Introduction
Most of the oncogenes have been formed by mutation,
amplification, and rearrangement of proto-oncogenes, which regulate
cell growth, cell differentiation, and apoptosis. Overexpression or
hyperactivation of these oncogenes drives uncontrolled cell
proliferation and resistance to apoptosis, which are the main
characteristics of cancer cells (1). c-Myc is one of the key oncogenes
responsible for many human cancers, and unlike normal cells, it is
aberrantly expressed in cancer cells (2–4). In
addition, c-Myc also induces many genes such as eIF-2, eIF-4E, p53,
cyclin D/E, CDK4, CDC25A, and p19/p14ARF, and facilitates
degradation of p27 (5,6). However, it is not known clearly
whether c-Myc is related to the expression of natural killer group
2 member D (NKG2D) ligands and the susceptibility of cancer cells
to NK cells. Since NK cells and γδ-T cells recognize cancer cells
through NKG2D ligands and eliminate them (7), induction of NKG2D ligands in cancer
cells is very critical to evoke NK cell-mediated anti-cancer
immunity. It was well known that normal cells, except activated
T-cells and some tissue cells, such as in the intestinal
epithelium, generally do not express NKG2D ligands (8,9);
besides, the abundant expression of MHC class molecules on their
surface also protect the normal cells from the activated NK cells
(10). However, the potency of
immune surveillance in patients is not sufficient to eliminate
transformed cells through the process called ‘lack of induction’,
which means insufficient induction of NK activating ligands
(11,12). Since aberrant expression of c-Myc
oncogenes changes the expression of many other genes, and maintains
malignancy in cancer cells, it was suspected that c-Myc might
disturb NK cell-mediated immune responses through the altered
expression of several genes, such as those encoding NKG2D ligands.
Previously, it was revealed that inhibition of several oncogenes,
such as EGFR, PI3K, Ras, NF-κB and BCR/ABL, could modulate the
expression of NKG2D ligands (13–16).
In this study, we investigated whether c-Myc modulates the
expression of NKG2D ligands and affects the susceptibility of
cancer cells to NK cells.
Materials and methods
Cell lines and reagents
The K562 chronic myeloid leukemia cell line was
available with the Korean Cell Line Bank (Seoul, Korea) originated
from ATCC (@CCL-243). These cells were maintained in RPMI medium
(Invitrogen; Thermo Fisher Scientific, Inc.) supplemented with 10%
fetal bovine serum (FBS) (Gibco; Thermo Fisher Scientific, Inc.), 2
mM L-glutamine, 100 µg/ml streptomycin, and 100 U/ml penicillin.
The NK-92 cell line was obtained from the American Type Culture
Collection and maintained in alpha-Minimum Essential Modified
medium supplemented with 12.5% (v/v) FBS, 12.5% (v/v) horse serum,
2 mM L-glutamine, 0.1 mM 2-mercaptoethanol, 200 U/ml of recombinant
human interleukin-2, 100 µg/ml streptomycin, and 100 U/ml
penicillin. All cell lines were maintained at 37°C in a humidified
atmosphere containing 5% CO2. c-Myc inhibitor, 10058-F4,
which prevented the binding of c-Myc/Max dimers to its DNA targets,
was purchased from Calbiochem.
Total RNA extraction and multiplex
reverse transcription (RT)-PCR
Total cellular RNA was extracted using
RNeasy® Mini Kit (Qiagen GmbH) by following the
manufacturer's protocol. cDNA was synthesized from 1 µg of
extracted total RNA using 100 pmol of random primers (Takara) and
100 units of M-MLV reverse transcriptase (Promega Corporation). The
synthesized cDNA was used in the PCR reaction with reagents in the
QIAGEN® Multiplex PCR kit (Qiagen GmbH). Seven pairs of
primer sets were used to investigate the expression of genes,
including ribosomal protein L19 (RPL19), MICA, MICB, ULBP1-3 and
β-actin (ACTB) (17). ACTB and
RPL19 were used as a loading control and degradation marker,
respectively. The PCR products were analyzed using ethidium
bromide-stained 2.0% agarose gel electrophoresis and quantitated by
image analyzing software, Quantity One (Bio-Rad Laboratories,
Inc.).
c-Myc silencing using siRNA
transfection
K562 cells were transfected with c-Myc targeting
small interfering RNA (siRNA) or scrambled RNA (scRNA) using
Oligofectamine™ Reagent (Life Technologies; Thermo Fisher
Scientific, Inc.) by following the manufacturer's protocol.
Chemically synthesized siRNA and scRNA were purchased from Bioneer.
The cells were treated with 200 nM final concentration of
siRNA/scRNA and harvested after incubation for 24, 48 and 72 h.
c-Myc overexpression in K562 cells
using pCMV6 vector
pCMV6 and pCMV6-myc vectors were purchased from
Origene. Each vector was transfected into K562 cells using Xfect™
Transfection Reagent (Clontech) by following the manufacturer's
protocol. After 24 h of incubation, cells were distributed into
12-well and selected by treatment with 0.8 mg/ml of G418
(Geneticin®; Life Technologies; Thermo Fisher
Scientific, Inc.). Positive cells were maintained in RPMI medium
containing 100 µg/ml of G418.
