Introduction
Sepsis is defined as a life-threatening organ
dysfunction caused by a dysregulated host response to an infection
(1). Sepsis is particularly lethal
as it often follows a linear continuum from systemic inflammatory
response syndrome through to septic shock and organ function
failure (2–4). Despite advancements in antibiotic
therapy, immunotherapy and resuscitative strategies, sepsis remains
the leading cause of death in intensive care units (5).
In the pathophysiological process of sepsis, certain
gene expression levels markedly change in vivo and
contribute to the outcome of the disease (6,7).
Gene microarray or gene profiling are molecular detection
techniques that have been used for a number of years. Gene
microarray analysis can quickly detect gene expression levels at
various time-points, which is particularly effective for the
screening of differentially expressed genes (DEGs).
MicroRNAs (miRNAs/miRs) are a group of small
non-coding RNAs that regulate gene expression at the
post-transcriptional level and serve as key regulators in the
progression of many types of disease (8,9). To
investigate the roles of miRNAs and their target genes in sepsis,
Chen et al (10) detected
the altered expression of specific miRNAs and their target genes in
patients with sepsis, and identified specific miRNA and target
genes involved in the activation of immune and inflammatory
responses. In addition, previous studies have also indicated that
the expression level of miRNA was correlated with the mortality of
sepsis patients and certain miRNAs in the blood can be used to
predict the prognosis of sepsis (11,12).
The aim of the present study was to obtain key sepsis-related
miRNA-mRNA pairs in mice by analyzing mRNA and miRNA microarray
datasets, in order to provide molecular targets for diagnostic or
therapeutic strategies.
Materials and methods
Microarray data
The sepsis and control group blood gene expression
profiles GSE74952 and GSE55238 in mice were downloaded from the
Gene Expression Omnibus (GEO) database of the National Center for
Biotechnology Information (www.ncbi.nlm.nih.gov/geo). The microarray data of
GSE74952 was based on GPL21136 Platforms (Multiplex Circulating
miRNA Assay) and the miRNA profiles of 5 sepsis samples and four
control samples were obtained. These 5 sepsis samples were obtained
from septic mice models that were created by cecal ligation and
puncture (CLP). The microarray data of GSE55238 was based on
GPL1261 Platforms (Affymetrix Mouse Genome 430 2.0 Array) and the
mRNAs profile of 4 sepsis samples and 4 control samples were
obtained. These 4 sepsis samples were also obtained from CLP septic
mice models. The present study chose these 4 sepsis samples as Day
1, which was the same time-point used in GSE74952. GSE74952 was
conducted at the Massachusetts General Hospital, Charlestown, USA;
GSE55238 was conducted at the University of Florida, Gainesvile,
USA.
Data preprocessing and screening
strategy
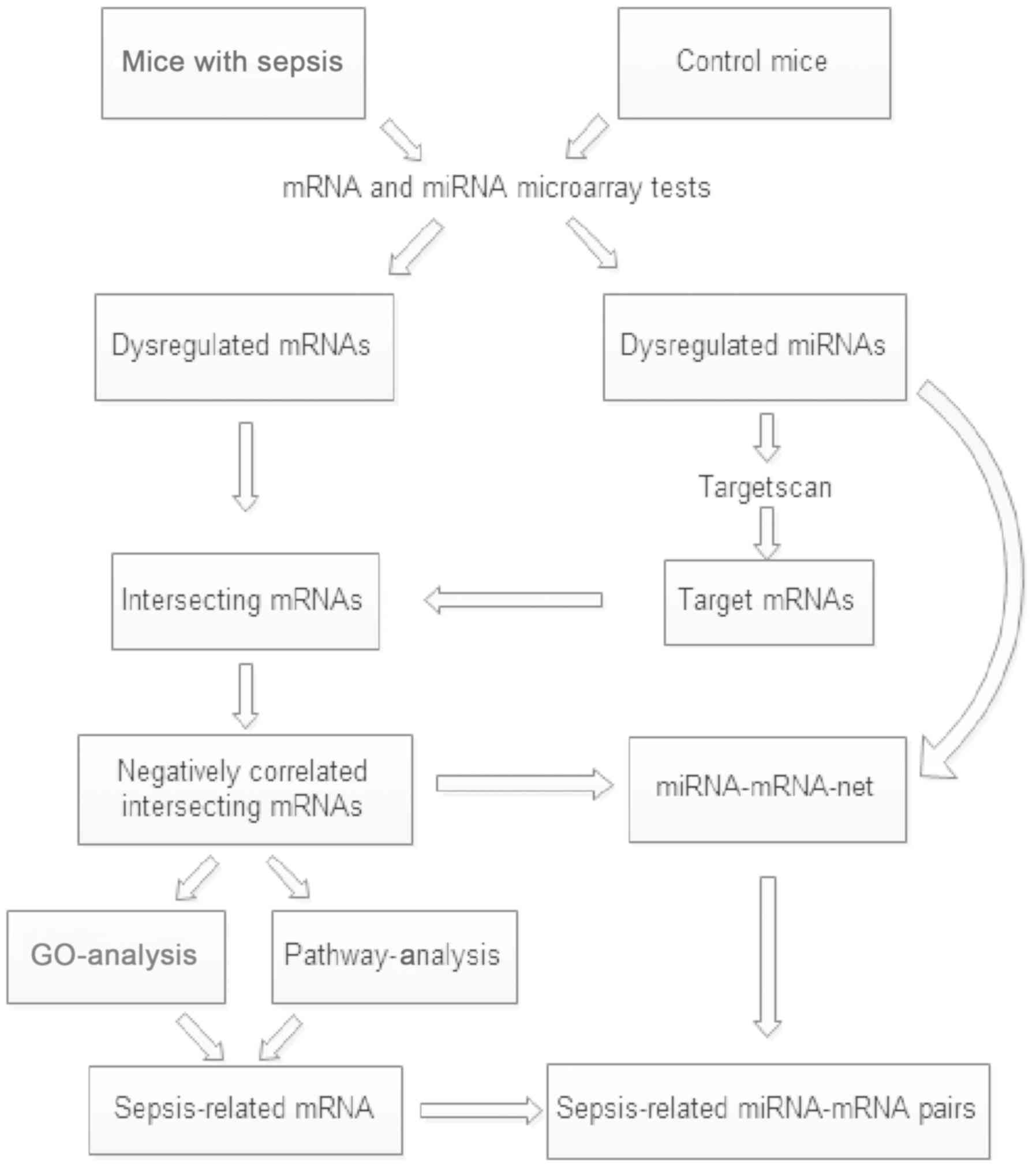
A multi-step strategy (Fig. 1) was used to identify genes
dysregulated in the sepsis model mice relative to the control
group. First, GEO2R was applied to identify the differentially
expressed miRNAs and mRNAs between sepsis and control samples.
GEO2R (www.ncbi.nlm.nih.gov/geo/geo2r/) is an interactive web
tool that is widely applied to detect DEGs by comparing two groups
of samples in a GEO series (11).
An adjusted P<0.05 and |log2 fold-change (FC)| >1 were set as
the cut-off criteria.
Second, the TargetScan database (version 7.2;
www.targetscan.org/mmu-72/) was used to
predict the target genes of dysregulated miRNAs and the overlap
between the negatively correlated target genes and differentially
expressed mRNAs was established. Gene sets data was processed using
VENNY 2.1 software (bioinfogp.cnb.csic.es/tools/venny/index.html).
Third, these interacting genes were classified
according to Gene Ontology (GO; http://geneontology.org/) and Kyoto Encyclopedia of
Genes and Genomes (KEGG; www.genome.jp/kegg) (13) pathways using Database for
Annotation, Visualization and Integration Discovery (DAVID)
software (david.ncifcrf.gov/). Based on the
enriched GO terms and significant KEGG pathways, the sepsis-related
mRNAs were screened.
Finally, Cytoscape software (version 3.6.0;
http://cytoscape.org/) was used to construct a
miRNA-mRNA network. Based on the sepsis-related mRNAs, the
sepsis-related miRNA-mRNA pairs were obtained.
Validation by reverse
transcription-quantitative (RT-q)PCR in mouse sepsis models
A total of 12 C57BL/6 male mice (aged ~8-10 weeks,
weighing 20–25 g) were purchased from the Experimental Animal
Center of Guangxi Medical University. All mice were housed in
specific pathogen-free facilities and acclimated for 1 week before
the operation. Mice were maintained at 22°C with 55% humidity under
a 12:12-h light/dark cycle for 1 week, with access to food and
water ad libitum. A total of 6 mice were employed to
establish the sepsis model using the CLP procedure according to a
previously reported method (12).
The remaining 6 mice were sham-operated to produce the control
group; control mice underwent the same procedure as the sepsis
model mice but without ligation and puncture of the cecum. All
operation procedures were under sterile conditions. After 1 day, ~1
ml blood was collected from the hearts of the mice. Red blood cells
were lysed using BD Pharm Lyse™ lysing buffer (Becton, Dickinson
and Company). Total RNA was extracted using RNAiso Plus (Takara
Bio, Inc.) and then reverse transcribed into cDNA using
PrimeScript™ RT reagent kit with gDNA Eraser (Takara Bio, Inc.) at
42°C for 2 min, 37°C for 15 min and 85°C for 5 sec according to the
manufacturer's protocol. qPCR was conducted using the SYBR Premix
Ex Taq™ II (Takara Bio, Inc.) according to the manufacturer's
protocol. The PCR primers were designed by the National Center for
Biotechnology Information (www.ncbi.nlm.nih.gov/) and provided by Sangon Biotech
Co., Ltd., (Table I). GAPDH was
selected as the internal reference for the genes. RT-qPCR
thermocycling conditions included: Pre-denaturation step at 95°C
for 30 sec, followed by 40 cycles of denaturing at 95°C for 5 sec
and extension at 60°C for 34 sec (Applied Biosystems 7500 Real Time
PCR System); each sample was performed in triplicate. Relative
expression values were calculated using the 2−∆∆Cq
method (14). The present study
was approved by the Animal Care Committee of Guangxi Medical
University (approval no. 201904001).
 | Table I.Primer sequences for reverse
transcription-quantitative PCR. |
Table I.
Primer sequences for reverse
transcription-quantitative PCR.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| CD8a |
TTCTGTCGTGCCAGTCCTTC |
TGGGACATTTGCAAACACGC |
| CD247 |
ATGGGTATTGACTCGCTCCG |
CAGGCTTCACCACTGAAATAAG |
| Zap70 |
ATCATGGCTTATGGCCGTGT |
CATGCACTCCCGGGTTAGAG |
| Ikbkb |
CCCACCCCTCCTCTCCTTAC |
TTCATACTGCCTCTGCGGTG |
| GAPDH |
CCCTTAAGAGGGATGCTGCC |
ACTGTGCCGTTGAATTTGCC |
Statistical analysis
Data were analyzed using SPSS version 17.0 (SPSS,
Inc.). Continuous variables are presented as the mean ± standard
deviation. A Student's t-test was applied for comparisons between
two groups. If the variance was not equal between two groups, a
Mann-Whitney U test was used for statistical analysis. P<0.05
was considered to indicate a statistically significant difference.
In addition to P-value, the false discovery rate value was also
used to rank the significance of the pathways.
