Introduction
Hypertrophic cardiomyopathy (HCM) is the most common
inherited cardiovascular disease, with a prevalence of 1 in 500 in
the population (1,2). A recent study reported that the
prevalence is most likely to be 1 in 200 (3). HCM is a common cause of sudden death
in adolescents and the leading cause of sudden death in young
athletes (4,5). Approximately 50% of HCM cases are
inherited, and this variant is referred to as familial HCM (FHCM)
(6). The underlying pathogenesis
in HCM is primarily attributed to cardiac sarcomere protein
mutations, with over 1,400 HCM mutations being reported in at least
20 genes (7,8). FHCM is also known as a sarcomere
disease. Moreover, FHCM is morphologically diverse, substantially
heterogeneous within families and shows considerable genetic
variability (9). HCM follows a
single-gene dominant inheritance, with approximately 7% of cases
having polygenic or compound mutations. Compared with patients with
single-gene mutations, those with polygenic or compound mutations
have a greater incidence of disease, with more severe clinical
manifestations and worse prognosis (10–12).
Although polygenic or compound mutations are mainly sarcomere
protein mutations, a recent study showed that the occurrence of HCM
is closely associated with calcium channel gene mutations (13).
Approximately 20–30% of all HCM cases are FHCM ones
with β-myosin heavy chain (MHC) 7 (MYH7) as the dominant
pathogenic gene (14). Patients
carrying compound heterozygous mutations of two alleles in the
MYH7 gene may have a more severe clinical phenotype.
However, the clinical phenotypes of monoallelic double mutations in
MYH7 or their severity has not yet been studied. The aim of
the present study was to determine the clinical phenotypes caused
by compound heterozygous and monoallelic double mutations of
MYH7 using next-generation sequencing.
Materials and methods
Subjects
This study conformed to the principles of the
Helsinki Declaration and was approved by the Ethics Committee of
Xijing Hospital, Fourth Military Medical University, China. The
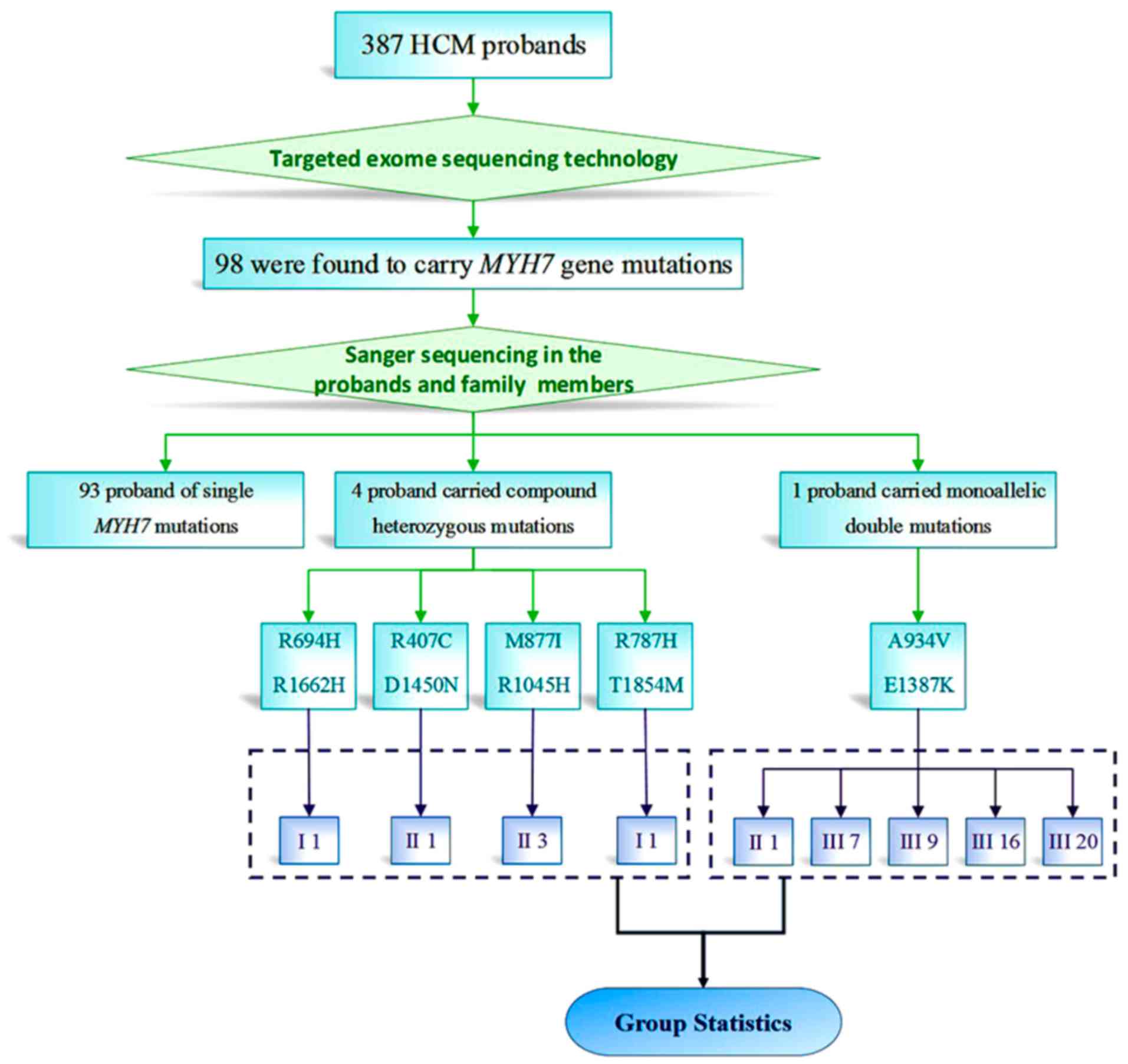
subjects were 387 HCM probands, with clinical data of medical
history, 12-lead electrocardiograms and echocardiograms. The
probands sought medical care in Xijing Hospital of the Fourth
Military Medical University (Fig.
1). We recalled the 4 patients with compound heterozygote
mutations and all family members carrying monoallelic double
mutations after confirming the mutation, and then acquired
ultrasound images and electrocardiographic data. Other patients
were not called back for data collection.
The diagnostic criteria were based on the 2011
ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic
Cardiomyopathy and the 2014 ESC Guidelines on Diagnosis and
Management of Hypertrophic cardiomyopathy (8,9). In
adults, HCM diagnosis requires a wall thickness ≥15 mm in one or
more left ventricular (LV) myocardial segments, measured by any
imaging technique including echocardiography, cardiac magnetic
resonance imaging or computed tomography. In children, HCM
diagnosis requires LV wall thickness to be ≥ predicted mean ±
standard deviation (z-score >2). On the other hand, among
first-degree relatives of patients with unequivocal disease, HCM is
defined as an unexplained increase in LV wall thickness ≥13 mm in
one or more LV myocardial segments, measured using any cardiac
imaging technique. The exclusion criteria for diagnosis are
ventricular wall hypertrophy resulting from hypertension, coronary
heart disease, coarctation of the aorta, valvular heart disease,
congenital heart disease or metabolic disorders and cardiac
hypertrophy in athletes.
Genomic DNA extraction and
high-throughput sequencing
The RelaxGene Blood DNA System (cat. no. DP319;
Tiangen Biotech Co., Ltd., Beijing, China) was used to extract the
genomes from white blood cells isolated from 10 ml of peripheral
vein blood of the proband, family members and healthy controls.
Using targeted exome-sequencing technology, the proband underwent
exon amplification and high-throughput sequencing for 96 genes
(15) (Table SI) related to FHCM. After
synonymous mutations were filtered, mutation loci >0.1% were
excluded through several databases including the 1000 Genomes
Project (http://browser.1000genomes.org), Exome Variant Server
(http://evs.gs.washington.edu/EVS) and
ExAC browser (http://exac.broadinstitute.org/terms). The associated
variants were subsequently verified by Sanger sequencing in blood
relatives of the proband. Meanwhile, 300 healthy individuals of the
same ethnic group as the probands (Han ethnic group) were checked
for the presence of the same variants.
Imaging studies
For echocardiography, the subjects were examined in
the left lateral decubitus position and with quiet breathing, with
simultaneous electrocardiogram recording. First, an S5-1 probe was
used to perform routine echocardiography, with two-dimensional
measurements being made for interventricular septal end-diastolic
dimension and LV end-diastolic wall thickness in a 16-segment
model. Simpson's rule was used to measure LV volume, LV
end-diastolic volume and LV end-systolic volume. Pulsed wave
Doppler (PWD) was used to measure the peak velocity flow in early
diastole (E) and late diastole (A) so as to calculate the E/A
ratio. Tissue Doppler imaging and PWD were used in combination to
measure peak velocity of early and late diastolic mitral annulus
(Ea and Aa, respectively), followed by the calculation of Ea/Aa and
E/Ea. Further, LV mass, LV mass index, stroke volume, LV volume
index and LV ejection fraction were determined.
Prediction of mutant protein
function
Using Swiss-PdbViewer 4.1 (16) (http://swissmodel.expasy.org/), three-dimensional (3D)
models of MYH7-associated proteins were created, and
conservation of sequence across species was analyzed.
Statistical analysis
Continuous variables are expressed as means ±
standard errors. Differences were analyzed using Student's t-tests.
P-values of <0.05 were considered to indicate statistical
significance.
Results
Gene mutation analyses
Among the 387 HCM probands included in the study, 98
were found to carry MYH7 mutations. Among 5 probands with
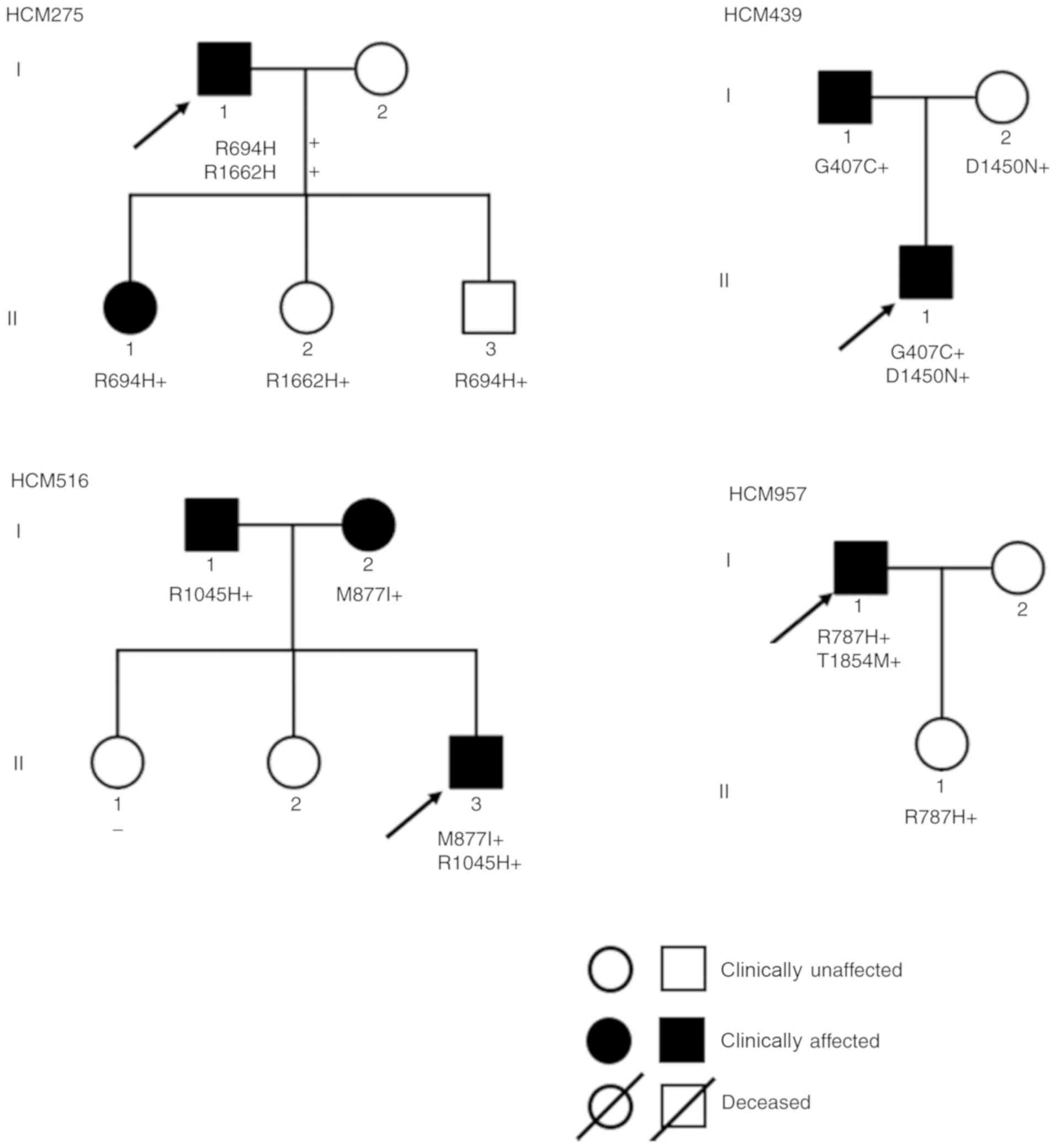
double MYH7 mutations, 4 carried compound heterozygous
mutations (Fig. 2) and 1 carried
monoallelic double mutations. Targeted exon sequencing was
performed in 387 HCM probands. These included 98 probands carrying
MYH7 mutations (25.3% of all probands) and 5 probands
carrying double MYH7 mutations (5.1% of all carrying
MYH7 probands) after other sarcomere gene mutations were
excluded. Among the 98 probands with MYH7 mutations, 4
(4.1%) had compound heterozygous double mutations and 1 (1%) had
monoallelic double mutations. In the family of the proband with
monoallelic double mutations, 3 patients were aged <30 years at
the time of initial diagnosis, 2 patients had LV outflow tract
obstruction, 2 had reduced diastolic function and 3 had syncope
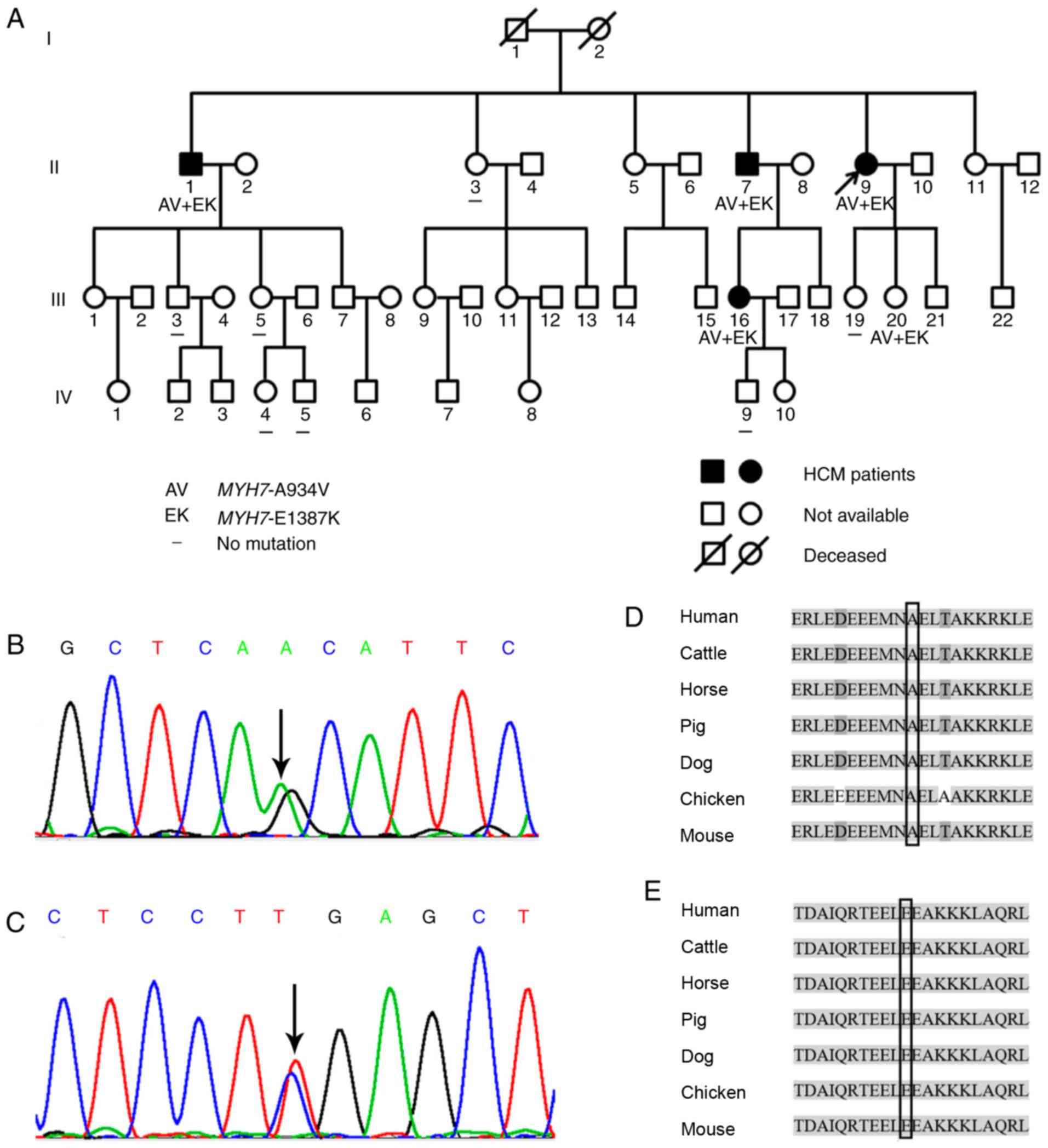
(Table I). The variants confirmed
in this family were MYH7-A934V and MYH7-E1387K
(Fig. 3A). MYH7-A934V is a
mutant caused by a C>T substitution in base 2801 of MYH7
exon 23, which results in a change from alanine (amino acid 934,
Fig. 3B) to valine.
MYH7-E1387K is a mutant caused by a G>A substitution in
base 4159 of MYH7 exon 30, which results in a change from
glutamic acid (amino acid 1387) to lysine (Fig. 3C).
 | Table I.Clinical and genetic characteristics
of the probands carrying compound heterozygous MYH7
mutations in the studied HCM cohort. |
Table I.
Clinical and genetic characteristics
of the probands carrying compound heterozygous MYH7
mutations in the studied HCM cohort.
|
|
|
|
|
|
|
Echocardiography |
|---|
|
|
|
|
|
|
|
|
|---|
| Patient ID | Gender/age
(years) | Symptoms | Mutation 1 | Mutation 2 | ECG | MLVWT (mm) | LVEF (%) | E/A ratio | LVOT-PG (mmHg) |
|---|
| HCM275 | M/59 | Syncope (4) | R694H | R1662H | Inverted T-waves,
ST-segment depression | 28 | 65 | 1.30 | 68 |
| HCM439 | M/15 | − | G407C | D1450N | Inverted T-waves,
ST-segment depression | 22 | 64 | 1.21 | 14 |
| HCM516 | M/23 | Chest tightness,
syncope (1) | M877I | R1045H | Inverted T-waves,
ST-segment depression | 28 | 67 | 0.64 | 44 |
| HCM957 | M/30 | Syncope (many
times) | R787H | T1854M | Inverted T-waves,
ST-segment depression, left anterior branch block | 28 | 58 | 1.25 | 7 |
Polymerase chain reaction primers were designed to
perform Sanger sequencing of MYH7-A934V and
MYH7-E1387K for the proband with monoallelic double
mutations and her blood relatives. Five family members, including
the proband (II9, II1, II7, III16 and III20), were revealed to be
carriers of MYH7-A934V and MYH7-E1387K, which were
not detected in other family members.
Clinical features
Table II
summarizes the clinical features and genotype characteristics. The
same 2 mutations were carried by II9, II1, II7, III16 and III20.
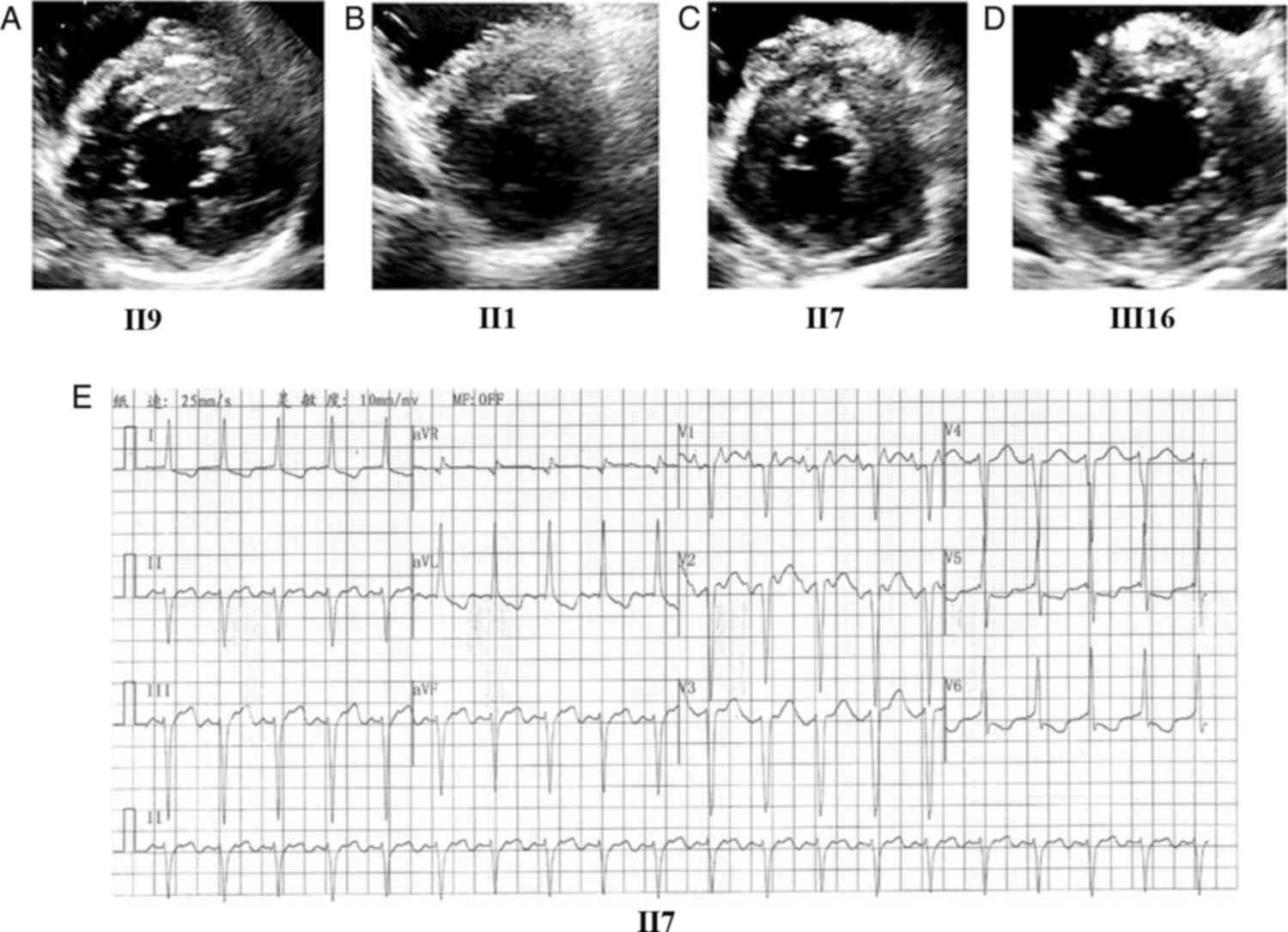
The proband (II9) was a 46-year-old female who presented with
palpitation, breathlessness and chest discomfort. Echocardiography
revealed the following: Maximum LV wall thickness, 22 mm (Fig. 4A); left atrium (LA) dimension, 44
mm; E/A, 1.29 and Ea/Aa, 0.74. Electrocardiography showed TI and
aVL (flattened and inverted) as well as left axis deviation (−48°).
II1 was a 59-year-old male without marked symptoms, for whom
echocardiography revealed the following: Maximum LV wall thickness,
15 mm, (Fig. 4B); E/A, 0.4 and
Ea/Aa, 0.5. Electrocardiography indicated clockwise rotation. The
elder brother of the proband, II7, a 55-year-old male, presented
with clear symptoms and the following echocardiography findings:
Maximum LV wall thickness, 28 mm (Fig.
4C) and LA dimension, 59 mm. Electrocardiography showed atrial
tachycardia, left axis deviation, LV fascicular block, inverted
T-waves and ST-segment depression (Fig. 4E). The proband's asymptomatic niece
(III16), a 26-year-old female, had borderline LV wall thickening
(Fig. 4D). Another carrier was the
proband's 14-year-old daughter (III20), who was asymptomatic and
had unremarkable physical examination findings. Among the family
members without the 2 mutations, none showed abnormal findings.
| Table II.Clinical and genetic characteristics
of the index family with single allele double MYH7 mutations
in the studied HCM cohort. |
Table II.
Clinical and genetic characteristics
of the index family with single allele double MYH7 mutations
in the studied HCM cohort.
|
|
|
|
|
|
|
Echocardiography |
|---|
|
|
|
|
|
|
|
|
|---|
| Patient ID | Gender/age
(years) | Symptoms | MYH7-
A934V | MYH7-
E1387K | ECG | MLVWT (mm) | LVEF (%) | E/A ratio | LVOT-PG (mmHg) |
|---|
| II-1 | M/59 | − | + | + | Indicated clockwise
rotation. | 15 | 60 | 0.40 | 5 |
| II-3 | F/57 | − | − | − | Normal | 10 | 63 | 1.31 | 1 |
| II-7 | M/55 | Chest
tightness | + | + | LV fascicular block
inverted T-waves, ST-segment depression | 28 | 56 | atrial
tachycardia | 4 |
| II-9 | F/46 | Chest tightness,
palpitation | + | + | T flattened and
inverted, and left axis deviation (−48°). | 22 | 63 | 1.29 | 4 |
| III-3 | M/32 | − | − | − | Normal | 12 | 52 | 0.69 | 2 |
| III −5 | F/31 | − | − | − | Nearly normal | 9 | 63 | 1.33 | 3 |
| III-16 | F/26 | − | + | + | Flat T waves in
leads I, aVL and V | 13 | 58 | 1.13 | 2 |
| III-19 | F/19 | − | − | − | Normal | 9 | 57 | 1.6 | 3 |
| III-20 | F/14 | − | + | + | Normal | 6 | 67 | 1.32 | 1 |
| IV-4 | F/6 | − | − | − | Normal | 4 | 61 | 1.54 | 2 |
| IV-5 | M/4 | − | − | − | Normal | 3 | 65 | 2.96 | 1 |
| IV-9 | M/4 | − | − | − | Normal | 4 | 62 | 1.3 | 2 |
Comparisons of compound heterozygous
and monoallelic double mutations
Statistical analysis of clinical ultrasound data
showed that the compound heterozygous mutation carriers had higher
LV mass (LVM) than those with monoallelic double mutations
(P=0.03). The compound heterozygous mutation carriers also showed
higher amplitudes in QRS, SV1 and RV5+SV1. On the other hand, there
were no significant differences in LV wall thickness, LA dimension
or other parameters (Table
III).
| Table III.Comparisons of the echocardiographic
parameters between the subgroups. |
Table III.
Comparisons of the echocardiographic
parameters between the subgroups.
| Variables | Compound
heterozygous mutations (n=4) | Monoallelic double
mutations (n=5) |
P-valuea |
|---|
| Age (years) | 32±19 | 40±19 | 0.543 |
| LVOT-PG (mmHg) | 33±28 | 3±2 | 0.123 |
| MLVWT (mm) | 27±3 | 17±8 | 0.068 |
| LA diameter
(mm) | 41±6 | 39±11 | 0.079 |
| LVMI
(g/m2) | 114±24 | 86±33 | 0.191 |
| LVM (g) | 222±32 | 135±57 | 0.03a |
| LVEF | 64±4 | 61±4 | 0.363 |
| E/A | 1.1±0.3 | 1.0±0.4 | 0.815 |
| E/e′ | 16.57±9.52 | 15.28±10.72 | 0.864 |
| HR (bpm) | 69±12 | 88±21 | 0.157 |
| QRS (msec) | 111±7 | 95±8 | 0.015a |
| PR (msec) | 161±26 | 172±32 | 0.578 |
| QT (msec) | 415±41 | 358±46 | 0.094 |
| QTC (msec) | 440±30 | 424±29 | 0.459 |
| RV5, mV | 1.64±0.67 | 1.00±0.23 | 0.149 |
| SV1, mV | 3.19±1.11 | 1.11±0.38 | 0.028a |
| RV5+SV1, mV | 4.84±1.74 | 2.11±0.56 | 0.047a |
Prediction analysis of mutant protein
function
Prediction for sequence
conservation
In the blocks shown (Fig. 3D and E), the sequences marked in
light grey, where sequence conservation is usually observed, were
identical. In addition, the mutation sites, i.e., MYH7-A934
(Fig. 3D) and MYH7-E1387
(Fig. 3E), which are highly
conserved among species, were located in the block, whereas
sequences indicated in light grey were exactly the same. Genetic
backgrounds of both novel MYH7 mutations identified in the
cohort are described in Table
IV.
| Table IV.Genetical background of 2 novel
MYH7 mutations identified in the study. |
Table IV.
Genetical background of 2 novel
MYH7 mutations identified in the study.
|
| Prediction | Frequency |
|
|---|
|
|
|
|
|
|---|
| Mutation | SIFT | PolyPhen-2 | 1000 Genomes | ExAC_EAS | ESP | Conservation
GERP++ |
|---|
|
MYH7-A934V | 0.001 (D) | 0.647 (P) | 0 | 0 | 0 | 5.33 |
|
MYH7-E1387K | 0 (D) | 1 (D) | 0 | 0 | 0 | 4.83 |
Protein structure and pathogenicity
prediction
Three-dimensional structure of amino acid 934 based
on the published MYH7 structure (PDB code, 2FXM) viewed with the
Swiss-PdbViewer 4.1 software showed that the replacement of alanine
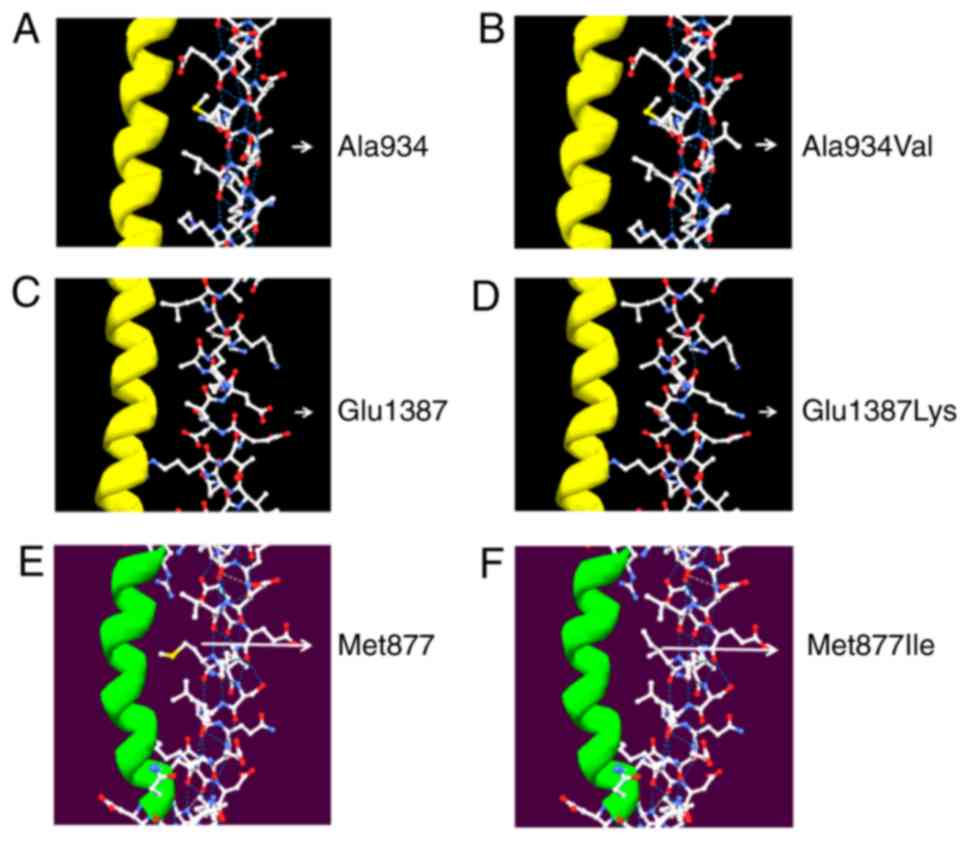
by valine at codon 934 (Fig. 5A)
led to a change in the side chains of the amino acid but did not
have a significant impact on the local intermolecular hydrogen
bonds or surface charge distribution. The structural changes on the
side chains may result in pathogenesis by affecting the flexibility
of the MHC neck domain (Fig. 5B).
On the other hand, the simulated structure of amino acid 1387 based
on the published MYH7 structure 4XA3 showed that the replacement of
the acidic glutamic acid by an alkaline lysine at codon 1387
(Fig. 5C) led to a significant
impact on surface charge distribution, resulting in a change in the
molecular surface, but had no effect on local intermolecular
hydrogen bonds. This mutation might change the local structure and
potential molecular interactions, contributing to pathogenesis by
affecting the reactivity of the MHC rod domain (Fig. 5D).
For amino acid 877, the simulation based on the MYH7
structure (2FXM) showed that the replacement of methionine by
isoleucine at codon 877 (Fig. 5E)
led to a change in the side chains of the amino acid but had no
significant impact on the local intermolecular hydrogen bonds or
surface charge distribution. These structural changes on the side
chains might result in pathogenesis by affecting the flexibility of
the MHC neck domain (Fig. 5F).
In accordance with the variant classification
criteria stated in the American College of Medical Genetics and
Genomics (ACMG) guidelines (17),
MYH7-A934V is considered in line with 1 type of PM (PM2) and
3 types of PP (PP1, PP3 and PP4) variants, and was therefore
identified as having uncertain significance; MYH7-E1387K,
considered in line with 1 type of PM (PM2) and 4 types of PP (PP1,
PP3, PP4 and PP5) variants, and was therefore identified as likely
pathogenic. Since the 2 variants existed in the same allele,
carrier status for both variants should be classified as likely
pathogenic; however, sufficient evidence to support this
classification is currently unavailable.
Discussion
In the present study cohort, there were a total of 5
hypertrophic cardiomyopathy (HCM) probands carrying double
MYH7 mutations. Among these, MYH7-V934A,
MYH7-E1387K and MYH7-M877I were identified as novel
MYH7 mutations causing HCM. We were unable to find these
mutations in the normal control population in searches of the ESP
database (http://evs.gs.washington.edu/EVS), the 1000 Genomes
and the ExAC browser. Predictive analyses using SIFT (http://sift.jcvi.org), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2)
and GERP++ (http://mendel.stanford.edu/sidowlab/downloads/gerp/index.html)
revealed that MYH7-M877I, MYH7-E1387K and
MYH7-M877I were all pathogenic mutations. More importantly,
our findings demonstrated a rare HCM family with monoallelic double
mutations in MYH7 that has not been reported previously.
Finally, statistical analysis revealed that the clinical phenotype
was less severe in the carriers with monoallelic double mutations
than in those with diallelic double mutations in MYH7.
Based on the published MYH7 structure 2FXM, the 3D
structure of the amino acids 934 and 877 simulated with the
Swiss-PdbViewer 4.1 showed that V934A and M877I could cause changes
in the side chains of the amino acids. These changes may contribute
to HCM pathogenesis by affecting the flexibility of the MHC neck
domain. Similarly, based on the published MYH7 structure 4XA3, the
simulated structure of the amino acid 1387 showed that E1387K could
result in a significant impact on surface charge distribution,
resulting in a change in the molecular surface, but would otherwise
have no effect on the local intermolecular hydrogen bonds. This
mutation would change the local structure and potential molecular
interactions, potentially contributing to the pathogenesis by
changing the reactivity of the MHC rod domain.
Molecular genetic studies have demonstrated that
40–60% of the pathogenic genes in patients with HCM are
attributable to mutations in sarcomere proteins and associated
proteins (9,18). MYH7 is the predominant
pathogenic gene in HCM (30–50% of all cases). The common symptoms
associated with the mutation are observed in malignant HCM
phenotypes and include early onset, high penetrance, high degree of
hypertrophy and high incidence of sudden death (18,19).
MYH7 is located between 14q11 and 14q12 and contains 40
exons, among which 38 participate in encoding 1935 amino acids. It
encodes β-MHC, which is the principal constituent of the thick
myofilament and is composed of globular head, neck and rod domains
(20).
The head is the location for the adenosine
triphosphatase activity locus as well as the binding sites for
actin and the essential light chain. This region thus serves a
vital function and mutations in this region often result in more
severe clinical phenotypes than those in the rod domain (21). The 2018 ACMG guidelines for the
Pathogenicity Analysis of MYH7 provide moderate pathogenic
evidence for the region comprising the amino acids 181–937
(22). However, these definitions
are for single-gene mutation sites, and currently there are no
guidelines for carriers with ≥2 MYH7 mutations, including
those with monoallelic double mutations.
The main pathogenic event in HCM cases with
MYH7 as the mutant gene is a single point mutation. No
studies have reported the proportion of compound heterozygous
MYH7 mutations in HCM families. Second-generation analysis
of 387 HCM families in the current study showed that the proportion
of compound heterozygous MYH7 mutations was 5.1%.
Furthermore, no study in China or elsewhere has elucidated
monoallelic double mutations in MYH7; the present study is
the first to provide data on the proportion of compound
heterozygous MYH7 mutations in Chinese families with HCM,
which should aid in the genetic counselling of HCM patients.
Regarding the pathogenesis associated with these
mutations, it is possible that the presence of the monoallelic
double mutations might be functionally compensated by the normal
allele; this might explain the normal clinical presentation of some
of the carriers in the present study. In probands with multiple
MYH7 gene mutations, using a familial cosegregation test to
identify specific infringement alleles will have a significant
impact on risk assessment and clinical intervention strategies in
carriers. The pathogenicity of single-gene MYH7 mutations
can be determined using the 2018 ACMG Guidelines for the
Pathogenicity Analysis of MYH7 (22). However, these guidelines do not
address the potential influence of carrying multiple genes on
disease severity. It is controversial whether the double mutations
or compound heterozygosity in MYH7 may affect HCM severity.
Viswanathan et al provided evidence that compound mutations
may not increase the severity of the HCM phenotype compared with
single mutations, but it may substantially increase the risk of
developing HCM (23). Wang et
al enrolled 529 Chinese HCM patients, and found that the HCM
patients with multiple rare sarcomere mutations had earlier age of
onset, more severe left ventricular hypertrophy and enlarged left
atrium, as well as higher risk of sudden cardiac death, as compared
to those with single rare mutation (11). The 2017 Guideline for Hypertrophic
Cardiomyopathy in China showed that approximately 7% patients have
complex mutations regardless of their genetic inheritance status
(10–12,24).
HCM onset in these patients is earlier than those with single-gene
mutations, and the clinical manifestations are more severe. In the
present study, such patients carried previously unreported
monoallelic double mutations. Carriers with compound heterozygous
mutations had significantly higher LVM by echocardiography and
higher amplitudes in QRS, SV1 and RV5+SV1 than those with
monoallelic double mutations.
Upon electrocardiography, QRS, RV5, RV5+SV1 were
positively correlated with LV voltage. Furthermore, LV voltage was
positively correlated with LV wall thickness, and higher LV
voltages were associated with more severe LV hypertrophy. The
degree of hypertrophy of the ventricular wall has been positively
correlated with the risk of sudden death in patients with HCM
(25,26). Therefore, this study suggests that
if patients undergoing clinical genetic analyses are detected with
2 mutations of the pathogenic gene, the proband will either carry
monoallelic double mutations or compound heterozygous double
mutations. The prognosis of mutation carriers among blood relatives
has a certain guiding significance and should have an important
impact on the clinical management of patients and those at risk.
However, our study lacks clinical indicators of the prognosis to
stratify the arrhythmic risks in both control and mutant groups,
such as Holter and stress test. This has been realized by us, and
relevant data will be collected in future studies.
Limitations to the present study must be addressed.
In the present study, we found differences between the two groups
of compound heterozygote mutations and monoallelic double
mutations. Yet, a statistically significant impact of the mutations
on clinical phenotypes in a single family with monoallelic double
mutations is not representative of all patients or families
carrying this type of variation. Whether our preliminary results
can be amplified into the population still requires verification by
large populations that will be carried out in future
investigation.
In conclusion, we identified several novel
MYH7 mutations that are potentially pathogenic in HCM.
Moreover, we reported the findings of a rare HCM family carrying
monoallelic MYH7 double mutations, which have not been
previously reported. Our findings suggest that the clinical
presentation of carriers with these monoallelic double mutations
may be less severe than that of those with diallelic double
mutations.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thanks Professor Yuan-Ming
Wu, Fourth Military Medical University, Xi'an, Shaanxi, China, for
his genetic data analysis.
Funding
This research was supported by the International
Science and Technology Cooperation Program of China (2014DFA31980),
the Natural Science Foundation of China (81671693, 81601498 and
81670304), Shaanxi Provincial Key Project (2017ZDXM-SF-058) and
Xijing Funded Project for New Technologies and Services
(417432A).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
LWL and DH conceived and designed the study. BW,
LFW, YH and QLY collected and analyzed genetic data. JW, FY, LX and
LZ collected and analyzed UCG data. WXL and HS collected and
analyzed ECG data. BW and JW wrote the paper. LFW reviewed and
edited the manuscript. All authors read and approved the manuscript
and agree to be accountable for all aspects of the research in
ensuring that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
This study conformed to the principles of the
Helsinki Declaration and was approved by the Ethics Committee of
Xijing Hospital, Fourth Military Medical University, China.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Maron BJ, Gardin JM, Flack JM, Gidding SS,
Kurosaki TT and Bild DE: Prevalence of hypertrophic cardiomyopathy
in a general population of young adults. Echocardiographic analysis
of 4111 subjects in the CARDIA study. Coronary artery risk
development in (young) adults. Circulation. 92:785–789. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Maron BJ and Maron MS: Hypertrophic
cardiomyopathy. Lancet. 381:242–255. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Semsarian C, Ingles J, Maron MS and Maron
BJ: New perspectives on the prevalence of hypertrophic
cardiomyopathy. J Am Coll Cardiol. 65:1249–1254. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Maron BJ: Contemporary insights and
strategies for risk stratification and prevention of sudden death
in hypertrophic cardiomyopathy. Circulation. 121:445–456. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Maron BJ, Thompson PD, Ackerman MJ, Balady
G, Berger S, Cohen D, Dimeff R, Douglas PS, Glover DW, Hutter AM
Jr, et al: Recommendations and considerations related to
preparticipation screening for cardiovascular abnormalities in
competitive athletes: 2007 update: A scientific statement from the
American heart association council on nutrition, physical activity,
and metabolism: Endorsed by the American college of cardiology
foundation. Circulation. 115:1643–1655. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ahmad F, Seidman JG and Seidman CE: The
genetic basis for cardiac remodeling. Annu Rev Genomics Hum Genet.
6:185–216. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Waldmüller S, Sakthivel S, Saadi AV,
Selignow C, Rakesh PG, Golubenko M, Joseph PK, Padmakumar R,
Richard P, Schwartz K, et al: Novel deletions in MYH7 and MYBPC3
identified in Indian families with familial hypertrophic
cardiomyopathy. J Mol Cell Cardiol. 35:623–636. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gersh BJ, Maron BJ, Bonow RO, Dearani JA,
Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, et
al: 2011 ACCF/AHA guideline for the diagnosis and treatment of
hypertrophic cardiomyopathy: executive summary: a report of the
American College of Cardiology Foundation/American Heart
Association Task Force on Practice Guidelines. Circulation.
124:2761–2796. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Authors/Task Force members, ; Elliott PM,
Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, Hagege
AA, Lafont A, Limongelli G, et al: 2014 ESC guidelines on diagnosis
and management of hypertrophic cardiomyopathy: The task force for
the diagnosis and management of hypertrophic cardiomyopathy of the
European society of cardiology (ESC). Eur Heart J. 35:2733–2779.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Maron BJ, Maron MS and Semsarian C: Double
or compound sarcomere mutations in hypertrophic cardiomyopathy: A
potential link to sudden death in the absence of conventional risk
factors. Heart Rhythm. 9:57–63. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang J, Wang Y, Zou Y, Sun K, Wang Z, Ding
H, Yuan J, Wei W, Hou Q, Wang H, et al: Malignant effects of
multiple rare variants in sarcomere genes on the prognosis of
patients with hypertrophic cardiomyopathy. Eur J Heart Fail.
16:950–957. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zou Y, Wang J, Liu X, Wang Y, Chen Y, Sun
K, Gao S, Zhang C, Wang Z, Zhang Y, et al: Multiple gene mutations,
not the type of mutation, are the modifier of left ventricle
hypertrophy in patients with hypertrophic cardiomyopathy. Mol Biol
Rep. 40:3969–3976. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Helms AS, Alvarado FJ, Yob J, Tang VT,
Pagani F, Russell MW, Valdivia HH and Day SM: Genotype-dependent
and -independent calcium signaling dysregulation in human
hypertrophic cardiomyopathy. Circulation. 134:1738–1748. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cam FS and Güray M: Hypertrophic
cardiomyopathy: Pathological features and molecular pathogenesis.
Anadolu Kardiyol Derg. 4:327–330. 2004.PubMed/NCBI
|
|
15
|
Wang L, Zuo L, Hu J, Shao H, Lei C, Qi W,
Liu Y, Miao Y, Ma X, Huang CL, et al: Dual LQT1 and HCM phenotypes
associated with tetrad heterozygous mutations in KCNQ1, MYH7,
MYLK2, and TMEM70 genes in a three-generation Chinese family.
Europace. 18:602–609. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guex N and Peitsch MC: SWISS-MODEL and the
Swiss-PdbViewer: An environment for comparative protein modeling.
Electrophoresis. 18:2714–2723. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lopes LR, Zekavati A, Syrris P, Hubank M,
Giambartolomei C, Dalageorgou C, Jenkins S, McKenna W, Uk10k
Consortium, Plagnol V and Elliott PM: Genetic complexity in
hypertrophic cardiomyopathy revealed by high-throughput sequencing.
J Med Genet. 50:228–239. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang S, Zou Y, Fu C, Xu X, Wang J, Song L,
Wang H, Chen J, Wang J, Huan T and Hui R: Worse prognosis with gene
mutations of beta-myosin heavy chain than myosin-binding protein C
in Chinese patients with hypertrophic cardiomyopathy. Clin Cardiol.
31:114–118. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Woo A, Rakowski H, Liew JC, Zhao MS, Liew
CC, Parker TG, Zeller M, Wigle ED and Sole MJ: Mutations of the
beta myosin heavy chain gene in hypertrophic cardiomyopathy:
Critical functional sites determine prognosis. Heart. 89:1179–1185.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Walsh R, Rutland C, Thomas R and Loughna
S: Cardiomyopathy: A systematic review of disease-causing mutations
in myosin heavy chain 7 and their phenotypic manifestations.
Cardiology. 115:49–60. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kelly MA, Caleshu C, Morales A, Buchan J,
Wolf Z, Harrison SM, Cook S, Dillon MW, Garcia J, Haverfield E, et
al: Adaptation and validation of the ACMG/AMP variant
classification framework for MYH7-associated inherited
cardiomyopathies: Recommendations by ClinGen's inherited
cardiomyopathy expert panel. Genet Med. 20:351–359. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Viswanathan SK, Sanders HK, McNamara JW,
Jagadeesan A, Jahangir A, Tajik AJ and Sadayappan S: Hypertrophic
cardiomyopathy clinical phenotype is independent of gene mutation
and mutation dosage. PLoS One. 12:e01879482017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Guidelines for the diagnosis and treatment
for Chinese adult patients with hypertrophic cardiomyopathy.
Zhonghua Xin Xue Guan Bing Za Zhi. 45:1015–1032. 2017.(In Chinese).
PubMed/NCBI
|
|
25
|
Elliott PM, Gimeno JR, Tomé MT, Shah J,
Ward D, Thaman R, Mogensen J and McKenna WJ: Left ventricular
outflow tract obstruction and sudden death risk in patients with
hypertrophic cardiomyopathy. Eur Heart J. 27:1933–1941. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Maron MS, Olivotto I, Betocchi S, Casey
SA, Lesser JR, Losi MA, Cecchi F and Maron BJ: Effect of left
ventricular outflow tract obstruction on clinical outcome in
hypertrophic cardiomyopathy. N Engl J Med. 348:295–303. 2003.
View Article : Google Scholar : PubMed/NCBI
|