Western blot analysis
Cells were washed with ice-cold phosphate buffer,
lysed in lysis buffer consisting of 1% (w/v) sodium dodecyl sulfate
(SDS), 1.0 mM sodium ortho-vanadate, and 10 mM Tris, pH 7.4,
followed by sonication for 5 sec. Proteins in the cell lysate were
quantified using a Bradford protein assay kit (Pierce). Proteins
were separated by 10% SDS-polyacrylamide gel electrophoresis
(SDS-PAGE) using a mini gel apparatus (Bio-Rad Laboratories, Inc.)
and were transferred onto nitrocellulose membranes (Hybond-ECL; GE
Healthcare). Each membrane was blocked with 5% skimmed milk in
Tris-buffered saline containing 0.05% Tween-20 (TBST). Protein
bands were probed with primary antibody, followed by labeling with
horseradish peroxidase-conjugated anti-mouse, anti-rabbit secondary
antibody (Cell Signaling Technology). The primary antibodies used
were: c-Myc (Epitomics), β-actin antibody (Sigma-Aldrich; Merck
KGaA). Bands were visualized by enhanced chemiluminescence
(Amersham Pharmacia Biotech) according to manufacturer's
instruction and the densities were measured by Multi Gauge v3.0
(Fujifilm Medical Systems Inc.).
Flow cytometry analysis of NKG2D
ligands
To determine the surface expression of the NKG2D
ligands on cancer cells, the cells were incubated with mouse
anti-MICA, anti-MICB, and anti-ULBP1-3 (R&D Systems), which
were NKG2D ligand-specific monoclonal antibodies (mAbs), and the
corresponding isotype controls at 10 µg/ml, followed by incubation
with the goat anti-mouse-PE conjugated (BD Pharmingen Inc.). The
analysis was performed on the FACSCalibur® system using
the CellQuest software (both from Becton-Dickinson), and the cell
surface expression was quantified from the value of the mean
fluorescence intensities (MFI) obtained with the specific mAbs.
NK cell-mediated cytotoxicity
assay
NK cell-mediated cytotoxicity was determined using
flow cytometry. Briefly, untreated, 10058-F4 treated, and c-Myc
upregulated K562 cells were harvested. The cells were stained with
50 µM CFSE for 30 min at 37°C and washed three times. NK-92 cells
and CFSE-stained K562 cells were co-cultured for 4 h. Propidium
iodide (PI) was added to the co-cultured samples to mark dead
cells. The proportion of dead cells was analyzed by formula:
(CFSE+PI+ cells/CFSE+ cells)
