Introduction
Sodium taurocholate cotransporting polypeptide
(NTCP) deficiency is an inborn error of bile acid metabolism caused
by biallelic mutations of the solute carrier family 10 member 1
(SLC10A1) gene. SLC10A1 is located in chromosome
14q24.2, contains 5 exons and has a total length of 23 kb (1,2). The
protein product NTCP is composed of 349 amino acid residues and has
a molecular weight of 38 kDa (1).
NTCP is a transporter protein expressed in the sinusoidal plasma
membrane of hepatocytes, where it is involved in the uptake of bile
salts from plasma into hepatocytes in a sodium-dependent manner,
playing a crucial role in the enterohepatic circulation of bile
acids (3,4).
The SLC10A1 gene was cloned by Hagenbuch
et al (1) in 1994 and the
function of NTCP has since been widely investigated (5,6).
Theoretically, NTCP deficiency was thought to cause hypercholanemia
(7); however, there was no
clinical description of such patients until 2015, when Vaz et
al (8) reported the first
patient with NTCP deficiency (8).
At the time of publication of the current study, only a limited
number of patients with such a condition have been reported in 10
publications (8–17). The genotypic and phenotypic
features of this newly described condition therefore require
further investigation.
The reported SLC10A1 variants causing NTCP
deficiency included c.755G>A(p.Arg252His) (8), c.615_618del (p.Ser206Profs*12)
(9), c.263T>C(p.Ile88Thr)
(10) and c.800C>T(p.Ser267Phe)
(12). Among them, the variant
p.Ser267Phe is rather prevalent, with the allele frequency of 7.4%
(23/312), 3.1% (9/294) and 9.2% (28/306) in Chinese, Korean and
Vietnamese populations (6),
respectively. Therefore, although patients with NTCP deficiency
were rarely reported in the past over 20 years, this condition may
not be rare, especially in East Asian population. The present study
described the identification of 4 new patients with NTCP deficiency
in 2 unrelated families and presented the clinical and genetic
findings of the aforementioned patients.
Materials and methods
Patients and ethical approval
A total of 2 pediatric patients, including a
27.7-month-old male and a 3.8-month-old female, from two unrelated
families and 2 adult patients, including a male aged 31 years and a
female aged 35 years, from family 1 with hypercholanemia as well as
their family members were included in the current study, which was
performed between May 2016 and July 2017 in The First Affiliated
Hospital of Jinan University. The clinical findings were collected
and described as case reports in the Results section of the present
study. In order to explore the allele frequency of the identified
novel SLC10A1 variant, 50 blood samples (1–2 ml in volume
for each sample, with a total of 100 SLC10A1 alleles) from
healthy volunteers were collected to serve as the controls.
The current study was approved by the Committee for
Medical Ethics, The First Affiliated Hospital of Jinan University
and written informed consent was obtained from the parents of the
patients and all the healthy controls.
Sanger sequencing
Genomic DNA was extracted using a DNA extraction kit
(Simgen) according to the manufacturer's protocol. The five
SLC10A1 exons and their flanking sequences, including the
5′- and 3′-untranslated regions as well as 309 base pairs upstream
of the transcriptional start site, were amplified by PCR as
described previously (10). The
PCR products were purified using a gel extraction kit (Omega
Bio-Tek, Inc.) and Sanger sequencing was subsequently performed on
a 96-capillary ABI 3730×l DNA Analyzer (Applied Biosystems; Thermo
Fisher Scientific, Inc.) with a BigDye Terminator v 3.1 Cycle
Sequencing kit (Thermo Fisher Scientific, Inc.).
The sequencing results were aligned with the
SLC10A1 gene sequence, which was available at Ensembl Genome
Browser (www.ensembl.org), using DNAman software
(version 5.2.2; Lynnon Biosoft Corporation) and analyzed using
Chromas software (version 2.6.6; Technelysium Pty, Ltd.). The
allele frequency of the identified novel SLC10A1 variant was
investigated in four population databases, including the 1000
Genomes Project (browser.1000genomes.org), the Exome Sequencing Project
(esp.gs.washington.edu/drupal), the
Exome Aggregation Consortium (exac.broadinstitute.org) and the Human Gene Mutation
Database (www.hgmd.cf.ac.uk/ac/index.php).
PCR-restriction fragment length
polymorphism (RFLP) approach
In addition to the PCR-RFLP approach described
previously to detect the pathogenic SLC10A1 variant
c.800C>T(p.Ser267Phe) (12), a
novel PCR-RFLP procedure was developed in the present study to
verify the SLC10A1 genotypes of the patient family members
and to screen for the novel SLC10A1 variant in 50 healthy
individuals. The nucleotide sequences of the forward and reverse
primers used in PCR were 5′-CCACCTCTGTTCCTCTCTATCC-3′ and
5′-GCAACAGAGTGAGACCCTTTC-3′, respectively (Invitrogen; Thermo
Fisher Scientific, Inc.). The target fragment was amplified using a
PCR kit (Takara Biotechnology Co., Ltd.) and the PCR thermocycling
conditions were: 94°C for 5 min, followed by 35 cycles at 94°C for
30 sec, 58°C for 40 sec and 72°C for 50 sec, and 72°C for 10 min.
The HpyCH4V restriction enzyme (Thermo Fisher Scientific, Inc.) was
used to digest the PCR products and the digested DNA products were
subsequently separated by electrophoresis in a 4% agarose gel. For
frequency calculation of the novel variant, the number of mutated
alleles detected in all 50 control samples was divided by 100 and
the quotient was multiplied by 100%.
Alignment of homologous peptides
The amino acid sequences for the peptides homologous
to human NTCP were identified in 99 species using the Ensembl
Genome Browser. The 99 species were then classified into five
taxonomy subgroups: Primates (24 species), rodents and lagomorphs
(26 species), other mammals (24 species), other vertebrates (21
species) and other species (4 species), and aligned using
BLAST/BLAT Ensembl software (Ensembl Release 97, July 2019:
www.ensembl.org/Multi/Tools/Blast?db=core).
In silico prediction of
pathogenicity
A total of four prediction programs were used to
predict the pathogenicity of the novel SLC10A1 variant.
PolyPhen-2 (genetics.bwh.harvard.edu/pph2) analysis identifies a
variant as ‘probably damaging’ if the probability is >0.85 and
as ‘possibly damaging’, if the probability >0.15 (18). MutationAssessor (mutationassessor.org) scores a mutation by global and
subfamily specific conservation patterns as low, medium or high
(19). Sorting Intolerant From
Tolerant (SIFT; sift.jcvi.org) classifies the variant
as being ‘deleterious’ if the prediction score is <0.05
(20). Moreover, the deleterious
annotation of genetic variants using neural networks (DANN;
cbcl.ics.uci.edu/public_data/DANN) may be used to
annotate the pathogenicity of genetic variants using neural
networks and higher values are associated with deleterious variants
(21).
Effect of the novel mutation on the
structure of the NTCP protein
The wild structural model of NTCP protein was built
using the online software SWISS-MODEL automated protein modeling
server (swissmodel.expasy.org). The tertiary
structures of the wild type and mutant NTCP protein were compared
by SWISS-Pdb Viewer 4.1.0 (www.Expasy.org/spdbv) to evaluate the effect of the
variant on structure of the NTCP molecule.
Results
Patient 1
A 27.7-month-old male was referred to The First
Affiliated Hospital of Jinan University due to sustained elevated
serum total bile acids (TBA). A health examination 1.2 months prior
to admission revealed that the patient had a TBA level of 103.8
µmol/l and elevated transaminases. A biochemistry test repeated 3
weeks after the initial test revealed persistent hypercholanemia
and elevated aspartate transaminase level (Table II). The patient was subsequently
referred to the First Affiliated Hospital of Jinan University for
further investigation and management.
 | Table II.Biochemical indices in family 1 with
NTCP deficiency. |
Table II.
Biochemical indices in family 1 with
NTCP deficiency.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Mother |
|
|
|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|---|
|
| Patient 1 | Father | AP | Gestational
ages | PP | Aunt | MGF |
|---|
|
|
|
|
|
|
|
|
|
|---|
| Indices (reference
range) | 26.5M | 27.2M | 27.7Ma | 28.7M | 31.3M | 41M | 4Y | 25Y | 26Y | 27Y | 28Y | 31Y | 31Y | 32Y | 23.5Y |
12+2W |
15+6W | 32W |
39+6W | 29.3Y | 35Y | 59Y |
|---|
| ALT (5–40 U/l) | 158 | 36 | 28 | 34 | 20 | 20 | 13 | 13 | 12 | 7 | 14 | 5 | 8 | 4 | 15 | 13 | 32 | 19 | 14 | 13 | 10.4 | 29 |
| AST (5–40 U/l) | 147 | 80 | 76 | 59 | 43 | 40 | 32 | 18 | 28 | 18 | 23 | 15 | 18 | 14 | – | 17 | 22 | 18 | 16 | 14 | 12.9 | 22 |
| GGT (8–50 U/l) | – | 24 | 20 | 13 | 14 | 15 | 12 | – | – | – | – | – | 21 | – | 12 | – | – | 9 | 10 | 12 | 52.6 | – |
| ALP (20–500
U/l) | 236 | 218 | 273 | 277 | 1939 | 204 | 190 | 110 | 123 | 94 | 91 | 74 | 100 | 89 | – | – | – | – | – | 49 | 12.9 | 61 |
| TBA (0–10
µmol/l) | 103.8 | 30.8 | 70.5 | 132.3 | 18.6 | 63.8 | 44.7 | 23.4 | 9.8 | 21.1 | 27.4 | 19.7 | 30 | 58.2 | – | 2.17 | 1.76 | 1.67 | 6.55 | 4 | 45.5 | 21.6 |
| Tbil (5.1–23
µmol/l) | 8.9 | 3.3 | 5.4 | 7 | 7.9 | 4.6 | 6.8 | 8.3 | 13.1 | 12.4 | 16.8 | 8.7 | 11.4 | 4.9 | 13.9 | – | – | 7.1 | 7.6 | 11.2 | 13.3 | 9.4 |
| Dbil (0.6–6.8
µmol/l) | 4 | 2 | 1.4 | 2.4 | 2.4 | 1.8 | 3 | 3.1 | 4.6 | 4.3 | 2.8 | 3.9 | 3.9 | 2.4 | 3.7 | – | – | 1.5 | 1.9 | 3 | 4.5 | 3.4 |
| Ibil (1.7–17
µmol/l) | 4.9 | – | 4 | 4.6 | 5.5 | 2.8 | 3.8 | 5.2 | 9 | 8.1 | – | 4.8 | 7.5 | 2.5 | 10.2 | – | – | 5.6 | 5.7 | 8.2 | 8.8 | 6 |
| TP (65–85 g/l) | 68.1 | 72.1 | 65.9 | 67.4 | 67.5 | 71.5 | 65.8 | 86.3 | 74.1 | 70.3 | 80 | 74.8 | 78 | 70 | 70.6 | – | – | 68.6 | 64.7 | 7.69 | 72.8 | 72.8 |
| ALB (40–55
g/l) | 48.4 | 48.8 | 47.1 | 47.6 | 47.9 | 50 | 47.3 | 54.9 | 51.3 | 53 | 51.3 | 48.8 | 53.3 | 50.3 | 44.2 | – | – | 39.9 | 37.6 | 48.2 | 47.6 | 41.5 |
| GLB (20–40
g/l) | 19.7 | 23.3 | 18.8 | 19.8 | 19.6 | 21.5 | 18.5 | 31.4 | 22.8 | 20.3 | 28.7 | 26 | 24.7 | 19.7 | 26.4 | – | – | 28.7 | 27.1 | 28.7 | 25.2 | 31.3 |
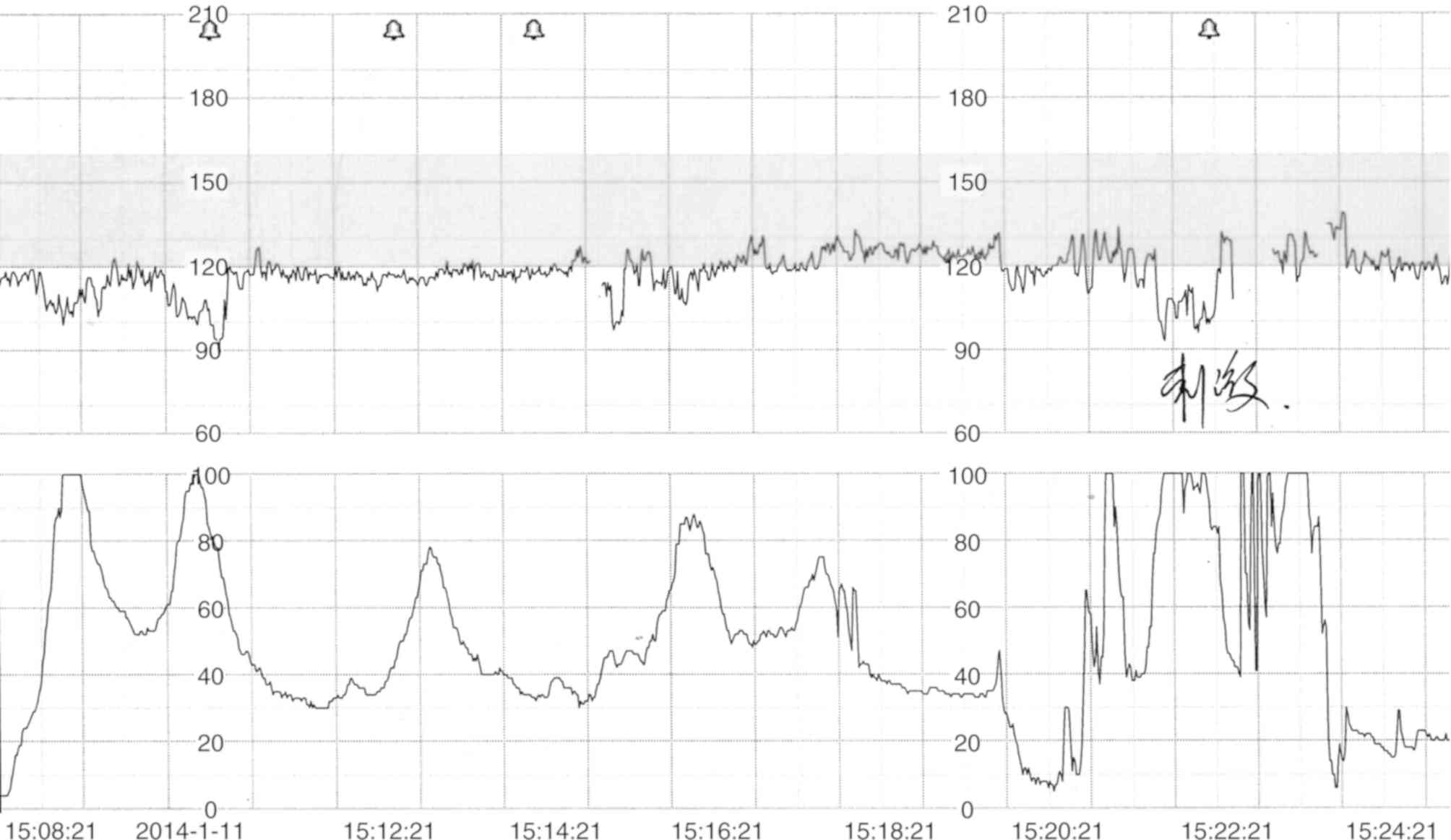
The patient had frequent early fetal heart
decelerations that were detected by cardiotocography during labor
at the gestational age of 39 weeks and 5 days (Fig. 1) and the patient was subsequently
delivered by a cesarean section. The patient had a birth weight of
3.0 kg and a body length of 47 cm. The patient's mother was healthy
during and after pregnancy; however, the father had occasional
hypercholanemia without any clinical manifestations. Additionally,
the patient's paternal aunt exhibited raised TBA levels without any
clinical symptoms. The TBA levels in the paternal grandparents and
maternal grandmother were in the normal range. The maternal
grandfather exhibited slightly elevated levels (Table II) and a liver ultrasonogram
revealed a 5×4 mm hemangioma in the right lobe.
A physical examination revealed that the patient had
a body weight of 11.5 kg, height of 85.5 cm and a head
circumference of 49 cm. No dysmorphic appearance or jaundice were
observed in the skin and sclera. No stridor, crackles or crepitus
were heard following auscultation of the lungs. The heart sounds
were normal without any murmurs. The liver and spleen were
nonpalpable. The tone of the body and limbs appeared normal.
Biochemical analysis of the patient revealed that
the levels of TBA and transaminase were elevated (Table II). The SLC10A1 gene was
analyzed to evaluate the possibility of NTCP deficiency.
Thereafter, no special treatment was administered to the patient
except vitamin D supplementation. Subsequent clinic follow-ups
revealed that the patient's aspartate transaminase gradually
decreased to normal levels; however, serum TBA was persistently
elevated and globulin levels exhibited an occasional decrease
(Table II). The patient exhibited
normal anthropometric social performance.
Patient 2
A 3.8-month-old female patient was referred to the
First Affiliated Hospital of Jinan University with increased TBA
levels that had persisted for 3.5 months. The patient was admitted
into the neonatal unit in the First Affiliated Hospital of Jinan
University as a preterm infant. The infant was delivered by a
cesarean section as the first-born of dichorionic diamniotic twins
at the gestational age of 32 weeks and 3 days (birth weight, 1.4
kg; body length, 40 cm). Her dizygotic twin sister, who had a birth
weight of 1.85 kg and body length of 45 cm, exhibited occasional
hypercholanemia (Table III).
| Table III.Biochemical indices in family 2 with
NTCP-deficient patient. |
Table III.
Biochemical indices in family 2 with
NTCP-deficient patient.
|
|
|
|
|
|
|
|
|
|
|
|
|
| Mother |
|
|
|
|
|
|
|
|
|
|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|---|
|
|
Patient
2 | Father | Gestational ages
(W) | Sister |
|---|
|
|
|
|
|
|
|---|
| Indices (reference
range) | 1D | 3D | 9D | 17D | 25D | 1.7M | 2.3M | 2.8M | 3.8Ma | 7M | 9.1M | 32Y |
24+6 |
26+4 |
29+6 |
30+4 |
31+2 | 1D | 8D | 16D | 24D | 1.7M | 1.9M | 2.2M | 2.5M | 2.6M |
|---|
| ALT (5–40 U/l) | 14 | 4 | 3 | 3 | 4 | 10 | 22 | 22 | 28 | 24 | 26 | 10 | 14 | 11 | 7 | 17 | 7 | 4 | 5 | 5 | 5 | 9 | 12 | 10 | 14 | 15 |
| AST (5–40 U/l) | 40 | 26 | 15 | 19 | 15 | 24 | 43 | 26 | 33 | 33 | 37 | 12 | 14 | 13 | 12 | 18 | 13 | 12 | 14 | 19 | 17 | 18 | 18 | 16 | 22 | 23 |
| GGT (8–50 U/l) | 121 | 75 | 61 | 56 | 86 | 272 | 225 | 74 | 46 | 13 | 13 | 18 | – | – | – | 12 | 10 | 97 | 49 | 95 | 155 | 67 | 77 | 66 | 63 | 58 |
| ALP (20–500
U/l) | 172 | – | – | – | 216 | 248 | 310 | – | 219 | 233 | 227 | 86 | – | – | – | 163 | 135 | 182 | – | – | 222 | 270 | 295 | 233 | 348 | 289 |
| TBA (0–10
µmol/l) | 8.4 | – | 35.4 | – | 81.5 | 176.4 | 260.5 | 214.8 | 256.5 | 86.2 | 77.8 | 8.1 | 1.7 | 4 | 0.9 | 2.4 | 2.7 | 3.9 | 9.7 | 10.8 | 12.4 | 13.6 | 12.5 | 9 | 9.5 | 19.2 |
| Tbil (5.1–23
µmol/l) | 36.5 | 106.3 | 158.1 | 141.8 | 57.9 | 26.3 | 13.5 | 7.9 | 5.6 | 3.6 | 3.9 | 12.2 | 6.1 | 4.9 | 5 | 5.2 | 5.1 | 33.4 | 166.3 | – | 20.8 | 15.6 | 6.7 | 6.1 | 5.9 | 6.6 |
| Dbil (0.6–6.8
µmol/l) | 10.4 | 11.4 | 11.6 | 13.3 | 13.2 | 13 | 8.5 | 4.1 | 1.7 | 1.3 | 1.5 | 1.6 | 0.8 | 1.2 | 0.7 | 1.3 | 0.8 | 13.1 | 11.8 | – | 7.5 | 4 | 4.5 | 2.5 | 1.3 | 3.5 |
| Ibil (1.7–17
µmol/l) | 26.1 | 94.9 | 146.5 | 128.5 | 44.7 | 13.3 | 5 | 3.8 | 3.9 | 2.3 | 2.4 | 10.6 | 5.3 | 3.7 | 4.3 | 3.9 | 4.3 | 20.3 | 154.5 | – | 13.3 | 11.6 | 2.2 | 3.6 | 4.6 | 3.1 |
| TP (65–85 g/l) | 51.3 | – | – | – | 47.8 | 48.6 | 59.8 | – | 59.5 | 60.8 | 61.5 | 72.8 | – | – | – | 57.6 | 57.4 | 45.3 | – | – | 46.7 | 47.2 | 48.5 | 48.5 | 47.5 | 49.9 |
| ALB (40–55
g/l) | 36.8 | 29.4 | 28.9 | 33.6 | 35.6 | 34.1 | 44.4 | – | 40.4 | 42.8 | 43.4 | 42 | – | – | – | 30.4 | 30.3 | 33.6 | 31.4 | 33.8 | 34.6 | 34.5 | 37.3 | 34 | 34.2 | 35.7 |
| GLB (20–40
g/l) | 14.5 | – | – | – | 12.2 | 14.5 | 15.4 | – | 19.5 | 18 | 18.1 | 30.7 | – | – | – | 27.2 | 27.1 | 11.7 | – | – | 12.1 | 12.7 | 11.2 | 14.5 | 13.3 | 14.2 |
The patient's parents were healthy and there was no
family history of genetic diseases. The patient had a slightly
increased serum TBA levels 9 days following birth. Additionally,
the patient developed hypoalbuminemia during hospitalization and
was treated with intravenous albumin. The patient was discharged
with unresolved hypercholanemia 25 days following birth. The
elevation of TBA was intractable on subsequent clinic follow-up,
with a peak level 260.5 µmol/l at the age of 2.3 months (Table III).
Physical examination revealed a body weight of 4.7
kg, height of 54 cm and a head circumference 36.5 cm. There was no
dysmorphic appearance or jaundice in the skin and sclera. The skull
and facial appearance were not malformed. No positive signs were
found in the lungs and the heart. The patient's liver and spleen
were not enlarged. The tone of the body and limbs appeared normal.
Biochemical analysis revealed elevated TBA levels, with a peak of
256.5 µmol/l and occasional decreases in globulin and albumin
levels. All other indices were normal (Table III).
As the patient exhibited persistent hypercholanemia,
SLC10A1 analysis was performed to evaluate the possibility
of NTCP deficiency. No special treatment other than vitamin D and
zinc supplements was given thereafter. The patient was followed-up
to the age of 9.1 months and exhibited normal development patterns.
During the follow-up, the patient had persistently elevated TBA
levels, while other liver indices were in the normal range
(Table III).
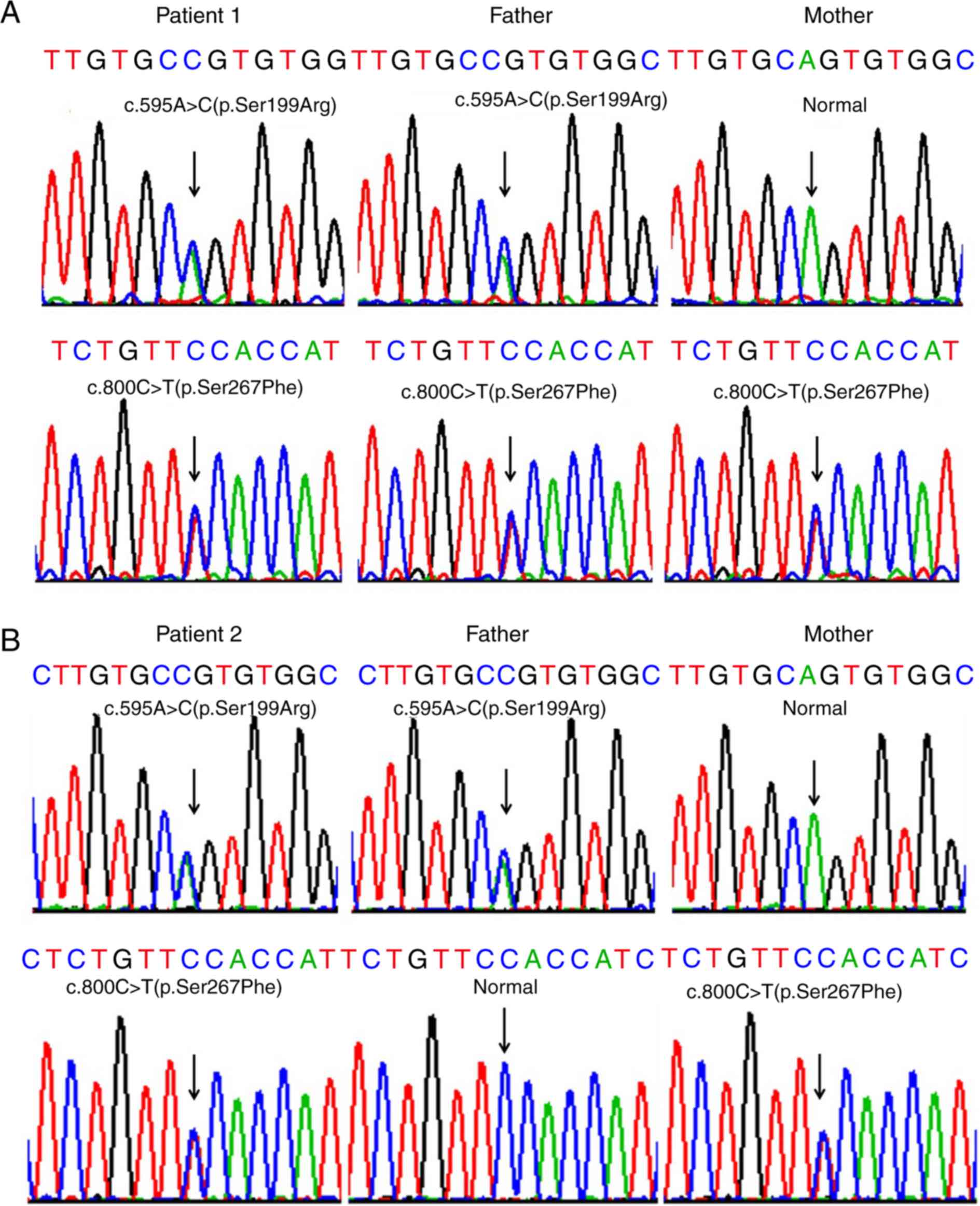
SLC10A1 genotypes
Sanger sequencing of the SLC10A1 gene in the
two families demonstrated that both index patients were compound
heterozygotes for the variants c.800C>T(p.Ser267Phe) and
c.595A>C(p.Ser199Arg). In family 1, the father shared the same
genotype with patient 1 while the mother was a carrier of the
variant 800C>T (p.Ser267Phe) (Fig.
2A). In family 2, the father was a carrier of the variant
c.595A>C(p.Ser199Arg), while the mother was a carrier of
c.800C>T(p.Ser267Phe) (Fig.
2B). To the best of our knowledge, the variant c.595A>C
(p.Ser199Arg) has not been previously reported by other studies in
the PubMed database (www.ncbi.nlm.nih.gov) and is not currently included in
the 1000 Genomes Project (browser.1000genomes.org), the Exome Sequencing Project
(esp.gs.washington.edu/drupal), and
the Human Gene Mutation Database (www.hgmd.cf.ac.uk/ac/index.php). The Exome Aggregation
Consortium (exac.broadinstitute.org) database has recorded the
variant c.595A>C (p.Ser199Arg), with an allele frequency of 0.3%
in East Asian populations and of 0.025% in all humans.
PCR-RFLP findings
A novel PCR-RFLP approach was developed to explore
the SLC10A1 genotypes in the two families and to investigate
the frequency of the novel variant c.595A>C(p.Ser199Arg) in
healthy controls. As presented in Fig.
3, in addition to the 3 patients diagnosed by Sanger
sequencing, the patient's aunt in family 1 was also affected by
NTCP deficiency, having an SLC10A1 genotype of
c.800C>T(p.Ser267Phe)/c.595A>C(p.Ser199Arg). Moreover, no
c.595A>C(p.Ser199Arg) carrier status was detected in 50 healthy
controls, indicating an allele frequency of <1%. Therefore, the
missense variant identified in the present study was a novel
SLC10A1 mutation and not a single nucleotide
polymorphism.
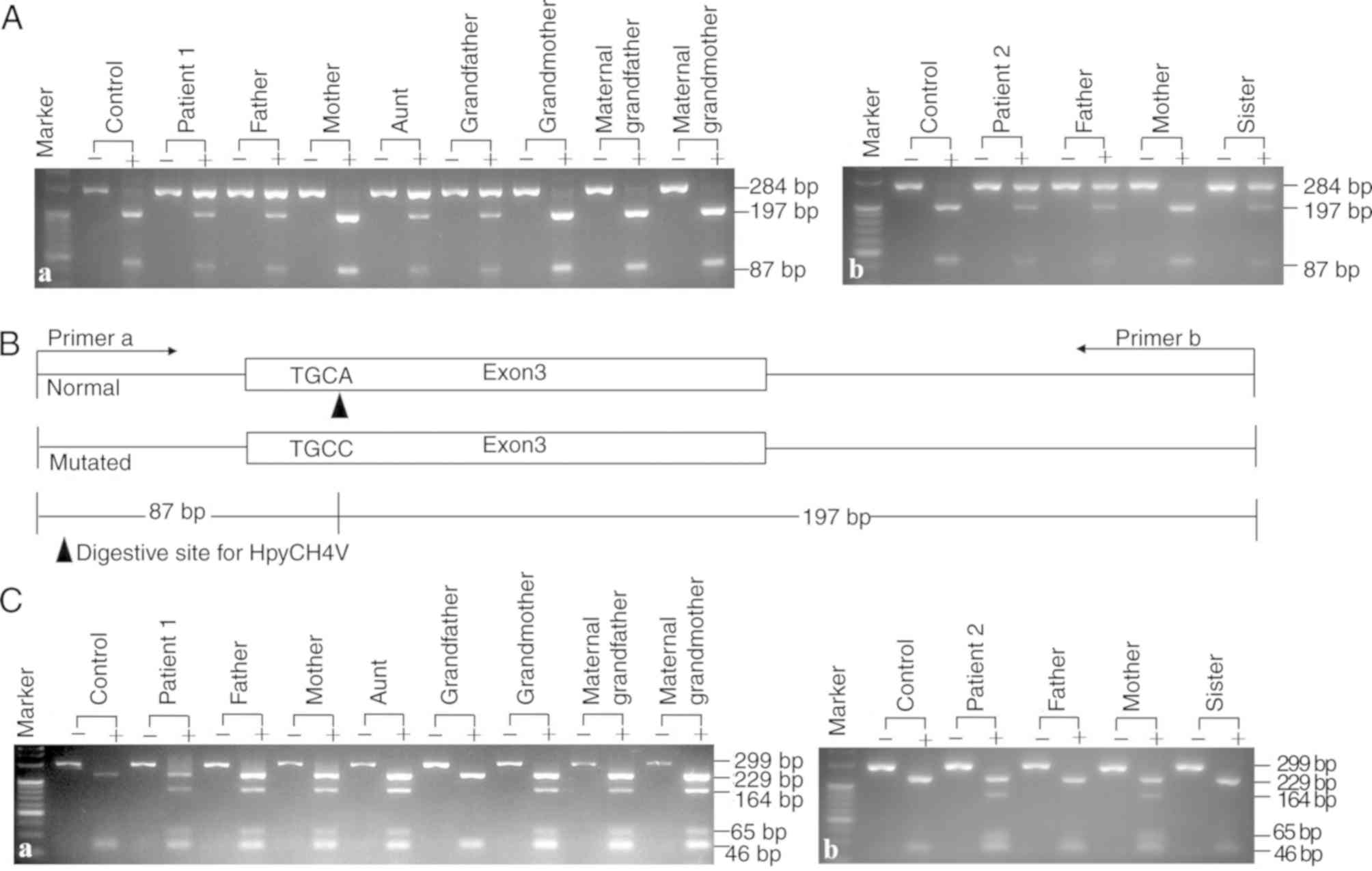 | Figure 3.PCR-RFLP protocols screening for the
variants c.800C>T(p.Ser267Phe) and c.595A>C(p.Ser199Arg).
Representative gel electrophoresis images of the digested products
by the newly-developed PCR-RFLP protocol for the detection of the
novel variant c.595A>C(p.Ser199Arg) in family (Aa) 1 and (Ab) 2.
The results showed that patient 1, his father, aunt and grandfather
and patient 2, her father and sister all harbored the
c.595A>C(p.Ser199Arg) variant. (B) A schematic diagram of the
PCR-RFLP approach. The wild-type SLC10A1 allele had a
HpyCH4V restriction enzyme site and produced the 87 and 197 bp
fragments from the 284 bp band following enzymatic digestion. A
representative gel electrophoresis image for PCR-RFLP screening for
the c.800C>T (p.Ser267Phe) variant in family 1 (Ca) and 2 (Cb).
The results showed that patient 1, his father, mother, aunt,
grandmother, maternal grandfather and maternal grandmother and
patient 2 and her mother harbored the c.800C>T(p.Ser267Phe)
variant. RFLP, restriction fragment length polymorphism;
SLC10A1, solute carrier family 10 member 1. |
Bioinformatics analysis
The amino acid sequences of the homologous peptides
in a total of 99 species were aligned to allow comparative
analysis. Although the amino acid p.Ser199 was not conserved in
other vertebrates (11/21) and other species (1/4) (data not shown),
this residue was relatively conserved in primates and mammals. In
fact, this residue was found in 23/24 of the primates including
humans (Table I), 21/26 of rodents
and lagomorphs and 16/24 of the other mammals (data not shown).
| Table I.Comparative alignment of the
homologous peptides in primates. |
Table I.
Comparative alignment of the
homologous peptides in primates.
| Primates | Peptide | From | Amino acid
sequence | To |
|---|
| Human |
ENSP00000216540 | 181 |
PQYMRYVIKGGMIIILLCSVAVTVLSAINVGKSIMFAMTPLLIATSSLMPFIGFLLGYVL | 240 |
| Angola colobus |
ENSCANP00000002782 | 181 |
PQYMRYVIKGGTIIILLCSVAVTVLSAINVGKSIMFAMTPLLIATSSLMPFIGFLLGYVL | 240 |
| