidified incubator with 5%
CO2 at 37°C.
Patient sample collection (esophageal cancer and
adjacent non-tumor samples) was approved by the Institutional
Ethics Committee of Shanghai Chest Hospital (Shanghai, China), and
all patients provided written informed consent. A total of 52
patients (32 males and 20 females, 45–78 years old with middle and
lower esophageal cancer) between May 1 2010 and June 24 2014 who
had not undergone radiotherapy and/or chemotherapy were recruited
for the present study.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from tissue or cell samples
with TRIzol® (Thermo Fisher Scientific, Inc.) and cDNA
was synthesized using an MMLV Reverse Transcription kit (Promega
Corporation, Madison, WI, USA) at 37°C for 1 h. qPCR was performed
with 2X SYBR Green mixture (Takara Biotechnology Co., Ltd., Dalian,
China) in triplicate. The thermocycling conditions were: 95°C for 2
min; then 45 cycles of 94°C for 15 sec, 55°C for 15 sec and 68°C
for 30 sec. The relative expression levels of target genes were
normalized to 18S rRNA (Applied Biosystems; Thermo Fisher
Scientific, Inc.) and were calculated using 2−∆∆Cq
(15). The primer sequences for
SREBP2 were: F, 5′-TGACTTGTTTTCAGAACAGC-3′ and R,
5′-AACTCTGCAAGTCAAGGTT-3′.
Immunohistochemistry
ESCC tissues and paired non-cancerous tissues were
fixed with 4% PFA overnight at 4°C for 8 h and embed in paraffin
wax. The paraffin-embed ESCC sections (5-µm-thick) were kept at
65°C for 30 min and then deparaffinized, washed in with xylene and
rehydrated with descending ethanol series (100, 95, 75 and 50%).
Endogenous peroxidase activity was blocked with 0.35%
H2O2 solution. Antigen retrieval was
performed using citrate solution (10 mM, pH 6.0) at 98°C for 20
min. Non-specific binding was blocked with 1% bovine serum albumin
solution (Sangon Biotech Co., Ltd., Shanghai, China) at room
temperature for 1 h. Sections were stained with an SREBP2 antibody
(1:100; HPA031962; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
at 4°C for 8 h. Then, the sections were washed with TBST and
incubated with the secondary antibody [Gene Tech (Shanghai) Co.,
Ltd., Shanghai, China] for 2 h at room temperature. Following
washing with TBST three times (5 min for each time), the sections
were developed with 3,3′-diaminobenzidine (2 min at room
temperature) and counterstained with hematoxylin (8 min at room
temperature). The sections were examined under an inverted
microscope and images acquired at the magnification, ×20.
Western blotting
Cellular proteins were harvested from tissues and
cells with radioimmunoprecipitation assay buffer (Thermo Fisher
Scientific, Inc.) containing a protease inhibitor (Sigma-Aldrich;
Merck KGaA). Cell lysates were placed on ice for 30 min and were
subsequently centrifuged at 12,000 × g for 10 min at 4°C. The
Bradford method was used to determine the protein concentration.
Equal amounts of proteins (20 µg/lane) were loaded to 10% SDS-PAGE
and were transferred to a polyvinylidene difluoride membranes.
Subsequently, the membranes were blocked (5% non-fat milk in 1X
Tris-buffered saline with Tween-20) for 1 h at room temperature.
The membranes were incubated with primary antibodies overnight at
4°C: SREBP2 (1:1,000; ab30682; Abcam, Cambridge, UK), c-Myc
(1:1,000; 12189; Cell Signaling Technology, Inc., Danvers, MA,
USA), HMGCR (1:1,000; HPA008338; Sigma-Aldrich; Merck KGaA) and
GAPDH (1:5,000; EMD Millipore, Billerica, MA, USA). Secondary
antibodies conjugated with peroxidase (1:1,000; 14708 and 4878;
Cell Signaling Technology, Inc.) were applied to membranes for 1 h
at room temperature. Following immunoblotting, the films were
scanned with a Bio-Rad imaging system (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
Mass spectrometry assay
The coding sequence for SREBP2 was inserted into
pCMV14/3×FLAG-Pax3 (25427; Adgene, Watertown, MA, USA) using the
restriction enzymes EcoR1 and EcoRV. Subsequently,
the plasmids (Flag-SREBP2) were transfected into 293T cells
(exogenous protein is highly expressed in this cell line) using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). The transfection efficiency was monitored by the
co-transfecting GFP. After 48 h, the cells were harvested, and
immunoprecipitation was performed as described below. The
immunoprecipitates were subjected to SDS-PAGE and stained with
silver solution at room temperature for 1 h. The differentially
expressed bands were collected and digested with trypsin followed
by mass spectrometry analysis (PTM Biolabs Inc., Hangzhou, China;
http://www.ptm-biolab.com.cn).
Plasmids and cell transfection
The SREBP2 sequence was generated by PCR using
KYSE180 cDNA and subcloned into the pCMVTag2B vector (11463;
Adgene). KOD polymerase (Takara Biotechnology Co., Ltd.) was used.
The primer sequences for SREBP2 were: F, 5′-ATGGACGACAGCGGCGAG-3′
and R, 5′-TCAGGAGGCGGCAATGGC-3′. The c-Myc sequence was generated
by PCR and subcloned into the pcDNA3.1 vector (containing HA tag).
Caes17 and KYSE180 cells were transfected with SREBP2 plasmids (1
µg) using Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. After
48 h, transfected cells were selected using G418 (600 µg/ml) for 2
weeks. After 2 weeks, G418-resistant cells were pooled.