×100.
Statistical analysis
To evaluate the altered level of gene expression,
the mean folds of gene expression were calculated. For comparison
of groups, one way ANOVA was performed using SPSS software (version
11.01; SPSS, Inc.). P<0.05 was considered to indicate a
statistically significant difference.
Results
c-Myc inhibitor increases the
transcription of MICB and ULBP1
Using multiplex-PCR reaction, the expression of 7
genes, including those for 5 NKG2D ligands and 2 housekeeping genes
were analyzed, and were normalized to the level of ACTB. K562 cells
were treated with 10058-F4, which selectively prevents the
c-Myc-Max complex from interaction (18). MICA was further transcribed by
treatment with 7.5 and 30 µM of 10058-F4. The levels of MICB and
ULBP1 transcripts were increased dose-dependently. The change in
the level of transcription of ULBP2 and ULBP3 was not significant
after treatment with 10058-F4 (Fig.
1). It was supposed that c-Myc would significantly affect the
expression of MICA, MICB, and ULBP1.
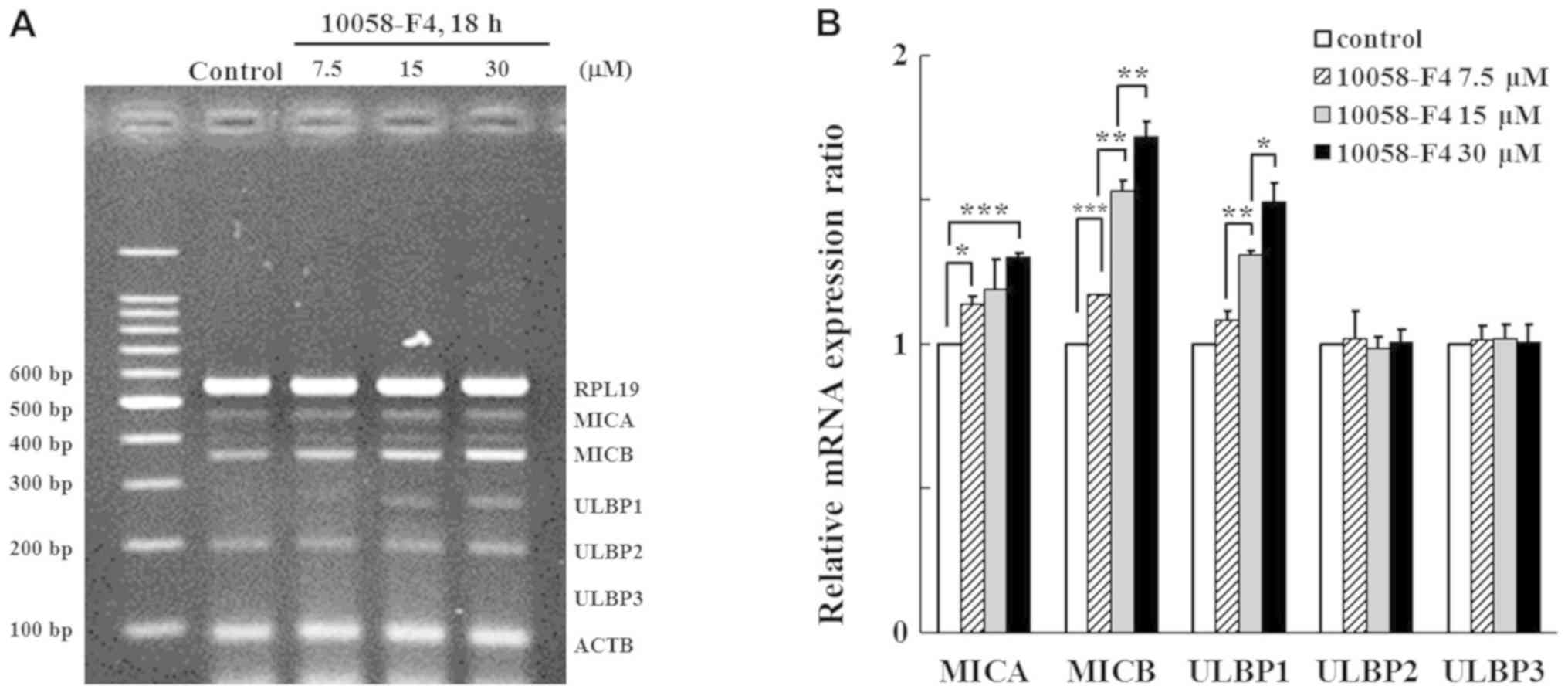 | Figure 1.Analysis of NKG2D ligand transcription
following treatment of K562 cells with 10058-F4. K562 cells were
treated with 10058-F4, a c-Myc inhibitor, at indicated doses, for
18 h. Subsequently, NKG2D ligand mRNA expression was analyzed using
(A) multiplex reverse transcription-PCR and (B) relative expression
ratios were compared with those of untreated control using image
analyzing software (Quantity One). *P<0.05, **P<0.01 and
***P<0.001. ACTB, actin-β; MIC, MHC class I polypeptide-related
sequence; NKG2D, natural killer group 2 member D; RPL19, ribosomal
protein L19; ULBP, UL16 binding protein. |
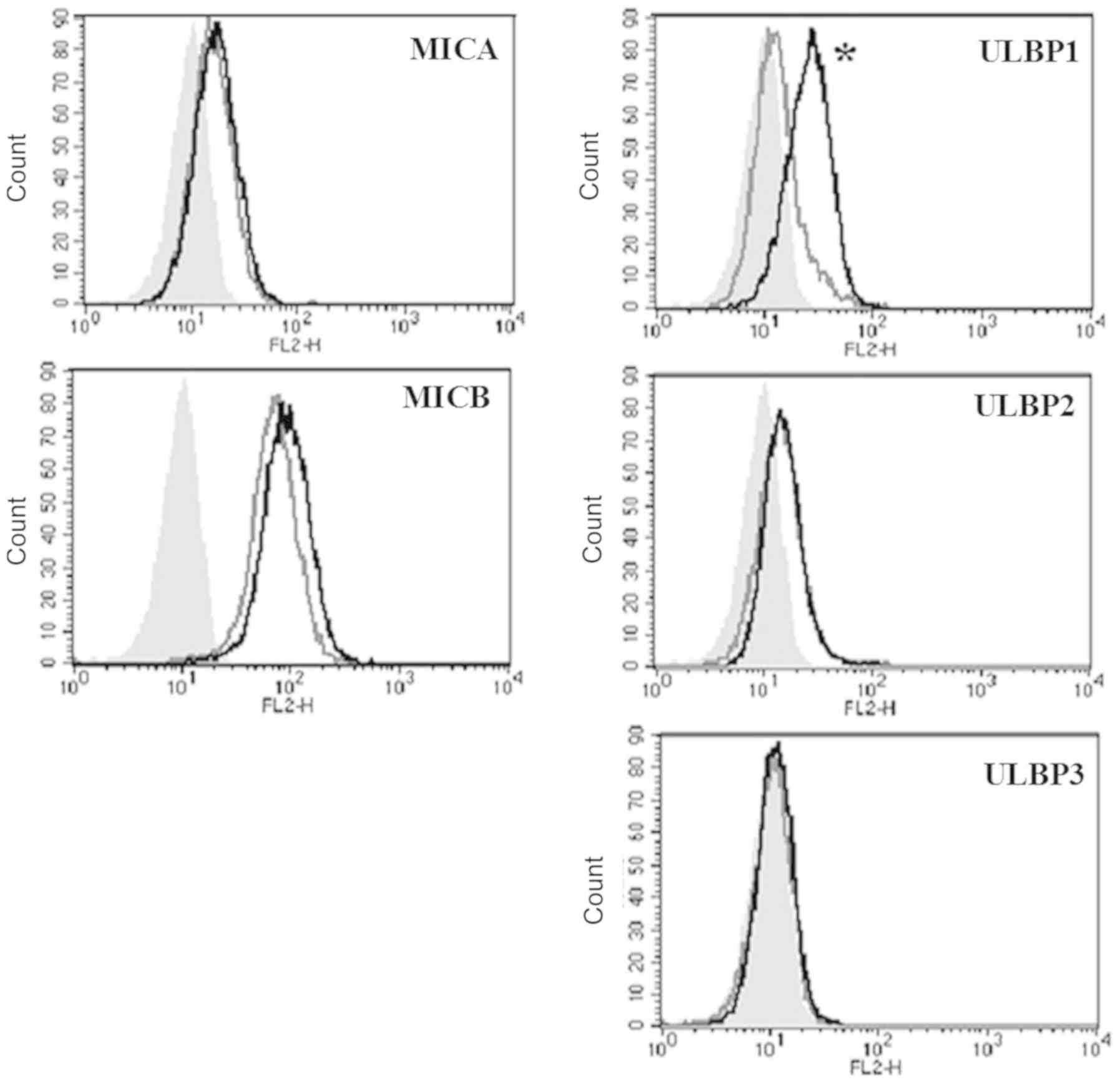
10058-F4 increases the surface
expression of MICB and ULBP1
To observe whether the inhibition of c-Myc could
induce the expression of surface proteins of NKG2D ligands, the
surface proteins were investigated using flow cytometry with
specific antibodies after treatment with 10058-F4 and c-Myc
inhibitor for 24 h. Histograms of 5 NKG2D ligands were presented
(Fig. 2). The expression of ULBP1
was prominently increased, whereas the expression of MICB was
slightly increased. The expression of MICA and ULBP2-3 on the
surface were not altered by treatment with 10058-F4.