Results
Identification of differentially
expressed miRNAs and mRNAs in sepsis model mice
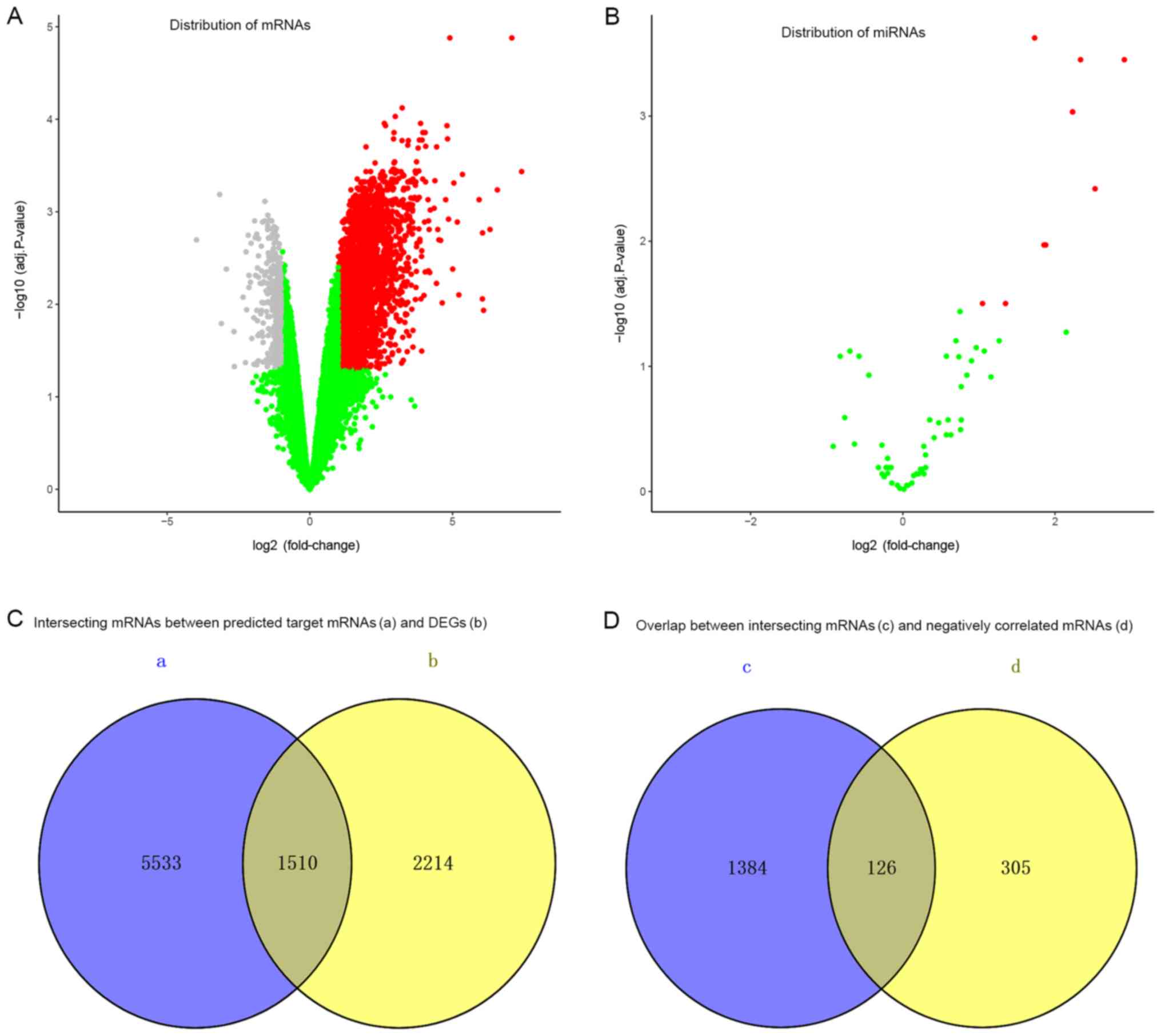
The gene expression levels of miRNAs and mRNAs were
download from the GEO database, respectively. Based on GEO2R
analysis, a total of 63 differentially expressed miRNAs and a total
of 21,765 differentially expressed mRNAs were identified in septic
mice compared with the control samples. There were 7,508
upregulated and 14,257 downregulated mRNAs in the GSE55238 dataset
(Fig. 2A), and 44 up- and 19
downregulated miRNAs in the GSE74952 dataset (Fig. 2B). Screened with the cut-off
criteria of adjusted P<0.05 and |log2 FC| >1, 10
differentially expressed miRNAs (Table II) and 3,724 differentially
expressed mRNAs were identified in sepsis model mice compared with
the control mice. These candidate targets included 10 upregulated
miRNAs and 3,293 up- and 431 downregulated mRNAs.
| Table II.Significantly dysregulated miRNAs
when comparing sepsis and control mice. |
Table II.
Significantly dysregulated miRNAs
when comparing sepsis and control mice.
| miRNA | Adjust P-value | LogFC | Differential
expression |
|---|
| miR-155-5p | <0.001 | 2.911 | Upregulated |
| miR-146a-5p | 0.004 | 2.527 | Upregulated |
| miR-21-5p | <0.001 | 2.337 | Upregulated |
| miR-370-3p | 0.001 | 2.234 | Upregulated |
| miR-28-5p | 0.001 | 2.231 | Upregulated |
| miR-590-5p | 0.011 | 1.879 | Upregulated |
| miR-29a-3p | 0.011 | 1.855 | Upregulated |
| miR-19a-3p | <0.001 | 1.731 | Upregulated |
| miR-103a-3p | 0.031 | 1.349 | Upregulated |
| let-7i-5p | 0.031 | 1.049 | Upregulated |
miRNA target gene prediction
Target mRNAs for differentially expressed miRNAs
were predicted using the TargetScan database. A total of 7,043
target genes were successfully predicted in 6 miRNAs, including
mmu-miR-155-5p, mmu-miR-146a-5p, mmu-miR-370-3p, mmu-miR-29a-3p,
mmu-miR-19a-3p and mmu-let-7i-5p.
Selection of disease-associated
miRNA-mRNA pairs
A total of 1,510 intersecting mRNAs between the
predicted target mRNAs and differentially expressed mRNAs were
selected (Fig. 2C), of which 126
mRNAs that were negatively correlated with their predicted miRNAs
matches were selected (Fig. 2D).
Based on differentially expressed miRNAs and their negatively
correlated mRNAs, a total of 147 miRNA-mRNA pairs were
predicted.
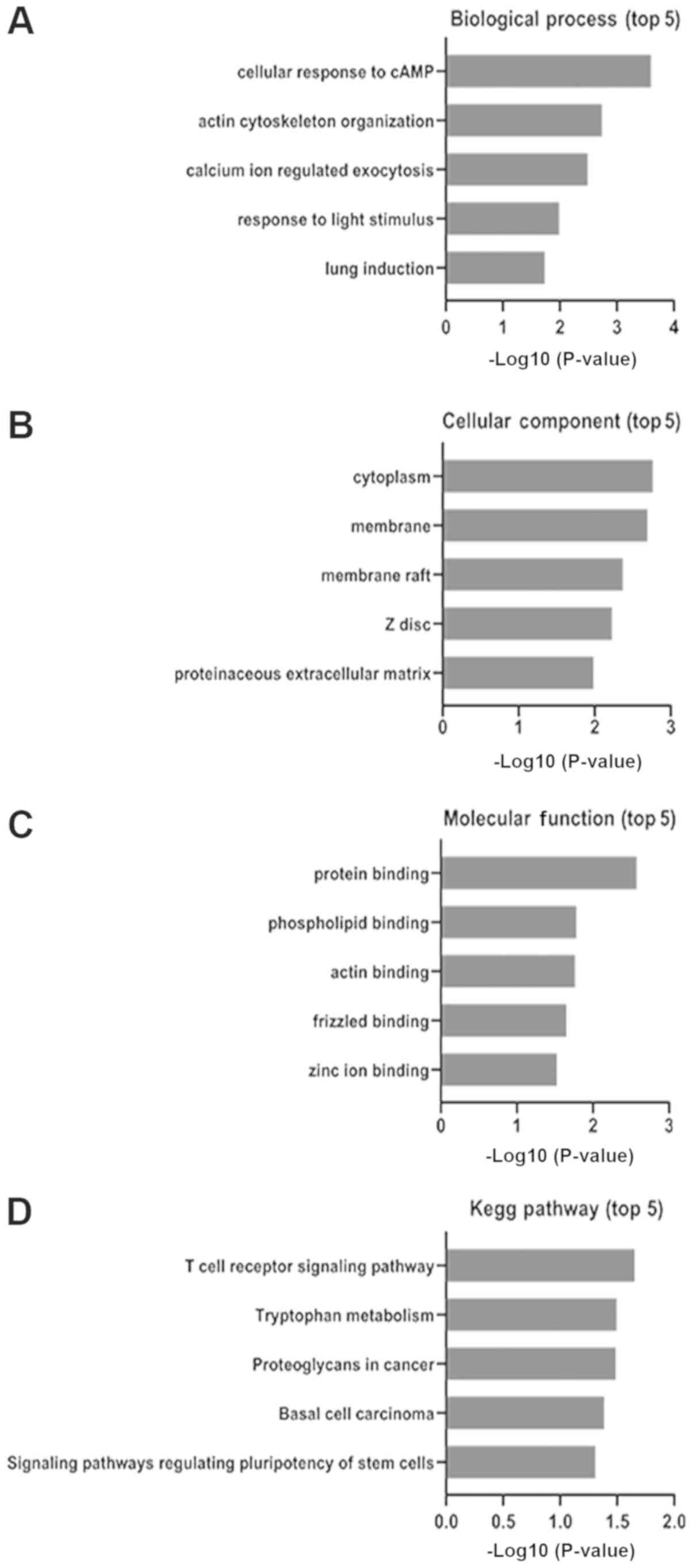
Selected mRNA ontology analysis in
sepsis model mice
The functional enrichment of 126 candidate DEGs were
analyzed using DAVID (david.ncifcrf.gov/; Table III). A total of three GO category
results are presented including biological processes, cellular
components and molecular functions. The biological process results
revealed that the selected DEGs were mainly enriched in ‘cellular
response to cAMP’, ‘actin cytoskeleton organization’ and ‘calcium
ion regulated exocytosis’ (Fig.
3A). The cellular component results revealed that selected DEGs
were mainly enriched in ‘cytoplasm’, ‘membrane’ and ‘membrane raft’
(Fig. 3B). The molecular function
analysis revealed that the selected DEGs mainly enriched in
‘protein binding’, ‘phospholipid binding’ and ‘actin binding’
(Fig. 3C). These results
demonstrated that the majority of the selected DEGs were enriched
in ‘binding’ and ‘cell signaling’.
| Table III.The 126 negatively correlated
intersecting genes with their predicted miRNAs. |
Table III.
The 126 negatively correlated
intersecting genes with their predicted miRNAs.
| Style | Genes name |
|---|
| Downregulated | Rnf123, Boc, Sox10,
Ebf1, 1700019N19Rik, Zdhhc7, Cbfa2t2, Cblc, Gjc1, Pip5k1b, Grid1,
Cacna1e, Arhgap17, Hip1r, Wnt2b, Fbln5, Cend1, Cd8a, Samd11,
Trp53i11, Haao, Phxr4, Aanat, Ralgps2, Gulp1, P2rx3, Mlxipl,
Hapln4, Naa40, Kcnk5, Ptgis, Fkbp8, Cgnl1, Dusp9, Fads1, R3hdm4,
Apobec2, Clip4, Spsb1, Tet3, Arhgef18, Ikbkb, Myo1e, Specc1,
Npepl1, Capn8, Polg, Klhl17, Nr5a1, 6330408A02Rik, Stx1a, Kmt2a,
Il5ra, Wnt7a, Lurap1, Brat1, Marf1, Lynx1, Pde11a, H2-Eb2, Dcun1d2,
Rnf165, Gpr50, Wnt2, Slc4a1, Ctnnd1, Wsb2, Zfp882, Dgcr6, Faim2,
Synpo2l, Fam222a, Hmg20a, Mark4, Bmp1, Bgn, Efna4, Unc45a, Ccr6,
Bcl2, Tgif2, Cmtr2, Smtnl2, Slc8a1, Asphd2, Tmem86b, Eef2k, Ermp1,
Ucp2, Adal, Snrnp70, Sumo3, Fam193a, 2-Mar, Atp9a, Dmtn, Dtnb,
Gorasp1, Vps13a, Kynu, Usp15, Ank1, Pmf1, Slc2a8, Snap47, Cers4,
Serpinh1, Mpp3, Plk2, Zap70, Igfbp5, Cd247, Akap4, Trappc12, Prrt4,
Enpp2, Slc6a2, Ehd2, Wasf3, Psd, Rapgef4, Usp14, Slc26a7, Sgip1,
Kl, Adipor1 |
Signaling pathway enrichment
analysis
The signaling pathway enrichment of 126 candidate
DEGs were analyzed using KEGG pathway online databases. The
selected DEGs were significantly enriched in ‘T-cell receptor
signaling pathway’, ‘tryptophan metabolism’ and ‘proteoglycans in
cancer’ (Fig. 3D). Based on a
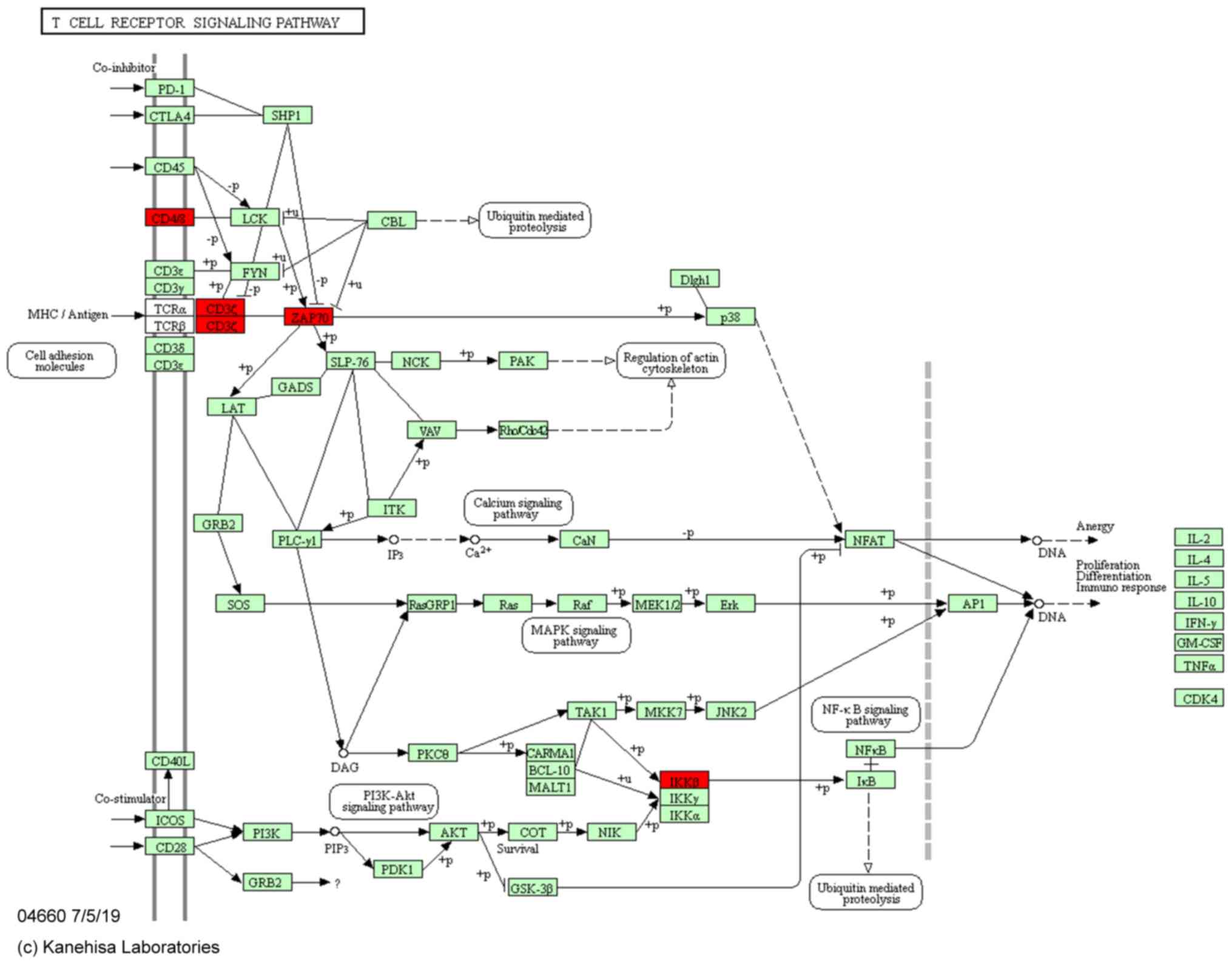
previous study (15), the T-cell
receptor signaling pathway is an important signaling pathway
associated with sepsis. In the present study, 4 sepsis-related
mRNAs were found in this pathway including CD8a, CD247, Zap70 and
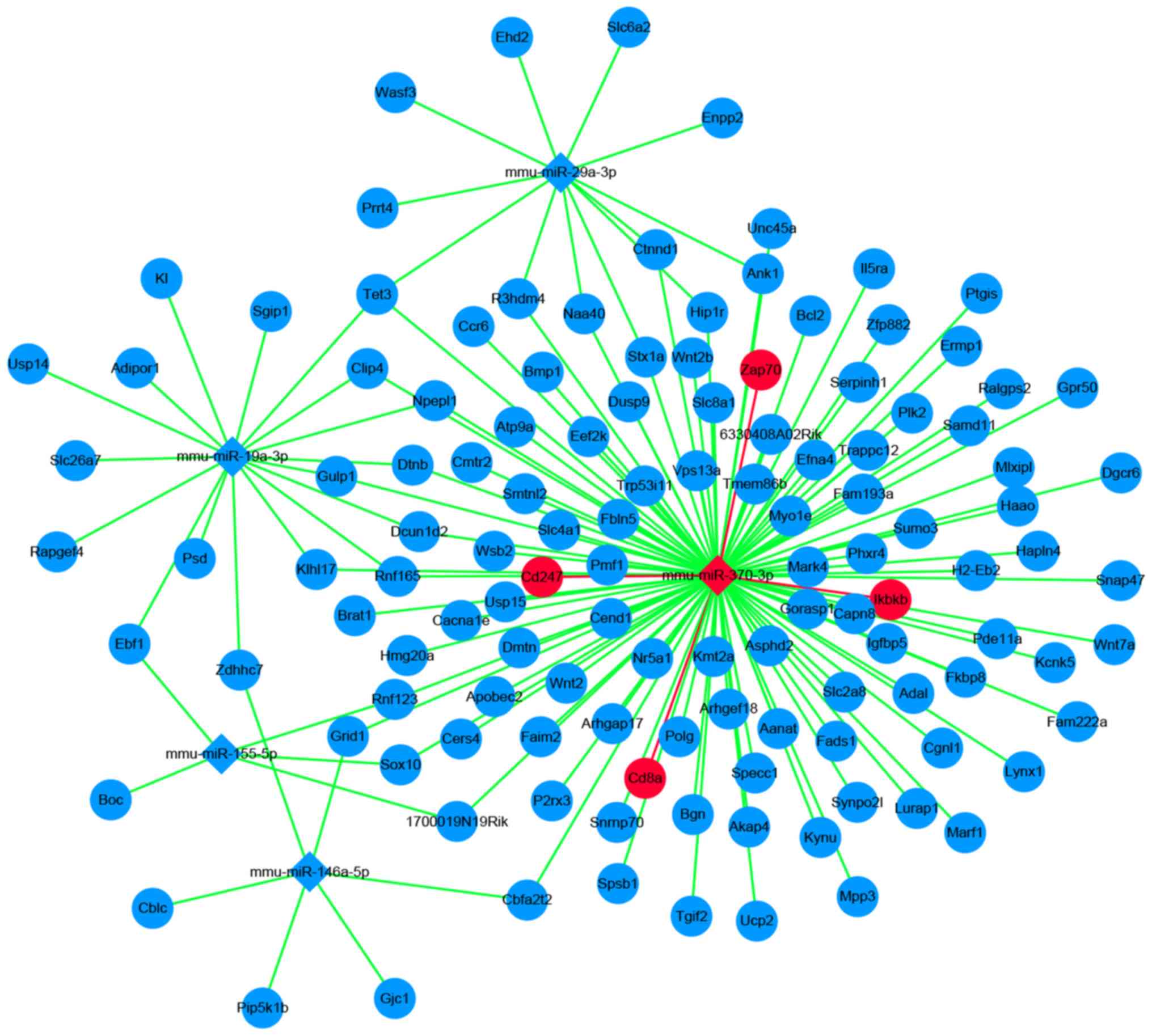
inhibitor of nuclear factor κ B kinase subunit β (Ikbkb; Table IV; Fig. 4).
| Table IV.The top 5 signaling pathways of
differentially expressed genes. |
Table IV.
