Introduction
Mutations in the STAM binding protein (STAMBP) gene
cause microcephaly-capillary malformation (MIC-CAP) syndrome (OMIM,
614261; ORPHA, 294016), which was described as a novel syndrome
several years ago (1,2). Elevated ubiquitin-conjugated protein
aggregation and apoptosis activation has previously been identified
in lymphoblastoid cell lines from patients with MIC-CAP caused by
STAMBP mutations. Additionally, elevated autophagosome
content, active and insensitive RAS-MAPK and PI3K-AKT-mTOR pathways
have also been found in this syndrome (1). The MIC-CAP syndrome is a rare
neurocutaneous disorder characterized by congenital microcephaly,
early-onset epilepsy, severe profound developmental delay and
diffuse cutaneous capillary malformations (1–3). In
total, 18 pathogenic mutations have been reported in 16 patients
from 8 ethnic groups (1–8).
To the best of our knowledge, the present study
reported the case of the first Chinese patient with MIC-CAP
syndrome caused by a novel compound heterozygous STAMBP
mutation. The present study also provided a review of relevant
previously published cases.
Patients and methods
Patients
Ethical approval for the present study was obtained
from the Institutional Review Board, Children's Hospital of
Chongqing Medical University (permit no. 2018-64). Informed consent
was obtained from the parents of the patient.
The case described in the present study is that of a
boy born after 40 weeks and 5 days of gestation, G2P1 (second
pregnancy and first successful birth of the mother), to
nonconsanguineous Chinese parents (father, 23 years old; mother, 22
years old). The boy was born through normal delivery, with a birth
weight of 3,600 g. The birth length and head circumference were not
known.
The patient was admitted (June 2018) to the
Department of Neurology, Children's Hospital of Chongqing Medical
University at the age of 1 year and 5 months. A routine examination
of general health and neurological evaluations were carried out.
Magnetic resonance imaging (MRI), electroencephalography and
metabolic screening were performed according to the manufacturer's
protocols. All the available clinical characteristics of the
patient along with the aforementioned auxiliary examination results
are summarized in the present study.
Genetic analysis
Peripheral blood samples (5 ml) from the proband and
the parents were collected into graded negative pressure vacuum
EDTA anticoagulant tubes. All sample preparation, whole-exome
sequencing and Sanger sequencing were performed by Beijing
Mygenostics Co, Ltd. Several online databases containing data from
different ethnic groups were used as following; dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/), ExAC
(http://exac.broadinstitute.org/faq),
Genome Aggregation Database (gnomAD, http://gnomad.broadinstitute.org/), Clinvar
(https://www.ncbi.nlm.nih.gov/clinvar/),
esp6500siv2_all (http://evs.gs.washington.edu/EVS/). 1000 genome
(http://www.1000genomes.org), Mutation
screening of STAMBP was performed using Sanger sequencing
with two primer pairs to amplify exon 9 (chr2-74087178) and exon 7
(chr2-74077603) of the STAMBP gene (NM_201647). The following
primers were used: Exon 9, forward 5′-CTCACAATGACCTCTGCCCT-3′,
reverse 5′ATTCCTGTCCCACACTGCTT-3′; and exon 7, forward
5′GAGCACCAGGGAATTGTGAC-3′ and reverse 5′AAGCCCTAAGTGTTCCCAGA-3′.
BigDye Terminator v3.1 Cycle Sequencing kit (Invitrogen; Thermo
Fisher Scientific, Inc.) was used and thermal cycling was performed
in a Mix Golden Star T6 Super PCR Mix (1.1×; TsingKe Biological
Technology Co.; Table I). The
segregation of the identified mutations was investigated in the
parents.
 | Table I.Thermocycling conditions for Sanger
sequencing. |
Table I.