Silencing of c-Myc using siRNA induces
the expression of MICB, ULBP1 and ULBP3 mRNA
c-Myc gene was successfully silenced by transfection
with c-Myc siRNA in a time dependent manner (Fig. 3A). c-Myc Silencing induced the
expression of MICB, ULBP1, and ULBP3 at the transcriptional level
(Fig. 3B). ULBP1 expression was
significantly induced after 48 h in a time-dependent manner. From
these c-Myc inhibition tests using 10058-F4 and siRNA, it could be
indicated that c-Myc might suppress the expression of MICB and
ULBP1.
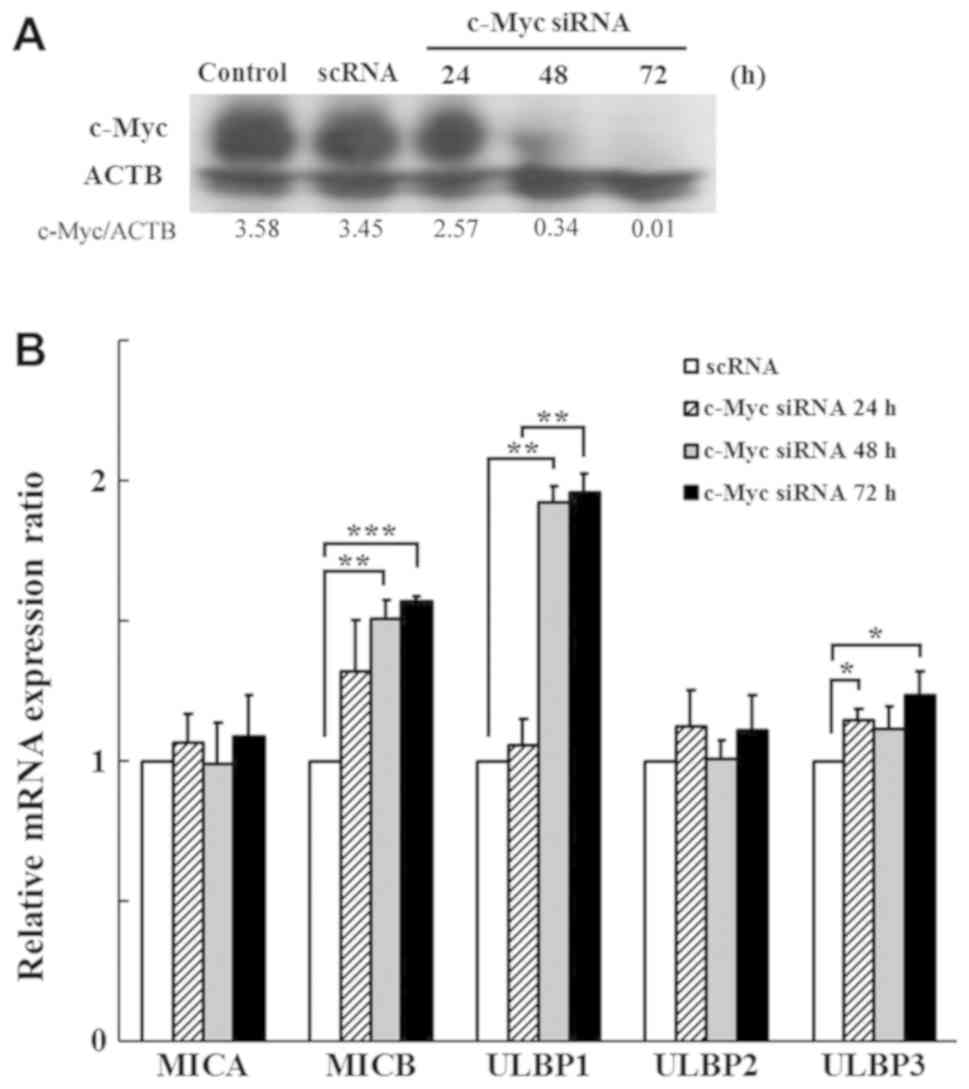 | Figure 3.Analysis of NKG2D ligand expression
following knockdown of c-Myc using siRNA in K562 cells. (A) K562
cells were transfected with scRNA and c-Myc siRNA. Efficiency of
c-Myc siRNA in K562 cells was confirmed by western blotting. The
densities were measured by Multi Gauge software and presented as
the ratio to ACTB. (B) Expression levels of NKG2D ligands were
analyzed using multiplex-PCR at 24, 48 and 72 h post siRNA
transfection. The altered expression levels were represented as
ratios relative to scRNA transfected cells. *P<0.05,
**P<0.01, ***P<0.001. ACTB, actin-β; c-Myc siRNA, c-Myc
targeted siRNA; MIC, MHC class I polypeptide-related sequence;
NKG2D, natural killer group 2 member D; scRNA, scrambled RNA;
siRNA, small interfering RNA; ULBP, UL16 binding protein. |
MICB expression is suppressed through
the upregulation of c-Myc and restored by c-Myc inhibition
To study whether the upregulation of c-Myc could
suppress the expression of NKG2D ligands, pCMV or pCMV6-Myc vector
was transfected into K562 cells and the transfected cells were
isolated by selection using G418. In c-Myc upregulated K562 cells,
surface proteins of all five NKG2D ligands have a trends of
reduction compared with that in pCMV transfected K562 cells.
Especially, MICB and ULBP 2 were decreased with statistically
significance (Fig. 4A). Although
the precise mechanisms are not clear, it was obvious that c-Myc
could inhibit the expression of NKG2D ligands. When c-Myc inhibitor
was added to the c-Myc upregulated cells, the expression of the
suppressed NKG2D ligands such as MICB and ULBP1/2 was restored
(Fig. 4B). Therefore, it could be
suggested that c-Myc negatively affected the expression of NKG2D
ligands.
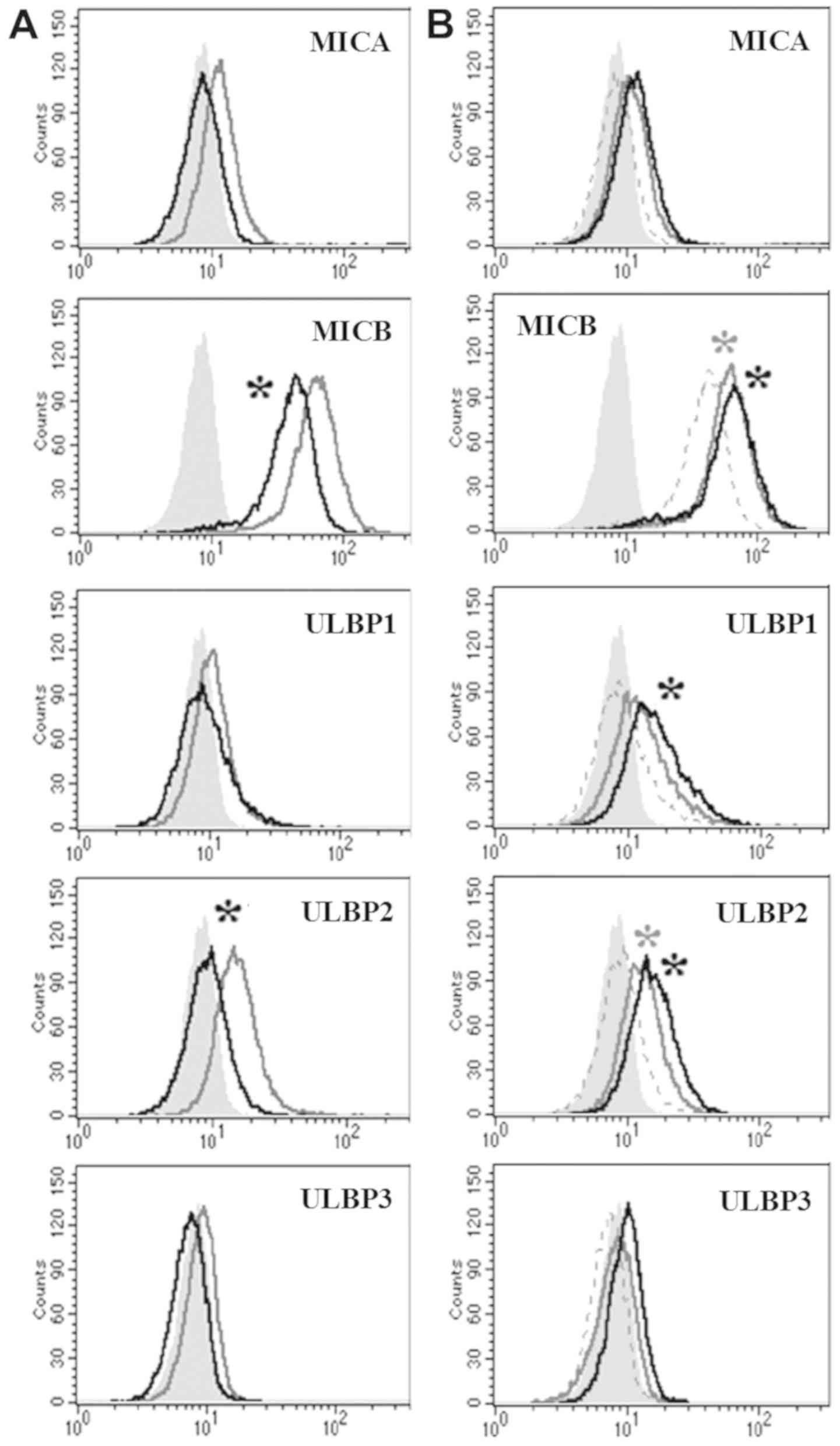 | Figure 4.Analysis of the expression levels of
NKG2D ligands in c-Myc-upregulated K562 cells. (A) Surface
expression levels of NKG2D ligands were analyzed using flow
cytometry at 48 h after pCMV-Myc vector transfection. Filled gray,
gray and black lines represent isotype, pCMV and pCMV-Myc
transfected cells, respectively. *P<0.05 pCMV6-empty transfected
cells vs. pCMV6-Myc transfected cells. (B) Once isolated, stable
c-Myc upregulated K562 cells were treated with 15 µM of 10058-F4
for 24 h and the surface expression of NKG2D ligands was analyzed
using flow cytometry. Filled gray, dot, gray and black lines
represent isotype, pCMV6-Myc transfected cells, pCMV6-Myc
transfected cells treated with 15 µM 10058-F4 and pCMV6-Myc
transfected cells treated with 30 µM 10058-F4, respectively.
*P<0.05 pCMV6-Myc transfected cells vs. pCMV6-Myc transfected
cells treated with 15 (gray) or 30 µM (black) 10058-F4. MIC, MHC
class I polypeptide-related sequence; NKG2D, natural killer group 2
member D; ULBP, UL16 binding protein. |
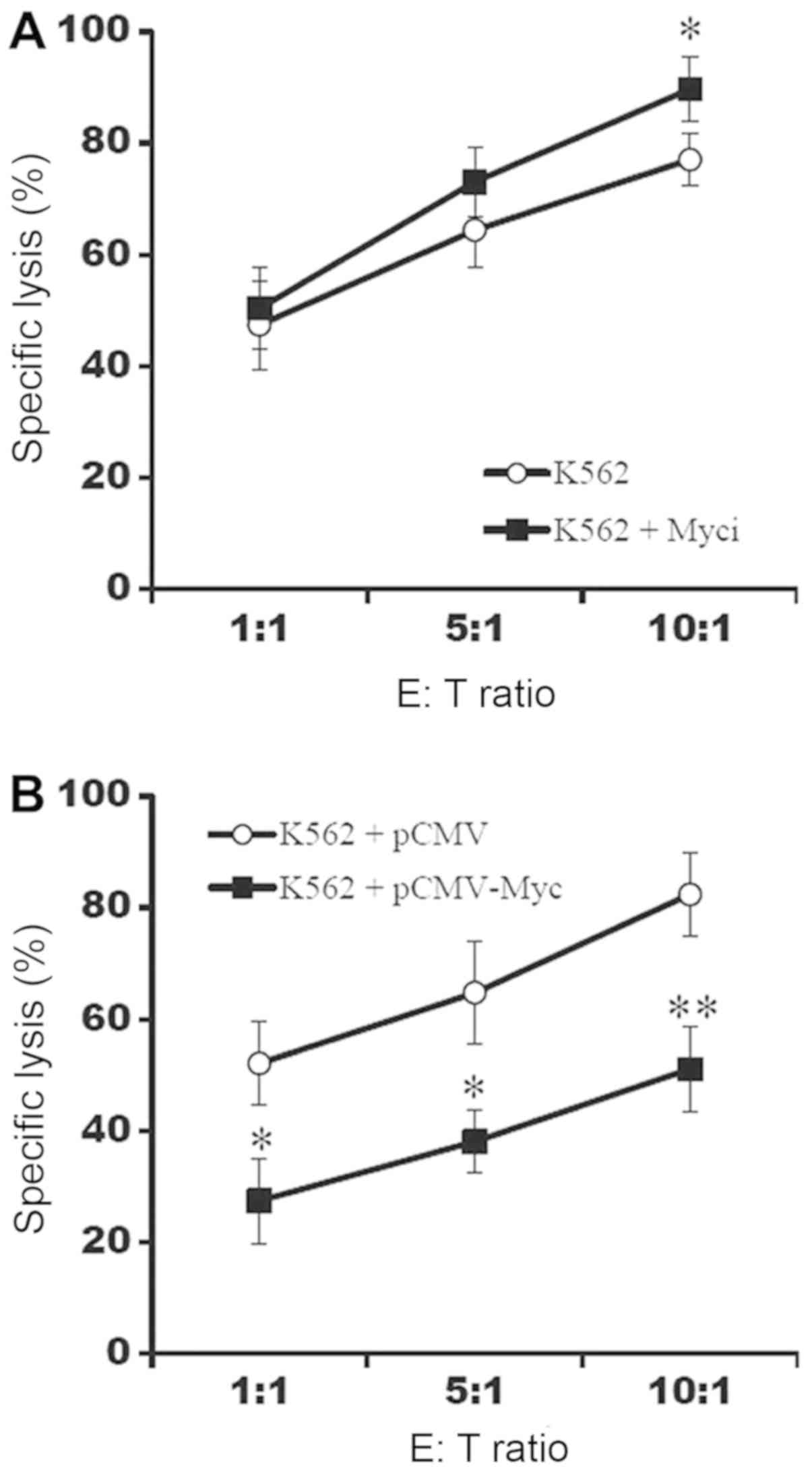
Upregulation of c-Myc suppresses the
susceptibility of K562 cells to NK-92 cells
To confirm whether c-Myc could affect the
susceptibility of K562 cells to NK-92 cells, K562 cells were
co-cultured with NK-92 cells and the proportion of dead cells was
determined through PI staining. The inhibition of c-Myc increased
the susceptibility of K562 cells to NK-92 cells (Fig. 5A) and the upregulation of c-Myc
decreased the susceptibility (Fig.
5). Therefore, it could be concluded that c-Myc suppressed the
susceptibility of K562 cells to NK-92, possibly through the
down-regulation of NKG2D ligand expression.
Discussion
c-Myc belongs to the MYC family, most members of
which are located on chromosome 8 and is related to cell
proliferation, cell growth, and apoptosis. It was reported that
malfunctioning of c-Myc leads to cancers of the breast, cervix,
colon, stomach, lungs, and leukocytes (2–4).
Since c-Myc influences the expression of many genes (5,6),
c-Myc overexpression in cancer cells is a factor behind poor
prognosis (19). It was suspected
that c-Myc might attenuate body defense against cancer and disturb
immune system through the altered expression of NKG2D ligands,
which are a kind of NK-cell activating ligands. In this study, we
investigated whether c-Myc affected the expression of NKG2D ligands
in chronic myeloid leukemia cells (K562 cells). The K562 cells
express BCR/ABL, a fusion oncoprotein, as well as abundant c-Myc
(20,21). Since K562 cells show impairment in
the functions of p53, which mediates Myc-induced apoptosis
(22,23), and since c-Myc transduction did not
enhance apoptosis of K562 cells (data not shown), it was thought
that K562 cells are adjustable to test viability and susceptibility
to NK cells in this study after alteration of c-Myc expression.
Although it was generally known that transformed
cells overexpressed NKG2D ligands, established cancer cells
acquired resistance against host immune systems and escaped immune
surveillance (24,25). Previously, it was demonstrated that
NKG2D ligand is not always upregulated in cancer cells when
compared with the adjacent normal cells (17). Therefore, we hypothesized that the
expression of NKG2D ligands on established cancer cells was
insufficient to evoke NK-cell-mediated anti-cancer immune
responses. It is already known that cancer cells use several immune
suppressive mechanisms to escape from NK cells, such as secretion
of TGF-beta and interleukin-6, increased extracellular shedding of
NKG2D ligands accompanying reduction in surface NKG2D ligands, and
increase of anti-apoptotic molecule (26–28).
Since successful NK-cell-based anti-cancer immunotherapy depends on
overcoming these immune escape mechanisms of cancer cells,
searching for molecules that affect NK-cell-mediated immune
responses was considered necessary.
Except BCR/ABL, which induces the expression of
NKG2D ligands (14), inhibition of
several oncogenes including Ras, EGFR, NF-κB, and Akt can induce
the expression of NKG2D ligands, therefore, it was thought that
these oncogenes might contribute to immune escape of cancer cells
(13,15,16).
However, it remains to be addressed whether c-Myc could modulate
the expression of NKG2D ligands. In the present study, we found
that overexpression of c-Myc decreased the expression of NKG2D
ligands including MICA, MICB, and ULBPs, and the inhibition of
c-Myc could restore the expression of NKG2D ligands. Depending on
the level of NKG2D ligands, the activity of NK cells was
altered.
Gasser et al (29) demonstrated that tumorigenesis of
ovarian epithelial cells by transduction with c-Myc did not induce
the expression of NKG2D ligands. Although these authors did not
assay inhibition of c-Myc in upregulated cells, they showed that
the transplanted cells had increased level of NKG2D ligands
(29). On the contrary, Nanbakhsh
et al (30) showed that
c-Myc had a role as a transcription factor in the expression of
ULBP1/3 in cytarabine-resistant acute myeloid leukemia cells. Since
cytarabine interferes with DNA synthesis and accumulates DNA
damage, DNA repair systems, which are key regulators of NKG2D
ligands, might complicate the results in the resistant cells.
Although it was not quite clear why c-Myc differently affected the
expression of NKG2D ligands in cytarabine-resistant acute myeloid
leukemia cells and K562 chronic myeloid leukemia cells, a variety
of functions of the hyperactivated c-Myc in tumorigenesis and
secondary reactions in varied cancer types might lead to the
differential expression of NKG2D ligands.
In conclusion, this study demonstrated that
inhibition of c-Myc induces NKG2D ligands in K562 cells, and
enhances their susceptibility to NK cells. Although there remain
many unsolved questions, inhibition of c-Myc might contribute to
better therapeutic outcome in the treatment of cancer patients when
combined with NK-cell-based cancer immunotherapy in future.
Acknowledgements
Not applicable.
Funding
This work was supported by the Dongnam Institute of
Radiological and Medical Sciences grant funded by the Korean
government (MSIT; grant no. 50595-2018).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YSL performed experiments, including PCR and flow
cytometry, and wrote the manuscript. WH performed experiments,
including western blotting and cytotoxicity assays. CHS performed
gene transfection. CDK developed the platform of multiplex PCR for
NKG2D ligands. YSP performed statistical analysis and
interpretation of data. JB designed and evaluated the study.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Authors' information
YSL: MS researcher at Department of Biochemistry,
Pusan National University School of Medicine; Department of
Molecular Cell Biology and Genetics, PNU BK21 Plus Biomedical
Science Education Center, Pusan National University School of
Medicine, Yangsan, Gyeongsangnam 50612, Republic of Korea. WH: MS
researcher at Department of Biochemistry, Pusan National University
School of Medicine; Department of Molecular Cell Biology and
Genetics, PNU BK21 Plus Biomedical Science Education Center, Pusan
National University School of Medicine, Yangsan, Gyeongsangnam
50612, Republic of Korea. CHS: PhD. Senior researcher at Department
of Research Center, Dongnam Institute of Radiological and Medical
Sciences, Gijang, Busan 46033, Republic of Korea. CDK: Professor at
Department of Biochemistry, Pusan National University School of
Medicine, Yangsan, Gyeongsangnam 50612, Republic of Korea. YSP: PhD
director at Department of Research Center, Dongnam Institute of
Radiological and Medical Sciences, Gijang, Busan 46033, Republic of
Korea. JB: Associate Professor at Department of Biochemistry, Pusan
National University School of Medicine; Department of Molecular
Cell Biology and Genetics, PNU BK21 Plus Biomedical Science
Education Center, Pusan National University School of Medicine,
Yangsan, Gyeongsangnam 50612, Republic of Korea.
References
|
1
|
Shortt J and Johnstone RW: Oncogenes in
cell survival and cell death. Cold Spring Harb Perspect Biol.
4(pii): a0098292012.PubMed/NCBI
|
|
2
|
Dang CV: MYC on the path to cancer. Cell.
149:22–35. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vennstrom B, Sheiness D, Zabielski J and
Bishop JM: Isolation and characterization of c-myc, a cellular
homolog of the oncogene (v-myc) of avian myelocytomatosis virus
strain 29. J Virol. 42:773–779. 1982.PubMed/NCBI
|
|
4
|
Zech L, Haglund U, Nilsson K and Klein G:
Characteristic chromosomal abnormalities in biopsies and
lymphoid-cell lines from patients with Burkitt and non-Burkitt
lymphomas. Int J Cancer. 17:47–56. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dang CV: c-Myc target genes involved in
cell growth, apoptosis, and metabolism. Mol Cell Biol. 19:1–11.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Grandori C and Eisenman RN: Myc target
genes. Trends Biochem Sci. 22:177–181. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bauer S, Groh V, Wu J, Steinle A, Phillips
JH, Lanier LL and Spies T: Activation of NK cells and T cells by
NKG2D, a receptor for stress-inducible MICA. Science. 285:727–729.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Diefenbach A, Jamieson AM, Liu SD, Shastri
N and Raulet DH: Ligands for the murine NKG2D receptor: Expression
by tumor cells and activation of NK cells and macrophages. Nat
Immunol. 1:119–216. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Groh V, Bahram S, Bauer S, Herman A,
Beauchamp M and Spies T: Cell stress-regulated human major
histocompatibility complex class I gene expressed in
gastrointestinal epithelium. Proc Natl Acad Sci USA.