The top 5 signaling pathways of
differentially expressed genes.
| KEGG pathway | Name | Count | P-value | FDR | Genes |
|---|
| mmu04660 | T-cell receptor
signaling pathway | 4 | 0.022 | 22.085 | CD8a, CD247, Zap70,
Ikbkb |
| mmu00380 | Tryptophan
metabolism | 3 | 0.032 | 30.270 | Kynu, Haao,
Aanat |
| mmu05205 | Proteoglycans in
cancer | 5 | 0.032 | 30.696 | Wnt2, Cblc, Ank1,
Wnt7a, Wnt2b |
| mmu05217 | Basal cell
carcinoma | 3 | 0.041 | 37.321 | Wnt2, Wnt7a,
Wnt2b |
| mmu04550 | Signaling pathways
regulating pluripotency of stem cells | 4 | 0.049 | 42.819 | Wnt2, Dusp9, Wnt7a,
Wnt2b |
miRNA-mRNA network construction
A total of 126 candidate mRNAs and 6 miRNAs were
analyzed using Cytoscape software. A total of 147 miRNA-mRNA pairs
were presented in the network. Based on this, 4 sepsis-related
mRNAs and 4 sepsis-related miRNA-mRNA pairs were obtained (Fig. 5).
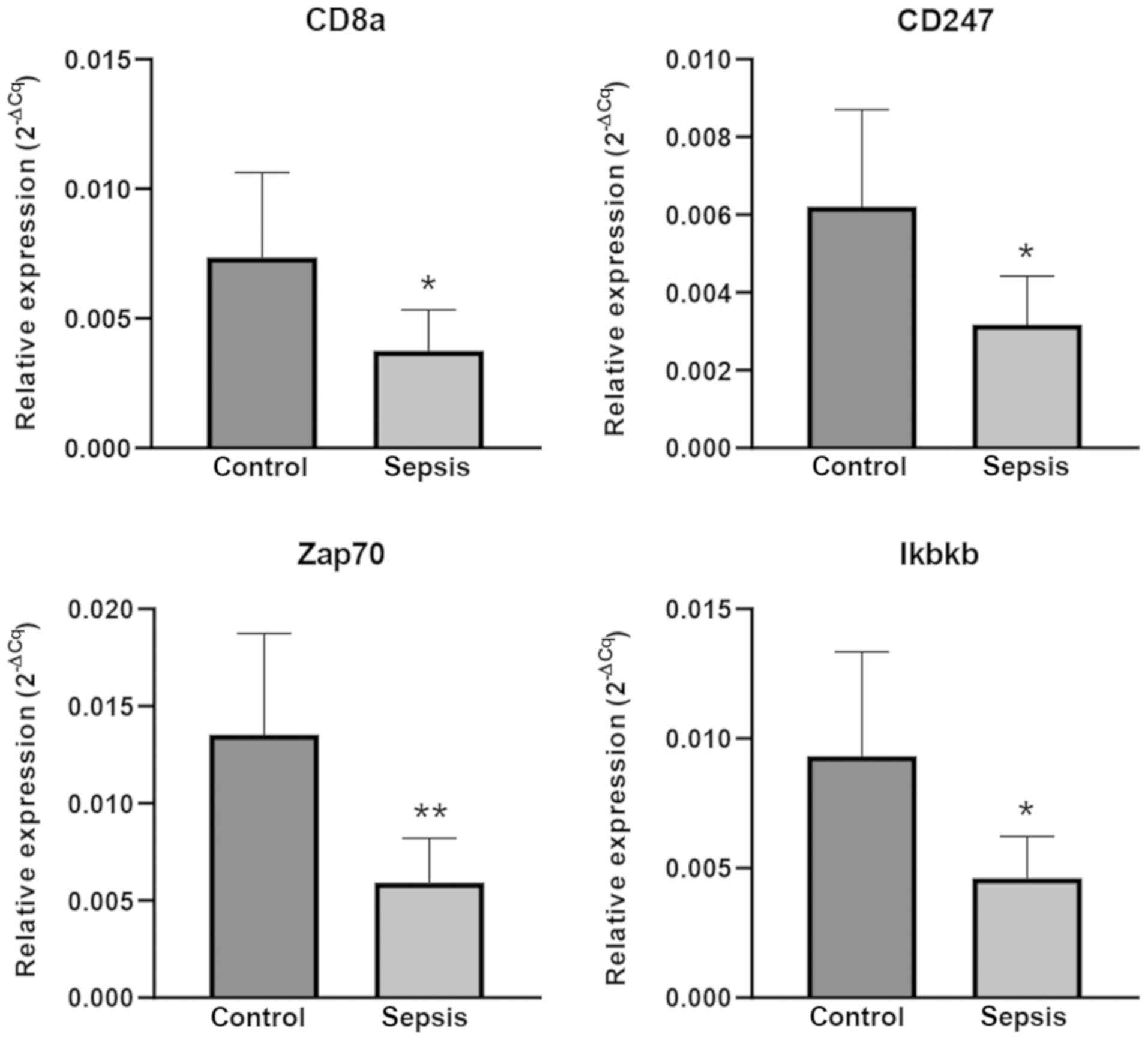
RT-qPCR validation
To confirm the reliability of the findings from the
bioinformatics microarray analysis, 4 mRNAs [cluster of
differentiation (CD)8a, CD247, Zap70 and Ikbkb] were selected for
validation by RT-qPCR in 6 pairs of matched sepsis and control
C57BL/6 mice. According to the experimental results, the relative
expression levels were similar between microarray and RT-qPCR
experiments. In sepsis model mice, the expression level of CD8a,
CD247, Zap70 and Ikbkb were significantly downregulated (P<0.05;
Fig. 6).
Discussion
Numerous studies have been performed to reveal the
underlying mechanisms of sepsis formation and progression in the
past several decades; however, the mortality rates of sepsis are
still very high worldwide (3,5). The
majority of previous studies have indicated that the high mortality
of sepsis is correlated with immune dysfunction, which may be due
to T-cell dysfunction (16,17).
However, which molecules change and how they interact during sepsis
are still unclear. The present study integrated two datasets
including miRNA and mRNA microarrays from different groups, using
bioinformatics methods to analyze data. Through DEG screening,
miRNA gene target prediction, GO terms and KEGG pathway analysis, 1
sepsis-related miRNA and 4 sepsis-related mRNAs including
mmu-miR-370-3p, CD8a, CD247, Zap70 and Ikbkb were obtained, of
which mmu-miR-370 was upregulated and the others were
downregulated. Subsequent RT-qPCR analysis validated this finding.
Through the construction of the miRNA-mRNA network, 4
sepsis-related miRNA-mRNA pairs were identified.