Thermocycling conditions for Sanger
sequencing.
| Step | Temperature (°C) | Time | Cycle |
|---|
| 1 | 95 | 10 min | 1 |
| 2 | 94 | 30 sec | 3 |
|
| 64 | 30 sec |
|
|
| 72 | 45 sec |
|
| 3 | 94 | 30 sec | 5 |
|
| 62 | 30 sec |
|
|
| 72 | 45 sec |
|
| 4 | 94 | 30 sec | 10 |
|
| 60 | 30 sec |
|
|
| 72 | 45 sec |
|
| 5 | 94 | 30 sec | 17 |
|
| 58 | 30 sec |
|
|
| 72 | 45 sec |
|
| 6 | 72 | 5 min | 1 |
| 7 | 4 | Holds |
|
The prediction of mutations was assessed using
software, including PolyPhen_2_Predict, PolyPhen_2 (http://genetics.bwh.harvard.edu/pph2/),
SIFT, SIFT_Predict (http://sift.jcvi.org/), SPIDEX (http://www.deepgenomics.com/spidex), MutationTaster,
MutationTaster_Predict (http://www.mutationtaster.org/ChrPos.html),
MCAP_score, MCAP_pred (http://bejerano.stanford.edu/MCAP/), GERP++_Predict,
GERP++ (http://mendel.stanford.edu/SidowLab/downloads/gerp/index.html)
and REVEL_score (https://sites.google.com/site/revelgenomics/). The
protein structure of STAMBP was modelled using the
SWISS-MODEL (3rzv.1, http://swissmodel.expasy.org/interactive)
Literature review
The PubMed (https://www.ncbi.nlm.nih.gov/pubmed/) and Wanfang
(http://www.wanfangdata.com.cn/index.html) databases
were used to retrieve previous studies using the keywords ‘STAMBP,
microcephaly, and capillary malformation’ until April 2019. The
publication language was limited to English and Chinese.
Results
Patient treatment
The child developed early-onset epilepsy after 3