93:12445–12450. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bakker AB, Phillips JH, Figdor CG and
Lanier LL: Killer cell inhibitory receptors for MHC class I
molecules regulate lysis of melanoma cells mediated by NK cells,
gamma delta T cells, and antigen-specific CTL. J Immunol.
160:5239–5245. 1998.PubMed/NCBI
|
|
11
|
Chang CC and Ferrone S: NK cell activating
ligands on human malignant cells: Molecular and functional defects
and potential clinical relevance. Semin Cancer Biol. 16:383–392.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hilpert J, Grosse-Hovest L, Grunebach F,
Buechele C, Nuebling T, Raum T, Steinle A and Salih HR:
Comprehensive analysis of NKG2D ligand expression and release in
leukemia: Implications for NKG2D-mediated NK cell responses. J
Immunol. 189:1360–1371. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bae JH, Kim JY, Kim MJ, Chang SH, Park YS,
Son CH, Park SJ, Chung JS, Lee EY, Kim SH and Kang CD: Quercetin
enhances susceptibility to NK cell-mediated lysis of tumor cells
through induction of NKG2D ligands and suppression of HSP70. J
Immunother. 33:391–401. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Boissel N, Rea D, Tieng V, Dulphy N, Brun
M, Cayuela JM, Rousselot P, Tamouza R, Le Bouteiller P, Mahon FX,
et al: BCR/ABL oncogene directly controls MHC class I chain-related
molecule A expression in chronic myelogenous leukemia. J Immunol.
176:5108–5116. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Heo W, Lee YS and Bae JH: Inhibition of
oncogenes affects the expression of NKG2D ligands in cancer cells.
J Life Sci. 23:1216–1222. 2013. View Article : Google Scholar
|
|
16
|
Peinado C, Kang X, Hardamon C, Arora S,
Mah S, Zhang H, Ngolab J and Bui JD: The nuclear factor-κB pathway
down-regulates expression of the NKG2D ligand H60a in vitro:
Implications for use of nuclear factor-κB inhibitors in cancer
therapy. Immunology. 139:265–274. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Park SW, Bae JH, Kim SD, Son YO, Kim JY,
Park HJ, Lee CH, Park DY, Kim JY, Lee MK, et al: Comparison of
level of NKG2D ligands between normal and tumor tissue using
multiplex RT-PCR. Cancer Invest. 25:299–307. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang MJ, Cheng YC, Liu CR, Lin S and Liu
HE: A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle
arrest, apoptosis, and myeloid differentiation of human acute
myeloid leukemia. Exp Hematol. 34:1480–1489. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Albajar M, Gómez-Casares MT, Llorca J,
Mauleon I, Vaqué JP, Acosta JC, Bermudez A, Donato N, Delgado MD
and León J: MYC in chronic myeloid leukemia: Induction of aberrant
DNA synthesis and association with poor response to imatinib. Mol
Cancer Res. 9:564–576. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Coulis CM, Lee C, Nardone V and Prokipcak
RD: Inhibition of c-myc expression in cells by targeting an
RNA-protein interaction using antisense oligonucleotides. Mol
Pharmacol. 57:485–494. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lozzio CB and Lozzio BB: Human chronic
myelogenous leukemia cell-line with positive Philadelphia
chromosome. Blood. 45:321–334. 1975.PubMed/NCBI
|
|
22
|
Law JC, Ritke MK, Yalowich JC, Leder GH
and Ferrell RE: Mutational inactivation of the p53 gene in the
human erythroid leukemic K562 cell line. Leuk Res. 17:1045–1050.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zindy F, Eischen CM, Randle DH, Kamijo T,
Cleveland JL, Sherr CJ and Roussel MF: Myc signaling via the ARF
tumor suppressor regulates p53-dependent apoptosis and
immortalization. Genes Dev. 12:2424–2433. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Campoli M and Ferrone S: Tumor escape
mechanisms: Potential role of soluble HLA antigens and NK cells
activating ligands. Tissue Antigens. 72:321–334. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dunn GP, Bruce AT, Ikeda H, Old LJ and
Schreiber RD: Cancer immunoediting: From immunosurveillance to
tumor escape. Nat Immunol. 3:991–998. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Eisele G, Wischhusen J, Mittelbronn M,
Meyermann R, Waldhauer I, Steinle A, Weller M and Friese MA:
TGF-beta and metalloproteinases differentially suppress NKG2D
ligand surface expression on malignant glioma cells. Brain.
129:2416–2425. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Groh V, Wu J, Yee C and Spies T:
Tumour-derived soluble MIC ligands impair expression of NKG2D and
T-cell activation. Nature. 419:734–738. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xu L, Chen X, Shen M, Yang DR, Fang L,
Weng G, Tsai Y, Keng PC, Chen Y and Lee SO: Inhibition of
IL-6-JAK/Stat3 signaling in castration-resistant prostate cancer
cells enhances the NK cell-mediated cytotoxicity via alteration of
PD-L1/NKG2D ligand levels. Mol Oncol. 12:269–286. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gasser S, Orsulic S, Brown EJ and Raulet
DH: The DNA damage pathway regulates innate immune system ligands
of the NKG2D receptor. Nature. 436:1186–1190. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nanbakhsh A, Pochon C, Mallavialle A,
Amsellem S, Bourhis JH and Chouaib S: c-Myc regulates expression of
NKG2D ligands ULBP1/2/3 in AML and modulates their susceptibility
to NK-mediated lysis. Blood. 123:3585–3595. 2014. View Article : Google Scholar : PubMed/NCBI
|