A septic event triggers high levels apoptosis in
immune cells such as T-cells, resulting in suppressed immune
functions, often inducing an increased susceptibility to secondary
infections (5,15,18,19).
The CLP mouse model of sepsis is considered the gold standard in
experimental sepsis research. However, sepsis induction varies
among different research groups, making it a challenge to compare
data among different studies. Despite the variation in sepsis
induction, the majority of studies have demonstrated that sepsis
leads to a significant decrease in immune factors. For instance,
Condotta et al (20)
demonstrated that polymicrobial sepsis exacerbated CD8+
T-cell exhaustion and reduced interferon-γ production. In addition,
Hotchkiss et al (21)
indicated that there is a significant loss of B and CD4 T-cells in
sepsis, which may contribute to immunosuppression.
In the pathological progression of sepsis, the host
response is disturbed by excessive inflammation and immune
suppression, and fails to return to normal homeostasis (22). The T-cell receptor signaling
pathway is an important pathway involved in immune responses.
Previous studies have suggested that the impairment of the T-cell
receptor signaling pathway was one of important factors for
immunosuppression in sepsis (23,24).
Based on KEGG pathway analysis, the hub genes
including CD8a, CD247, Zap70 and Ikbkb were enriched in the T-cell
receptor signaling pathway. These genes play an important role in
immunity to fight against foreign pathogens. CD8a encodes the CD8α
chain of the dimeric CD8 protein. CD8 is a coreceptor for the
T-cell receptor (TCR)-mediated recognition of major
histocompatibility complex class I, which involved cytotoxic T
lymphocyte activation and target cell lysis (25). Therefore, CD8+ T-cells
play an important role in the control and eradication of invading
pathogens (26). CD8 deficiency
increases susceptibility to infection (27). Harland et al (28) revealed that CpG methylation of the
CD8a locus has a potential role in the downregulation of CD8.
However, whether this epigenetic modification is involved in the
process of sepsis is still unclear. CD247 (also known as CD3z) is a
part of the TCR complex, which plays a key role in receptor
expression and signaling associated to T-cell functions (29,30).
Eldor et al (31) reported
that CD247 downregulation is associated with immunosuppression in
T-cells and was correlated with disease progression and severity in
patients with type 2 diabetes. Zap70 was enriched in the TCR
signaling pathway. The functional deletion of Zap70 can lead to a
serious immunodeficiency (32). In
these patients, T-cells are inadequately supervised by the thymus
microenvironment, exhibit decreased apoptosis and cannot
differentiate into Th2 T-cells (33). Huang et al (34) reported that the downregulation of
Zap70 accelerated disease progression of neonatal sepsis. IKBKB
encodes IKB kinase 2 (also known as IKKβ /IKK2), a component of the
nuclear factor-κB signaling pathway. IKKβ can phosphorylate and
degrade the inhibitors of κB, and then improve target gene
transcription (35,36). A number of studies have indicated
that a mutation in IKBKB can lead to a lack of IKKβ and IKKβ
deficiency leads to an inadequate response to stimuli in immune
cells, finally resulting in immune impairment (37,38).
In the present study, using mRNA and miRNA
microarray datasets and bioinformatics analysis, 4 sepsis-related
miRNA-mRNA pairs were obtained, which were enriched in the TCR
signaling pathway. The differences in the expression levels also
reflect functional changes in sepsis, which may contribute to
immune deficiency. These results could improve understanding of the
underlying molecular events in sepsis and these candidate
miRNA-mRNA pairs may be predictors of, or therapy targets for
sepsis. However, there were certain limitations in the present
study. Firstly, the number of samples in the two datasets was
small. Secondly, the CLP model mice of the two datasets were not
established by the same research group. Thirdly, mmu-miR-370-3p was
not verified and the causal regulatory relationship between miRNA
and mRNA was not further confirmed. This causal regulation
relationship may be verified using miR-mimics, in vitro
inhibitors or mutant generation and adenoviral-mediated miRNA
delivery in vivo. These experiments are important for future
studies.
In conclusion, using miRNA and mRNA microarray
datasets and multi-step bioinformatics analysis, four significant
miRNA-mRNA pairs were found in sepsis model mice. These hub genes
were enriched in the TCR signaling pathway, played an important
role in immunity and may be markers or therapeutic targets for
sepsis.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant. no. 81860334) and
Fujian Provincial Natural Science Foundation (grant. no.
2019J01585).
Availability of data and materials
The datasets analyzed (GSE74952 and GSE55238) during
the present study are available in the Gene Expression Omnibus
database.
Authors' contributions
JC analyzed the microarray data, performed the
experiments and was a major contributor in writing the manuscript.
ML downloaded the datasets and performed statistical analysis. SZ
designed the study and edited the manuscript. All authors have read
and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Animal Care
Committee of Guangxi Medical University (approval no.
201904001).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
miRNA
|
microRNA
|
|
DEGs
|
differentially expressed genes
|
|
DAVID
|
Database for Annotation, Visualization
and Integration Discovery
|
|
GEO
|
Gene Expression Omnibus
|
|
CLP
|
cecal ligation and puncture
|
|
GO
|
Gene Ontology
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
FC
|
fold-change
|
|
TCR
|
T-cell receptor
|
References
|
1
|
Singer M, Deutschman CS, Seymour CW,
Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche
JD, Coopersmith CM, et al: The third international consensus
definitions for sepsis and septic shock (Sepsis-3). JAMA.
315:801–810. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cecconi M, Evans L, Levy M and Rhodes A:
Sepsis and septic shock. Lancet. 392:75–87. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gobatto AL, Besen BA and Azevedo LC: How
can we estimate sepsis incidence and mortality? Shock 47 (1S Suppl
1). S6–S11. 2017.
|
|
4
|
Martin GS: Sepsis, severe sepsis and
septic shock: Changes in incidence, pathogens and outcomes. Expert
Rev Anti Infect Ther. 10:701–706. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mayr FB, Yende S and Angus DC:
Epidemiology of severe sepsis. Virulence. 5:4–11. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vincent JL: Individual gene expression and
personalised medicine in sepsis. Lancet Respir Med. 4:242–243.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Maslove DM and Wong HR: Gene expression
profiling in sepsis: Timing, tissue, and translational
considerations. Trends Mol Med. 20:204–213. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ullah S, John P and Bhatti A: MicroRNAs
with a role in gene regulation and in human diseases. Mol Biol Rep.