months, with a generalized tonic-clonic seizure, which progressed
to clusters of infantile spasms (2–10 clusters/day) 1 month later.
The patient was treated successively with levetiracetam (40–50
mg/kg/day), topiramate (6–7 mg/kg/day), valproic acid (30
mg/kg/day) and corticosteroids at the outpatient clinic. The spasms
decreased and became myoclonic, but the epilepsy remained
refractory.
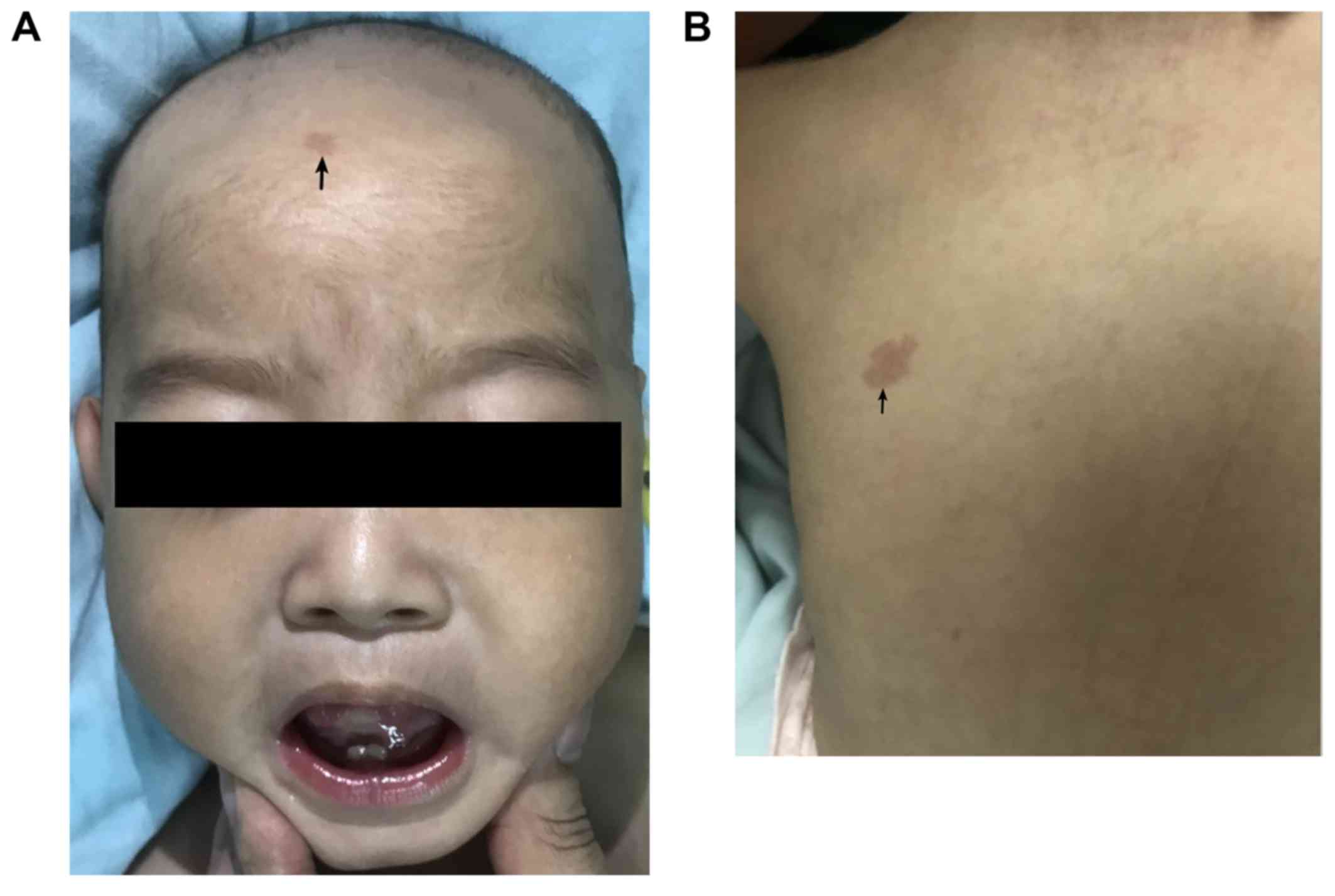
The patient was admitted to the neurologic ward
after 1 year and 2 months. The patient had drooping mouth corners,
a short nose and neck, and sporadic, multiple, small capillary
malformations (Fig. 1A and B).
Referring to the World Health Organization Anthro standards
(https://www.who.int/childgrowth/software/en/), the
patient was 8 kg [Z-score, −2.8 Standard Deviation (SD)] in
weight and 63 cm (Z-score, −6.8 SD) in length. The head
circumference was 39.5 cm (Z-score, −5.8 SD). The boy could
not hold his head and had prominent dyskinesia of the whole body,
particularly involuntary movement of the tongue and mouth (Video S1).
The patient was on a ketogenic diet during the
inpatient stay and received immunoglobulin intravenously as a
result of recurrent pneumonia, 22 days later, he was discharged.
During the follow-up, the seizures were still not well controlled,
although the ketogenic diet ratio was modified from 2:1 to 4:1 (4 g
fat/l g combined protein, carbohydrate). After 1 month, the patient
received vigabatrin (60 mg/kg/day) and the seizures reduced by 80%