Boyden chamber assays
Caes17 and KYSE180 cells (1×105
cells/well) were seeded on the top layer of the chamber. Medium
supplemented with serum was used as a chemoattractant in the bottom
chamber for the migration assay. The cells were incubated at 37°C
for 8 h. The same protocol was used for the invasion assay;
however, the membrane was pre-coated with 50 µg Matrigel. The
non-migratory cells in the top chambers were removed with cotton
swabs. The migrated and invaded cells on the lower membrane surface
were fixed in 100% methanol for 5 min at room temperature,
air-dried, stained with eosin for 5 min at room temperature and
subsequently counted under an inverted microscope (magnification,
×20).
Cell viability assay
An MTT assay was performed to evaluate the viability
of the cells. Cells were counted and placed in 96-well plates
(1,000 cells/well) in triplicate and incubated with fresh medium.
At 4 h prior to the analysis of cell viability at the indicated
time points (day 2, 4, 6 and 8), cells were incubated with MTT (200
µl/ml medium) for 4 h. Following the MTT incubation, the formazan
crystals were dissolved using DMSO. Subsequently, growth curves
were generated by analyzing the optical density at 540 nm using a
microplate reader.
Soft agar assay
Caes17 and KYSE180 cells (1×103 cells
/well) were seeded in 1 ml 0.5% noble agar in complete DMEM
overlaid with 2 ml 1% agar in the same medium. After 2 weeks of
culture, cell colonies were examined under a microscope.
SREBP2, HMGCR and c-Myc knockdown in
ESCC cell lines
SREBP2, HMGCR and c-Myc expression in ESCC cell
lines was downregulated via lentiviral transduction. The lentivirus
was purchased from Shanghai GeneChem Co., Ltd. (Shanghai, China).
Cells were seeded in 6-well plates (5×105 cells/well)
and incubated with the lentivirus for 24 h. The lentivirus
expressed GFP independent of siRNA. GFP-positive cells were sorted
by cytometry. The sequences for the siRNAs are: Si con:
5′-AATCTGAGTCCTTGCTTAATG-3′, Si SREBP2 1#:
5′-AATCAAGTGGGAGAGTTCCCT-3′, Si SREBP2 2#:
5′-AAGCTAAGACCAGCGCCCGGG-3′, Si HMGCR 1#:
5′-AAGTCATAGTGGGGACAGTGA-3′, SiHMGCR 2#:
5′-AACCAGAAATGTGATTCAGTA-3′, Si c-Myc 1#:
5′-AACGTTAGCTTCACCAACAGG-3′ and Si c-Myc 2#:
5′-AACCAGAGTTTCATCTGCGAC-3′.
Glutathione S-transferase (GST)
pull-down assay
The GST-Myc fusion protein was purified using GST
Protein Interaction Pull-Down kit (21516; Pierce; Thermo Fisher
Scientific, Inc.). Caes17 cells were harvested using RIPA buffer.
Then, the cell lysates were centrifugated (12,000 × g at 4°C for 20
min). Subsequently, 10 µg GST-Myc protein was incubated with the
lysates of Caes17 cells at 4°C overnight. Sepharose 4B beads were
added to the supernatant and incubated for 4 h at 4°C. Protein
binding to GST-Myc was immunoprecipitated and analyzed by 8%
SDS-PAGE with an anti-GST antibody (1:5,000, ab19256, Abcam). The
blots were examined using a Bio-Rad imaging system.
HMGCR promoter assay
The promoter region (−2000bp-1bp) of HMGCR was
cloned into pGL3 basic vectors using Mlu I and Bgl II. For the
reporter assay, cells in each well were transfected with 0.05 µg
HMGCR promoter, 0.01 µg TK as control, and 0.25 µg c-Myc expression
vectors. After 24 h later, cells were harvested using the lysis
buffer, and the reporter activity were measured using a
dual-luciferase kit (Promega Corporation) according to the
manufacturer's protocols.
Immunoprecipitation assay
Caes 17 cells (6×105) were seeded into a
10 cm dish. After 24 h, the cells were lysed with
immunoprecipitation lysis buffer (9806; Cell Signaling Technology,
Inc.). The cell lysates were incubated with Flag (1:1,000;
ab205606; Abcam) and c-Myc (1:500; 12189; Cell Signaling
Technology, Inc.) primary antibodies at 4°C overnight. Protein A
Sepharose CL-4B antibody purification resin (GE Healthcare Life
Sciences, Little Chalfont, UK) were added to the supernatant and
incubated for 4 h at 4°C. The beads were washed three times using
lysis buffer (0.05% Tween-20). The bound protein was
immunoprecipitated and analyzed by 8% SDS-PAGE (Sangon, Shanghai).
The blots were examined using Bio-Rad image system.
Chromatin immunoprecipitation (ChIP)
assay
ChIP assays were performed according to the
manufacturer's protocol (Cell Signaling Technology, Inc.). KYSE180
cells were cross-linked with 1% formaldehyde for 10 min at room
temperature. Subsequently, 1.0 M glycine was added to a final
concentration of 125 mM to terminate the cross-linking reaction via
incubation for 5 min at room temperature. The cells were collected,
resuspended and sonicated (100 Hz) six times for 5 sec with the 1
min interval between each sonication, to shear the chromatin into
200-to 1,000-bp fragments; one-third of the lysate was used as the
input following cross-link reversal, phenol/chloroform extraction
and ethanol precipitation. The remaining two-thirds of the lysate
were subjected to immunoprecipitation with an anti-Myc antibody
(1:1,000; 12189) or IgG (1:1,000; 14708; both Cell Signaling
Technology, Inc.) at 4°C overnight. The immunoprecipitated
complexes were collected using Protein G-Sepharose beads. The
precipitates were sequentially subjected to cross-link reversal in
300 mM NaCl at 65°C for 8 h, phenol/chloroform (1:1 mixture)
extraction to remove the protein by centrifugation at 12,000 × g
and 4°C for 20 min and 75% ethanol precipitation on ice for 4 h.
Following centrifugation (12,000 × g for 15 min), the supernatant
was removed, and the DNA was resuspended in 40 µl double-distilled
water for qPCR analysis (2X SYBR-Green mixture, Takara
Biotechnology Co., Ltd.). The thermocycling conditions were: 95°C
for 2 min; then 45 cycles of 94°C for 15 sec, 55°C for 15 sec and
68°C for 30 sec. The primers were: F, 5′-CAAGGTCGGGAGTGATGATG-3′
and R, 5′-TTCCTGTGCGAACCTTAC-3′. 18S was used as an internal
control. The expression of SREBP2 was expressed as Ct
(18S-SREBP2).
Statistical analysis
The results are presented as the mean ± standard
deviation. The experiments were repeated three times. Differences
between values were statistically analyzed using a Student's t-test
or one-way analysis of variance (ANOVA) using SPSS 15.0 (SPSS,
Inc., Chicago, IL, USA). Turkey was used as the post hoc test for
ANOVA. P<0.05 was considered to indicate a statistically
significant difference.
Results
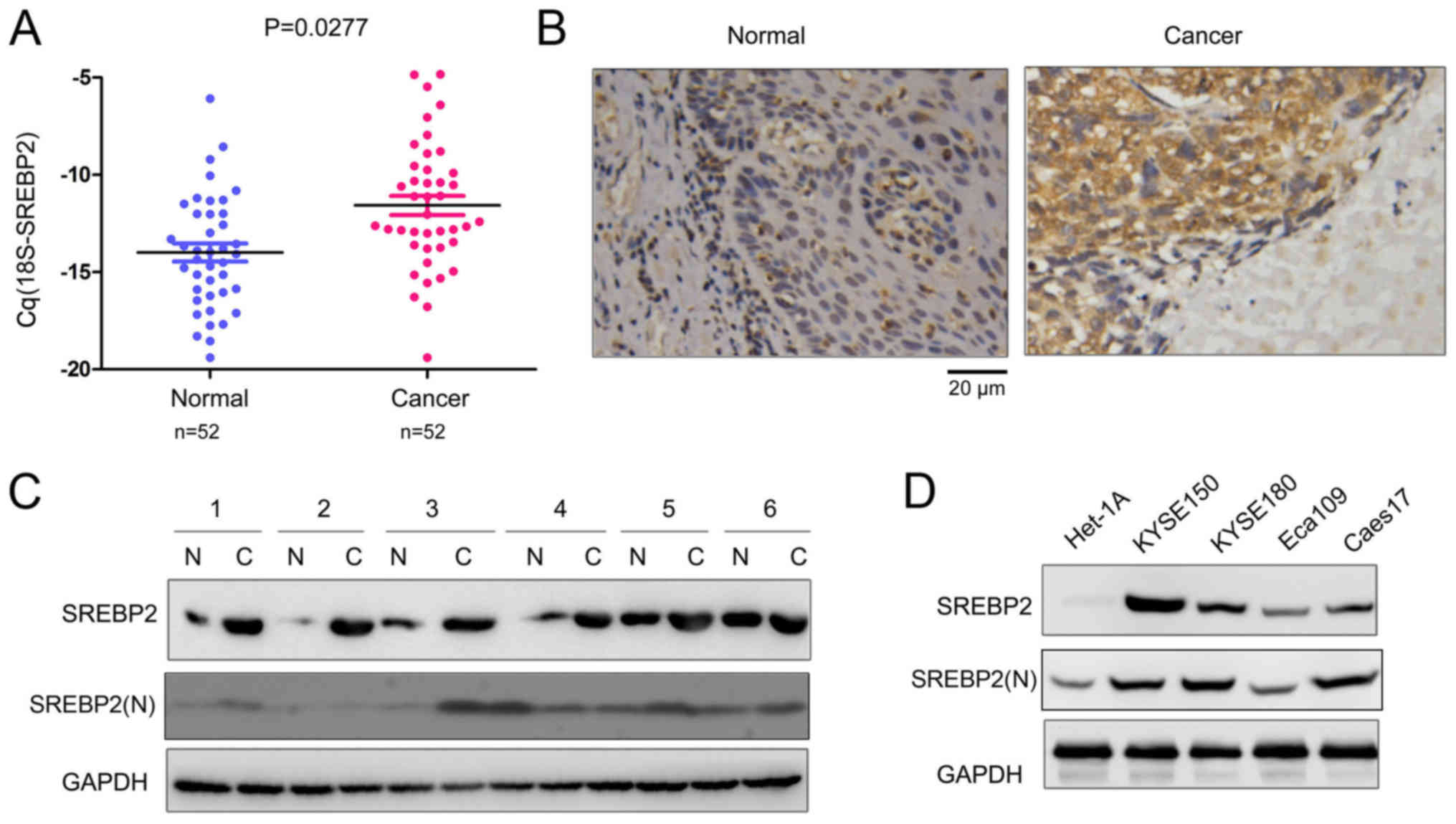
SREBP2 expression is increased in
ESCC
Our previous study demonstrated that HMGCR was
upregulated in ESCC, and statins, inhibitors of HMGCR, inhibited
the growth and migration of ESCC cells (13). SREBP2 is the master regulator of
HMGCR. Therefore, the expression profile of SREBP2 in ESCC tissues
was investigated in the present study. The mRNA expression levels
of SREBP2 in 52 ESCC tissues and paired non-cancerous tissues were
detected; as presented in Fig. 1A,
significantly increased SREBP2 mRNA expression levels were
identified in the ESCC tissues compared with the non-cancerous
tissues. In addition, the protein expression levels of SREBP2 in
ESCC tissues were analyzed via immunohistochemistry and western
blotting. SREBP2 protein expression levels were markedly increased
in ESCC tissues compared with normal samples (Fig. 1B and C). Furthermore, SREBP2
expression in normal esophageal epithelial cells (Het-1A) and ESCC
cells (KYSE150, KYSE180, Eca109 and Caes17) was investigated. As
presented in Fig. 1D, decreased
SREBP2 expression was detected in the normal Het-1A cells. The
N-terminus of SREBP2 [SREBP2(N)] serves as a transcriptional factor
following cleavage (16).
Therefore, the expression levels of the active form of SREBP2 in
ESCC tissues and cell lines were additionally detected. As
presented in Fig. 1C and D,
SREBP2(N) was markedly upregulated in ESCC cell lines with the
exception of the Eca109 cells; however, only a few ESCC tissue
samples exhibited upregulated SREBP2(N) expression levels. These
data suggested that SREBP2 is upregulated in ESCC.
SREBP2 promotes ESCC cell viability,
colony formation and migration
To study the biological functions of SREBP2 in ESCC,
SREBP2 was overexpressed in KYSE180 and Caes17 cells, which had low
expression levels of SREBP2 (Fig.
1D, Fig. 2A). SREBP2
overexpression significantly increased the KYSE180 and Caes17 cell
migration and invasion in Boyden chamber assays and Transwell
assays, respectively (Fig. 2B and
C). Additionally, SREBP2 significantly promoted the growth and
colony formation of KYSE180 and Caes17 cells in liquid culture and
on the soft agar compared with the empty vector, P23 (Fig. 2D and E). Lovastatin (10 mg/l), an
HMGCR inhibitor (17),
significantly decreased the promoting effects of SREBP2 on the
anchorage-independent growth of Caes17 and KYSE180 cells (Fig. 2E). Upregulated HMGCR expression
levels were observed in Caes17 and KYSE180 cells overexpressing
SREBP2; however, expression levels of HMGCR were decreased in cells
treated with lovastatin (Fig. 2E).
To investigate the effects of HMGCR expression on the malignant
phenotype associated with SREBP2, HMGCR expression was
downregulated in the present study. As presented in Fig. 2F, knockdown of HMGCR expression
inhibited the anchorage-independent growth of KYSE180 cells
compared with the control cells. These results suggested that
SREBP2 promoted ESCC cell tumorigenesis by enhancing HMGCR
activity.
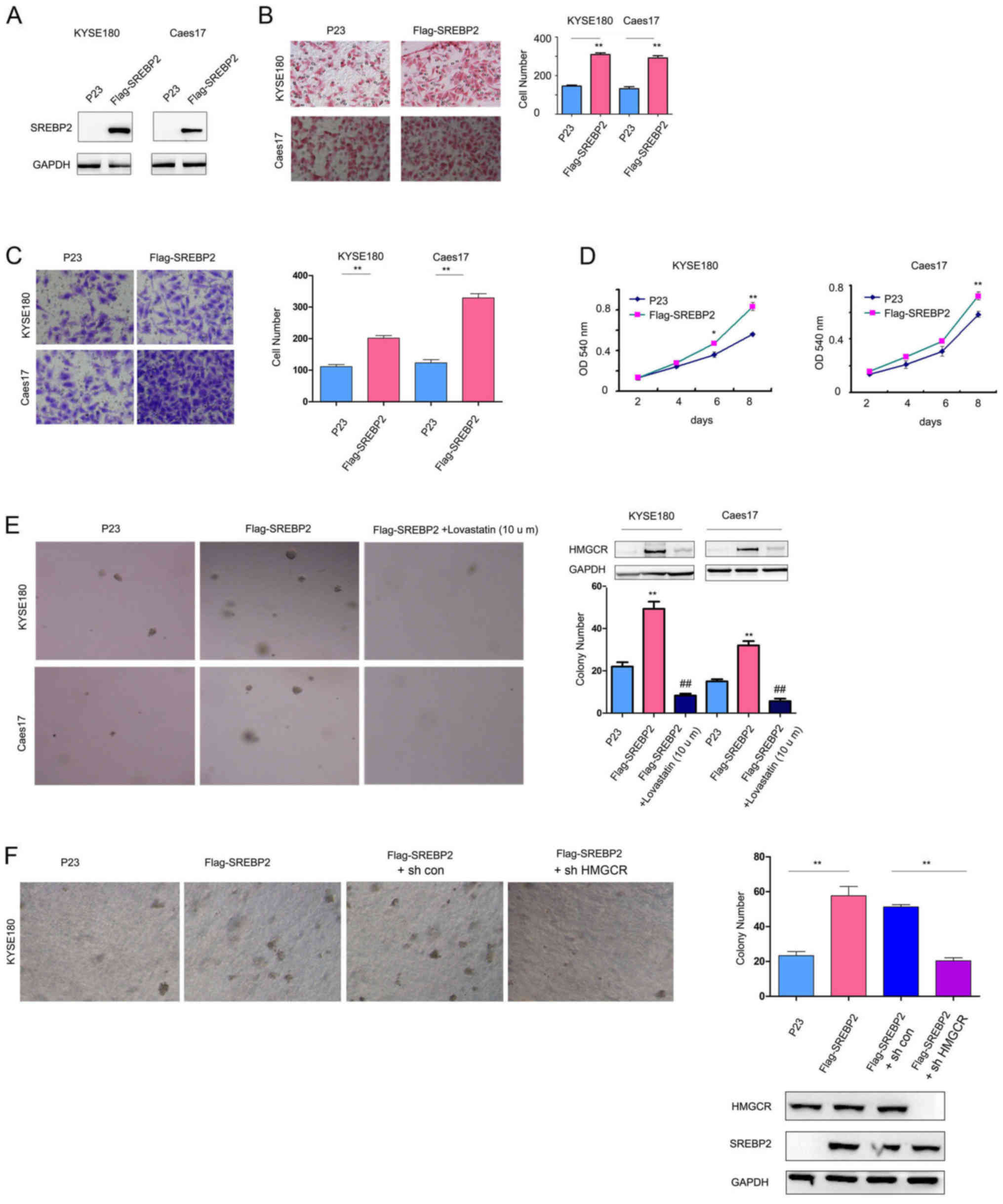 | Figure 2.SREBP2 promotes ESCC cell viability,
invasion and migration. (A) Constitutive SREBP2 expression in
Caes17 and KYSE180 cells. Effects of SREBP2 on ESCC cell (B)
migration and (C) invasion were evaluated using Boyden chamber and
Transwell assays. Magnification, ×20. (D) Effects of SREBP2 on ESCC
cell viability were evaluated via MTT assays. (E) Effects of SREBP2
on the anchorage-independent growth of ESCC cells were evaluated by
a soft agar assay; treatment with lovastatin rescued the effects of
SREBP2. Magnification, ×20. (F) Roles of HMGCR in the
anchorage-independent growth of ESCC cells induced by SREBP2.
Magnification, ×20. *P<0.05, **P<0.01 vs. respective P23;
##P<0.01 vs. respective Flag-SREBP2. sh, short
hairpin RNA; SREBP2, sterol regulatory element-binding protein 2;
ESCC, esophageal squamous cell carcinoma; HMGCR, 3-hydroxy
3-methylglutaryl coenzyme A reductase; OD, optical density. |
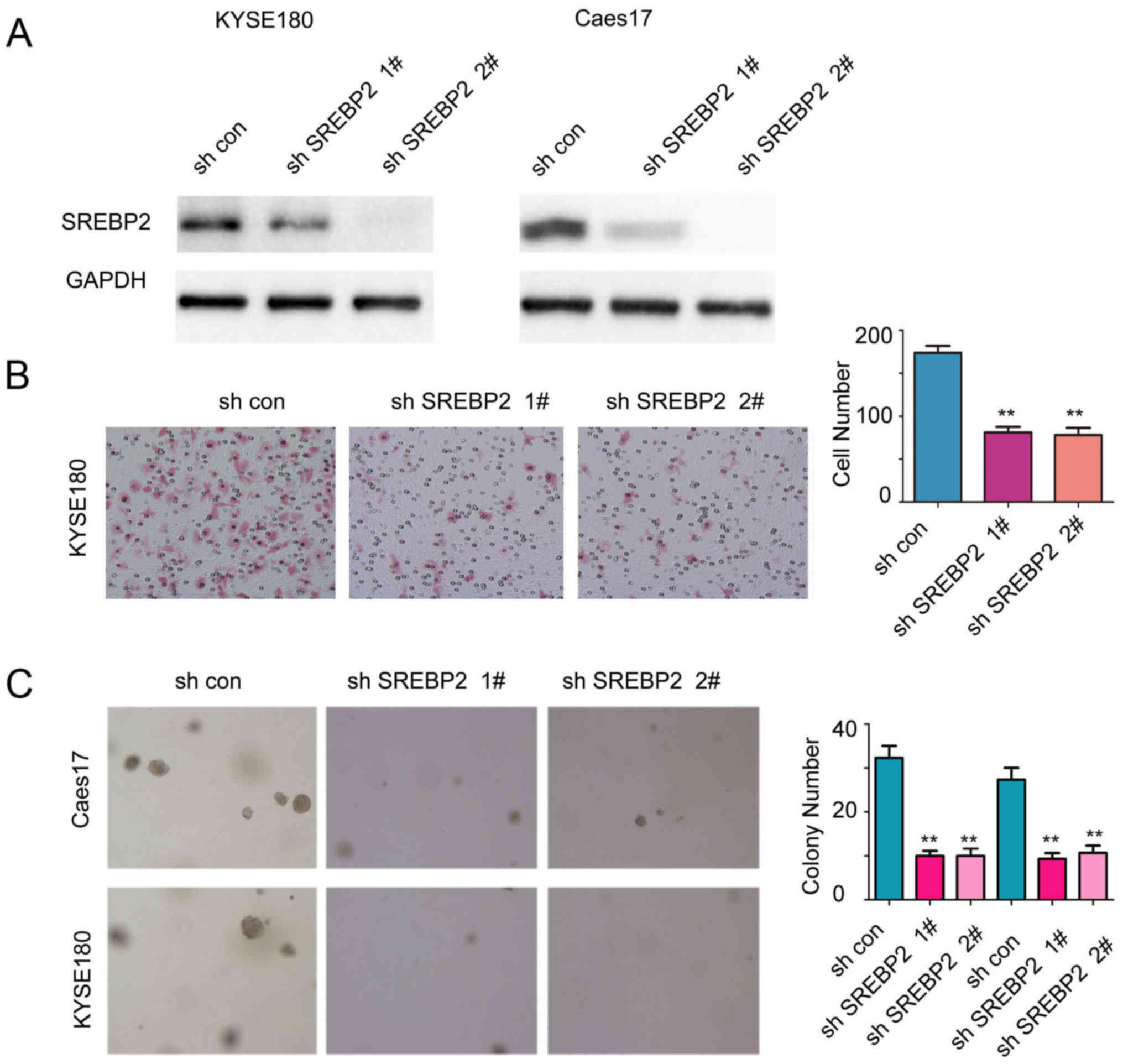
To further examine the functions of endogenous
SREBP2, the expression of SREBP2 was downregulated in Caes17 and
KYSE180 cells (Fig. 3A). As
presented in Fig. 3B and C, SREBP2
knockdown significantly inhibited the anchorage-independent growth
and migration of ESCC cells compared with the control (Fig. 3B and C; P<0.01). Collectively,
these results demonstrated that SREBP2 may promote ESCC cell growth
and migration.
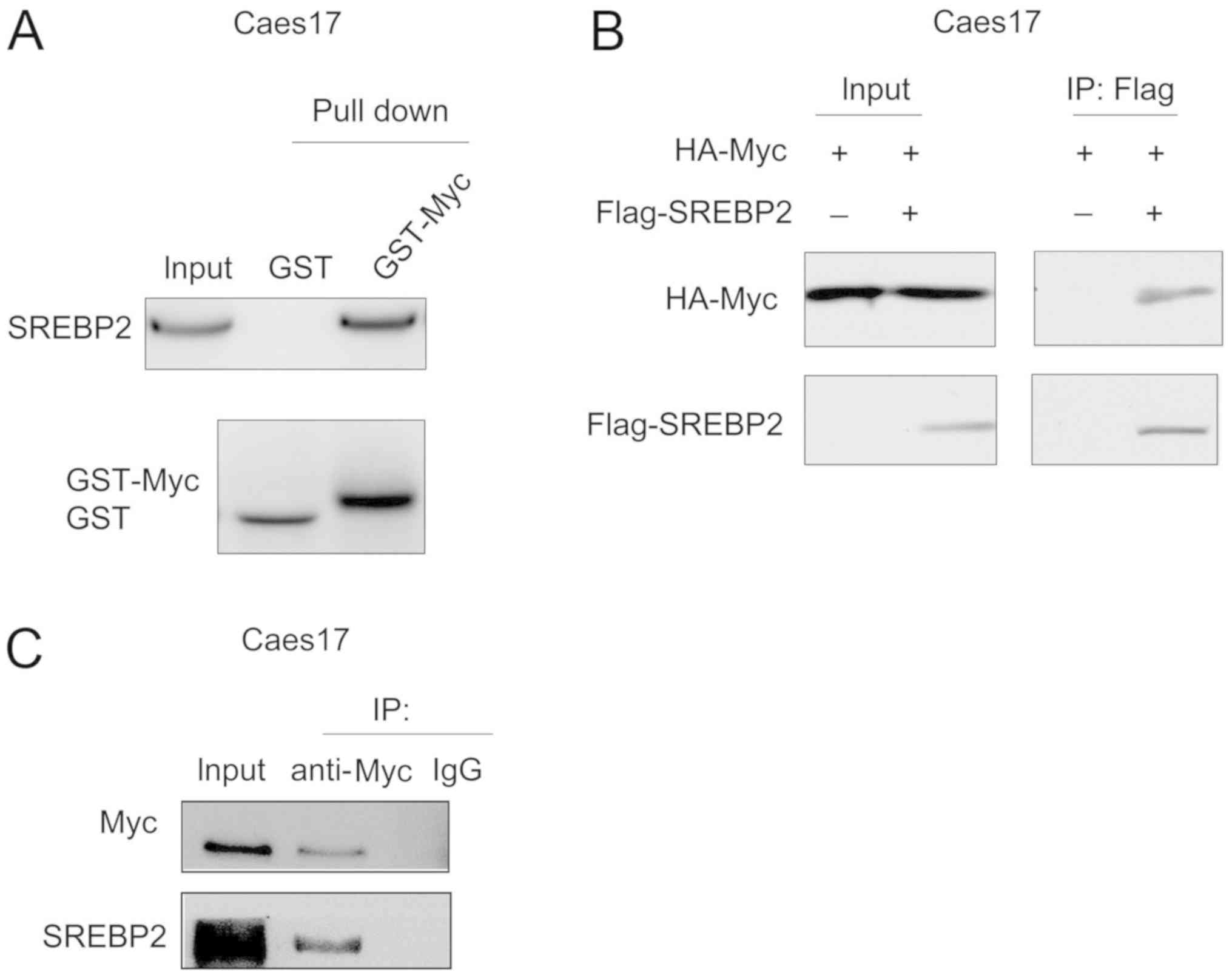
SREBP2 interacts with c-Myc in ESCC
cells
The present study screened the binding partners of
SREBP2 in ESCC cells via mass spectrometry (data not shown). c-Myc
was identified as a potential binding partner for SREBP2.
Therefore, the interaction between c-Myc and SREBP2 was
investigated via GST pull-down assays. As presented in Fig. 4A, the fusion protein GST-Myc bound
to SREBP2 in Caes17 cells. In addition, exogenously expressed c-Myc
[hemagglutinin (HA)-Myc] and SREBP2 (Flag-SREBP2) co-transfected
with Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) formed a complex in Caes17 cells (Fig. 4B). Furthermore, endogenously
expressed c-Myc and SREBP2 interacted with each other (Fig. 4C). Collectively, these data
suggested that c-Myc and SREBP2 form a complex.
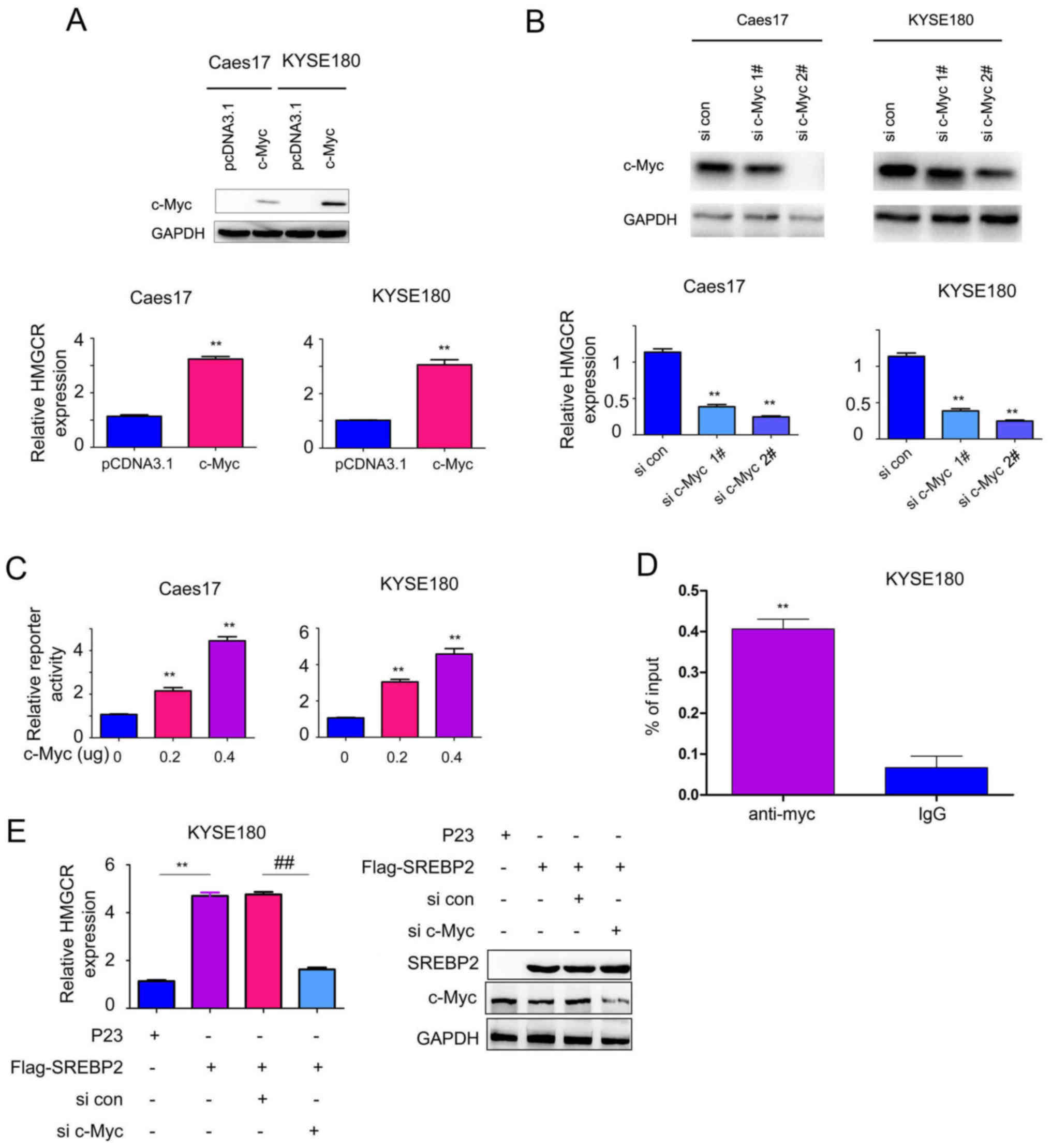
C-Myc is essential for SREBP2-induced
HMGCR expression
To further investigate the function of the
interaction between c-Myc and SREBP2, the effects of c-Myc on HMGCR
expression were studied. Overexpression of c-Myc significantly
increased the mRNA expression levels of HMGCR in Caes17 and KYSE180
cells compared with the control (Fig.
5A; P<0.01), whereas, knockdown of c-Myc expression
significantly inhibited HMGCR expression compared with the control
(Fig. 5B; P<0.01).
Additionally, whether c-Myc activates HMGCR promoter activity was
investigated in the present study. As presented in Fig. 5C, c-Myc significantly activated the
HMGCR promoter in a dose-dependent manner compared with the control
group (P<0.01) in KYSE180 cells. Additionally, c-Myc was
determined to directly bind the HMGCR promoter via ChIP assays
(Fig. 5D). Collectively, these
data suggested that c-Myc induced HMGCR expression.
Based on the interaction between c-Myc and SREBP2,
the present study subsequently investigated whether c-Myc and
SREBP2 cooperate to regulate HMGCR expression. As presented in
Fig. 5E, downregulation of c-Myc
impaired the effects of SREBP2 in promoting HMGCR transcription.
Collectively, these observations suggested that c-Myc is essential
for SREBP2-induced HMGCR expression.
Discussion
Dysregulation of the MVA pathway has been
demonstrated in numerous types of cancer (8,18,19).
In our previous study, it was demonstrated that ESCC cells were
very sensitive to treatment with statin (12). HMGCR, the target of statin, is
upregulated in ESCC samples and promotes epithelial cell migration
and transformation (13). In the
present study, it was demonstrated that the expression of SREBP2, a
regulator of HMGCR, was increased in ESCC. SREBP2 promoted the
migration and invasive abilities, viability and
anchorage-independent growth of ESCC. Additionally, the present
study demonstrated that c-Myc is a binding partner of SREBP2 and
cooperates with SREBP2 to enhance the expression of HMGCR.
Collectively, these results suggested that the MVA pathway may be
activated in ESCC, thus making ESCC sensitive to treatment with
statin. These results potentially suggest a novel therapeutic
target for ESCC.
A notable result of the present study is the
identification of c-Myc as a binding protein of SREBP2. Recently,
numerous transcriptional factors have been determined to be binding
partners of the SREBP protein family (16). For example, mutant P53 interacts
with SREBP and promotes the activation of the MVA pathway in breast
cancer cells (20), suggesting
that the MVA pathway is altered by oncogenes and tumor suppressors.
A number of previous studies demonstrated the overexpression of
c-Myc in ESCC (21–25). Therefore, it may be hypothesized
that the upregulation of c-Myc cooperated with SREBP2 to activate
the MVA pathway and promote tumorigenesis. In addition, numerous
previous studies demonstrated the key roles of c-Myc in cancer
metabolism (26–28). The results of the present study
suggested that c-Myc regulates cholesterol synthesis by interacting
with SREBP2.
In summary, the present study demonstrated the
oncogenic roles of SREBP2 in ESCC-associated tumorigenesis,
suggesting that SREBP2 may be a therapeutic target for ESCC.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Science Foundation of China (grant no. 81401933) and Shanghai
Science and Technology Committee (grant no. 13ZR1461300; Shanghai,
China).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CZ, LF, FY and HZ made substantial contributions to
the design of the present study. CZ, LF, FY and ZL performed the
experiments. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Ethics Committee of Shanghai Chest Hospital (Shanghai, China). All
patients provided written informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schwartz L, Supuran CT and Alfarouk KO:
The warburg effect and the hallmarks of cancer. Anticancer Agents
Med Chem. 17:164–170. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Munkley J and Elliott DJ: Hallmarks of
glycosylation in cancer. Oncotarget. 7:35478–35489. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Goldstein JL and Brown MS: Regulation of
the mevalonate pathway. Nature. 343:425–430. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Landau BR and Brunengraber H: Shunt
pathway of mevalonate metabolism. Methods Enzymol. 110:100–114.
1985. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Weinberger C: A model for farnesoid
feedback control in the mevalonate pathway. Trends Endocrinol
Metab. 7:1–6. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bathaie SZ, Ashrafi M, Azizian M and
Tamanoi F: Mevalonate pathway and human cancers. Curr Mol
Pharmacol. 10:77–85. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Swanson KM and Hohl RJ: Anti-cancer
therapy: Targeting the mevalonate pathway. Curr Cancer Drug
Targets. 6:15–37. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang T, Seah S, Loh X, Chan CW, Hartman M,
Goh BC and Lee SC: Simvastatin-induced breast cancer cell death and
deactivation of PI3K/Akt and MAPK/ERK signalling are reversed by
metabolic products of the mevalonate pathway. Oncotarget.
7:2532–2544. 2016.PubMed/NCBI
|
|
10
|
Madison BB: Srebp2: A master regulator of
sterol and fatty acid synthesis. J Lipid Res. 57:333–335. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ma K, Malhotra P, Soni V, Hedroug O,
Annaba F, Dudeja A, Shen L, Turner JR, Khramtsova EA, Saksena S, et
al: Overactivation of intestinal SREBP2 in mice increases serum
cholesterol. PLoS One. 9:e842212014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shi J, Zhu J, Zhao H, Zhong C, Xu Z and
Yao F: Mevalonate pathway is a therapeutic target in esophageal
squamous cell carcinoma. Tumour Biol. 34:429–435. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhong C, Fan L, Yao F, Shi J, Fang W and
Zhao H: HMGCR is necessary for the tumorigenecity of esophageal
squamous cell carcinoma and is regulated by Myc. Tumour Biol.
35:4123–4129. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xie JJ, Xu LY, Wu JY, Shen ZY, Zhao Q, Du
ZP, Lv Z, Gu W, Pan F, Xu XE, et al: Involvement of CYR61 and CTGF
in the fascin-mediated proliferation and invasiveness of esophageal
squamous cell carcinomas cells. Am J Pathol. 176:939–951. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mukherjee M, Basu Ball W and Das PK:
Leishmania donovani activates SREBP2 to modulate macrophage
membrane cholesterol and mitochondrial oxidants for establishment
of infection. Int J Biochem Cell Biol. 55:196–208. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kajinami K, Takekoshi N, Brousseau ME and
Schaefer EJ: Pharmacogenetics of HMG-CoA reductase inhibitors:
exploring the potential for genotype-based individualization of
coronary heart disease management. Atherosclerosis. 177:219–234.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Caruso MG and Notarnicola M: Biochemical
changes of mevalonate pathway in human colorectal cancer.
Anticancer Res. 25:3393–3397. 2005.PubMed/NCBI
|
|
19
|
Andela VB, Pirri M, Schwarz EM, Puzas EJ,
O'Keefe RJ, Rosenblatt JD and Rosier RN: The mevalonate synthesis
pathway as a therapeutic target in cancer. Clin Orthop Relat Res
(415 Suppl). S59–S66. 2003. View Article : Google Scholar
|
|
20
|
Freed-Pastor WA, Mizuno H, Zhao X,
Langerød A, Moon SH, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li
W, Polotskaia A, et al: Mutant p53 disrupts mammary tissue
architecture via the mevalonate pathway. Cell. 148:244–258. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gomes TS, Noguti J, Forones NM, Lima FO,
Dobo C, Fernandes Junior JA, Oshima CT and Ribeiro DA: Correlation
analysis of c-myc, p21(WAF/CIP1), p53, C-erbB-2 and COX-2 proteins
in esophageal squamous cell carcinoma. Pathol Res Pract. 209:6–9.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brown J, Bothma H, Veale R and Willem P:
Genomic imbalances in esophageal carcinoma cell lines involve Wnt
pathway genes. World J Gastroenterol. 17:2909–2923. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang W, Xue L and Wang P: Prognostic value
of β-catenin, c-myc, and cyclin D1 expressions in patients with
esophageal squamous cell carcinoma. Med Oncol. 28:163–169. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ishizuka T, Tanabe C, Sakamoto H, Aoyagi
K, Maekawa M, Matsukura N, Tokunaga A, Tajiri T, Yoshida T, Terada
M and Sasaki H: Gene amplification profiling of esophageal squamous
cell carcinomas by DNA array CGH. Biochem Biophys Res Commun.
296:152–155. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mandard AM, Hainaut P and Hollstein M:
Genetic steps in the development of squamous cell carcinoma of the
esophagus. Mutat Res. 462:335–342. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shukla SK, Gunda V, Abrego J, Haridas D,
Mishra A, Souchek J, Chaika NV, Yu F, Sasson AR, Lazenby AJ, et al:
MUC16-mediated activation of mTOR and c-Myc reprograms pancreatic
cancer metabolism. Oncotarget. 6:19118–19131. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Miller DM, Thomas SD, Islam A, Muench D
and Sedoris K: c-Myc and cancer metabolism. Clin Cancer Res.
18:5546–5553. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gordan JD, Thompson CB and Simon MC: HIF
and c-Myc: Sibling rivals for control of cancer cell metabolism and
proliferation. Cancer cell. 12:108–113. 2007. View Article : Google Scholar : PubMed/NCBI
|