41:225–232. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Eledge MR and Yeruva L: Host and pathogen
interface: MicroRNAs are modulators of disease outcome. Microbes
Infect. 20:410–415. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen J, Jiang S, Cao Y and Yang Y: Altered
miRNAs expression profiles and modulation of immune response genes
and proteins during neonatal sepsis. J Clin Immunol. 34:340–348.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Barrett T, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, Holko M, et al: NCBI GEO: Archive for functional
genomics data sets-update. Nucleic Acids Res. 41:D991–D995. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rittirsch D, Huber-Lang MS, Flierl MA and
Ward PA: Immunodesign of experimental sepsis by cecal ligation and
puncture. Nat Protoc. 4:31–36. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kanehisa M, Sato Y, Furumichi M, Morishima
K and Tanabe M: New approach for understanding genome variations in
KEGG. Nucleic Acids Res. 47:D590–D595. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Borken F, Markwart R, Requardt RP,
Schubert K, Spacek M, Verner M, Rückriem S, Scherag A, Oehmichen F,
Brunkhorst FM and Rubio I: Chronic critical illness from sepsis is
associated with an enhanced TCR response. J Immunol. 198:4781–4791.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Patil NK, Bohannon JK, Luan L, Guo Y,
Fensterheim B, Hernandez A, Wang J and Sherwood ER: Flt3 ligand
treatment attenuates T cell dysfunction and improves survival in a
murine model of burn wound sepsis. Shock. 47:40–51. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ramonell KM, Zhang W, Hadley A, Chen CW,
Fay KT, Lyons JD, Klingensmith NJ, McConnell KW, Coopersmith CM and
Ford M: CXCR4 blockade decreases CD4+ T cell exhaustion and
improves survival in a murine model of polymicrobial sepsis. PLoS
One. 12:e01888822017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Oami T, Watanabe E, Hatano M, Sunahara S,
Fujimura L, Sakamoto A, Ito C, Toshimori K and Oda S: Suppression
of T cell autophagy results in decreased viability and function of
T cells through accelerated apoptosis in a murine sepsis model.
Crit Care Med. 45:e77–e85. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cao C, Chai Y, Shou S, Wang J, Huang Y and
Ma T: Toll-like receptor 4 deficiency increases resistance in
sepsis-induced immune dysfunction. Int Immunopharmacol. 54:169–176.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Condotta SA, Khan SH, Rai D, Griffith TS
and Badovinac VP: Polymicrobial sepsis increases susceptibility to
chronic viral infection and exacerbates CD8+ T cell
exhaustion. J Immunol. 195:116–125. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hotchkiss RS, Tinsley KW, Swanson PE,
Schmieg RE Jr, Hui JJ, Chang KC, Osborne DF, Freeman BD, Cobb JP,
Buchman TG and Karl IE: Sepsis-induced apoptosis causes progressive
profound depletion of B and CD4+ T lymphocytes in humans. J
Immunol. 166:6952–6963. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
van der Poll T, van de Veerdonk FL,
Scicluna BP and Netea MG: The immunopathology of sepsis and
potential therapeutic targets. Nat Rev Immunol. 17:407–420. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Boomer JS, To K, Chang KC, Takasu O,
Osborne DF, Walton AH, Bricker TL, Jarman SD II, Kreisel D,
Krupnick AS, et al: Immunosuppression in patients who die of sepsis
and multiple organ failure. JAMA. 306:2594–2605. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Inoue S, Suzuki K, Komori Y, Morishita Y,
Suzuki-Utsunomiya K, Hozumi K, Inokuchi S and Sato T: Persistent
inflammation and T cell exhaustion in severe sepsis in the elderly.
Crit Care. 18:R1302014. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gil D, Schrum AG, Daniels MA and Palmer E:
A role for CD8 in the developmental tuning of antigen recognition
and CD3 conformational change. J Immunol. 180:3900–3909. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Harty JT, Tvinnereim AR and White DW: CD8+
T cell effector mechanisms in resistance to infection. Annu Rev
Immunol. 18:275–308. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dumontet E, Osman J, Guillemont-Lambert N,
Cros G, Moshous D and Picard C: Recurrent respiratory infections
revealing CD8α Deficiency. J Clin Immunol. 35:692–695. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Harland KL, Day EB, Apte SH, Russ BE,
Doherty PC, Turner SJ and Kelso A: Epigenetic plasticity of Cd8a
locus during CD8(+) T-cell development and effector differentiation
and reprogramming. Nat Commun. 5:35472014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Irving BA and Weiss A: The cytoplasmic
domain of the T cell receptor zeta chain is sufficient to couple to
receptor-associated signal transduction pathways. Cell. 64:891–901.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
D'Oro U, Munitic I, Chacko G, Karpova T,
McNally J and Ashwell JD: Regulation of constitutive TCR
internalization by the zeta-chain. J Immunol. 169:6269–6278. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Eldor R, Klieger Y, Sade-Feldman M, Vaknin
I, Varfolomeev I, Fuchs C and Baniyash M: CD247, a novel T
cell-derived diagnostic and prognostic biomarker for detecting
disease progression and severity in patients with type 2 diabetes.
Diabetes Care. 38:113–118. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Picard C, Dogniaux S, Chemin K,
Maciorowski Z, Lim A, Mazerolles F, Rieux-Laucat F, Stolzenberg MC,
Debre M, Magny JP, et al: Hypomorphic mutation of ZAP70 in human
results in a late onset immunodeficiency and no autoimmunity. Eur J
Immunol. 39:1966–1976. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Roifman CM, Dadi H, Somech R, Nahum A and
Sharfe N: Characterization of ζ-associated protein, 70 kd
(ZAP70)-deficient human lymphocytes. J Allergy Clin Immunol.
126:1226–1233.e1. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huang L, Qiao L, Zhu H, Jiang L and Yin L:
Genomics of neonatal sepsis: Has-miR-150 targeting BCL11B functions
in disease progression. Ital J Pediatr. 44:1452018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hayden MS and Ghosh S: Shared principles
in NF-kappaB signaling. Cell. 132:344–362. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Perkins ND: Integrating cell-signalling
pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol.
8:49–62. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pannicke U, Baumann B, Fuchs S, Henneke P,
Rensing-Ehl A, Rizzi M, Janda A, Hese K, Schlesier M, Holzmann K,
et al: Deficiency of innate and acquired immunity caused by an
IKBKB mutation. N Engl J Med. 369:2504–2514. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mousallem T, Yang J, Urban TJ, Wang H,
Adeli M, Parrott RE, Roberts JL, Goldstein DB, Buckley RH and Zhong
XP: A nonsense mutation in IKBKB causes combined immunodeficiency.
Blood. 124:2046–2050. 2014. View Article : Google Scholar : PubMed/NCBI
|