a week later. Unfortunately, the patient suddenly succumbed 3 weeks
later; no definitive causes were found as no autopsy was
performed.
The MRI scans showed slightly dilated lateral
ventricles and increased extra-axial spaces. Interictal
electroencephalography showed hypsarrhythmia and slow wave
background with bioccipital spike-slow wave during waking (Fig. 1C and D). Blood and urinary
metabolic screening indicated normal results.
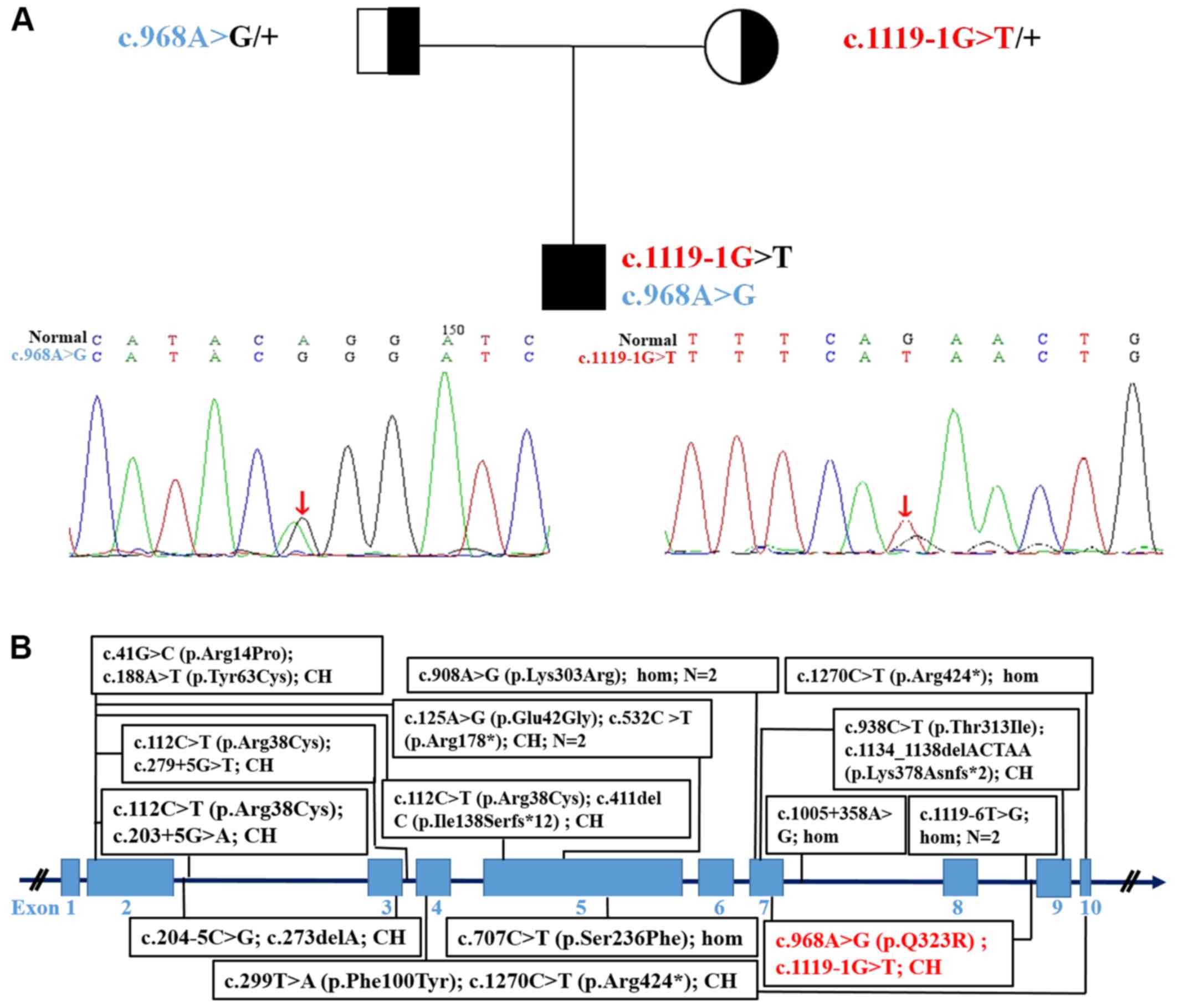
Exome sequencing
Exome sequencing revealed intronic (c.1119-1G>T)
and exonic mutations in the proband and their parents, which was
confirmed via Sanger sequencing (Fig.
2A). The mutations segregated according to a strictly recessive
model with full penetrance. The parents were found to be
heterozygous carriers, (mother, c.1119-1G>T; father,
c.968A>G; Fig. 2A). The protein
structures of wild-type and mutated STAMBP were modelled and
predicted, respectively, as shown in Fig. S1. The mutations affected the amino
acid side chain (p.Gln323Arg) and were predicted to be pathogenic.
The splice site variant (c.1119-1G>T) was predicted to generate
aberrant splicing of the STAMBP mRNA.
The clinical features and pathogenic variants of 16
previously reported cases of MIC-CAP are summarized in Table II and Fig. 2B.
| Table II.Summary of the patient and 16
previously reported MIC-CAP cases caused by STAMBP mutations. |
Table II.
Summary of the patient and 16
previously reported MIC-CAP cases caused by STAMBP mutations.
| Patients | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|
| Basic
characteristics |
|
Reference | This study |
|
|
| McDonell et al
2013 |
|
|
|
|
Gender | M | F | M | M | M | M | F | M | F |
| Age | 1 year 3 months | 2 years | 9 months | 12 months | 2 years | 22 days | 5 years 4 months | 2 months | 28 months |
|
Ethnicity | Chinese | African-American | ED | European | European | European | European | European |
|
| GA, BW
(weeks, SD or g) | 40+5,
3,600 | 39, −1.5 | 39, −1.5 | 36+5,
−1.5 | 36, −2 | 37, −2 | Term, +1.8 | 36, −1.5 | 37+2,
−4 |
| Symptoms and
signs |
|
Microcephaly | + | + | + | + | + | + | + | + | + |
|
Capillary malformations | + | + | + | + | + | + | + | + | + |
|
Dysmorphic
appearancea |
+1,4 | +3 |
+3 | − |
+3 |
+3 |
+3 |
+3 |
+3 |
| IE (age
of onset) | +(3 months) | +(NA) | +(NA) | +(NA) | +(NA) | +(NA) | +(NA) | +(NA) | + |
|
Infantile spasms | + | + | − | + | − | − | − | − | NA |
|
Myoclonus | + | − | + | + | + | − | − | + | + |
|
Developmental delay | + | + | + | + | + | + | + | + | + |
| Spastic
quadriparesis | + | + | + | + | + | + | − | − | + |
| Optic
atrophy | NA | + | + | + | + | + | NA | NA | + |
|
Dyskinesia | + | NA | NA | NA | NA | NA | NA | NA | NA |
| Auxiliary
examination |
|
Neuroimaging
featuresb |
+d |
+c–e |
+c–e |
+c–e |
+c,d |
+c–e | − |
+c–e |
+c–e |
| EEG
anomalies | + |
Presumed+f | Presumed + | Presumed + | Presumed + | Presumed + | Presumed + | Presumed + | Presumed + |
|
| Patient | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | Total |
|
| Basic
characteristics |
|
Reference | McDonell et
al 2013 | Faqeih et al
2015 | Naseer et al
2016 | Hori et al
2018 | Demikova et
al 2018 |
|
|
Gender | F | M | M | M | M | M | M | F | 5 F/12 M |
|
Age | 8 months | 15 months | 8 years 6
months | 5 years | NA | NA | 2 years | 6 months |
|
|
Ethnicity | Polynesian | ED | Arabic |
| Egyptian |
| Japanese | Russia | 9 |
| GA, BW
(weeks, SD or g) | 37+6,
−1.5 | 35, −1.5 | Term. 2,600 | NA, 2,800 | NA | NA | 37, 2,680 | 30, 2,250 |
|
| Symptoms and
signs |
|
Microcephaly | + | + | + | + | + | + | + | + | 17 (100%) |
| CM | + | + | + | + | + | + | + | + | 17 (100%) |
|
Dysmorphic appearance |
+3 |
+3 |
+1,2 |
+1,2,3 |
+1,2,3 |
+1,2,3 |
+1,2,4 |
+1,4 | 17 (100%) |
| IE (age
of onset) | + | + | +(7 months) | +(7 months) | +(Infancy) | +(Infancy) | +(7 months) | +(2 months) | 17 (100%) |
|
Infantile spasms | + | + | NA | NA | NA | NA | + | + | 7
(41.2%) |
|
Myoclonus | NA | + | NA | NA | NA | NA | NA | + | 8
(47.1%) |
|
Developmental delay | + | + | + | + | + | + | + | + | 17 (100%) |
| Spastic
quadriparesis | + | + | + | + | NA | NA | − | + | 12
(70.1%) |
| Optic
atrophy | − | − | + | + | + | + | − | NA | 10
(58.8%) |
|
Dyskinesia | + | NA | NA | NA | NA | NA | NA | NA | 2
(11.2%) |
| Auxiliary
examination |
|
Neuroimaging features |
+c–e |
+c,d |
+d |
+d |
+c,d |
+c,d |
+d |
+d | 16
(94.1%) |
| EEG
anomalies | Presumed + | Presumed + | + | + | + | + | + | + | 17
(100%) |
Discussion
So far as we know, the present study presented the
first case of a Chinese patient affected by refractory epilepsy,
microcephaly, severe developmental delay and diffuse cutaneous
capillary malformations. These four symptoms were also observed in
all of the patients studied in previous reports (100%) (1–8). The
patient in the present study also had spastic quadriparesis,
without optic atrophy; however, both optic atrophy and spastic
quadriparesis were common in previous cases (1–8).
Moreover, dyskinesia was more prominent in the present study and
was infrequent in previous cases, except in one patient (1). Although the patient in the present
study responded well to vigabatrin, the long-term efficacy of the
drug could not be evaluated due to the early death of the
patient.
A novel compound heterozygous co-segregating
mutation in STAMBP [c.1119-1G>T (splicing, exon9,
maternal allele) and c.968A>G (p.Gln323Arg, exon7, paternal
allele); Fig. 2A] was identified
through exome sequencing. The parents of the patient were healthy.
The variant frequency of the aforementioned mutation has not been
reported in the Chinese Reference Genome Database or any public
sequence database, to the best of our knowledge. In silico
analysis indicated that this splice site mutation generated
aberrant splicing of the STAMBP mRNA, and c.968A>G was also
predicted to be pathogenic. Therefore, a definite diagnosis for
this patient with MIC-CAP could be made due to the autosomal
recessive inheritance patterns, according to the previous reports
(1–8).
To date, only 16 cases (11 male and 5 female) from
13 families in 8 ethnic groups have been reported, with 18
different STAMBP mutations (1–8). All
the parents were from unrelated families, except for two
consanguineous relationships (4–6). A
further two patients had causative mutations (c.753_754insT,
c.1119-1G>T) (6,8), which have been omitted from the
figure as further details of these patients could not be found. In
total,~20 STAMBP mutations are listed online without further
patient details; therefore, their pathogenicity could not be
confirmed (ClinVar).
In conclusion, to the best of our knowledge, the
present study reported the first case of a Chinese patient with
refractory epilepsy as an initial symptom of MIC-CAP. Additionally,
novel pathogenic compound heterozygosity of the STAMBP was
identified. The results of the present study may improve
understanding of STAMBP mutations and ethnic background in
cases MIC-CAP.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported in part by a grant
from the Children's Hospital of Chongqing Medical University for
Special Project on Difficult and Rare Diseases (grant no.
HJYN2013-4).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
FW, YD, YW and MZ conceived and designed the study,
and analyzed the data; JW, MC, XL, PY, SL and LJ collected the
clinical information; JC and LY performed the EEG analysis; FW, YD
and MZ prepared the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Ethical approval for the present study was obtained
from the Institutional Review Board, Children's Hospital of
Chongqing Medical University (grant no. 2018-64).
Patient consent for publication
Informed consent was obtained from the parents of
the patient.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
McDonell LM, Mirzaa GM, Alcantara D,
Schwartzentruber J, Carter MT, Lee LJ, Clericuzio CL, Graham JM Jr,
Morris-Rosendahl DJ, Polster T, et al: Mutations in STAMBP,
encoding a deubiquitinating enzyme, cause microcephaly-capillary
malformation syndrome. Nat Genet. 45:556–562. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Carter MT and Boycott KM:
Microcephaly-capillary malformation syndrome: A story of rapid
emergence of a new recognizable entity. Am J Med Genet A.
155A:2078–2079. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Carter MT, Geraghty MT, De La Cruz L,
Reichard RR, Boccuto L, Schwartz CE and Clericuzio CL: A new
syndrome with multiple capillary malformations, intractable
seizures, and brain and limb anomalies. Am J Med Genet A.
155A:301–306. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pavlovic M, Neubauer D, Al Tawari A and
Heberle LC: The microcephaly-capillary malformation syndrome in two
brothers with novel clinical features. Pediatr. Neurol. 51:560–565.
2014.
|
|
5
|
Faqeih EA, Bastaki L, Rosti RO, Spencer
EG, Zada AP, Saleh MA, Um K and Gleeson JG: Novel STAMBP mutation
and additional findings in an Arabic family. Am J Med Genet A.
167:805–809. 2015. View Article : Google Scholar
|
|
6
|
Naseer MI, Sogaty S, Rasool M, Chaudhary
AG, Abutalib YA, Walker S, Marshall CR, Merico D, Carter MT,
Scherer SW, et al: Microcephaly-capillary malformation syndrome:
Brothers with a homozygous STAMBP mutation, uncovered by exome
sequencing. Am J Med Genet A. 170:3018–3022. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Demikova NS, Kakaulina VS, Pechatnikova
NL, Polyakova NA, Zakharova EY, Krylova TD and Zubkova MV: First
report of microcephaly-capillary malformations syndrome in Russia.
Egypt J Med Hum Genet. 19:147–150. 2018. View Article : Google Scholar
|
|
8
|
Hori I, Miya F, Negishi Y, Hattori A, Ando
N, Boroevich KA, Okamoto N, Kato M, Tsunoda T, Yamasaki M, et al: A
novel homozygous missense mutation in the SH3-binding motif of
STAMBP causing microcephaly-capillary malformation syndrome. J Hum
Genet. 63:957–963. 2018. View Article : Google Scholar : PubMed/NCBI
|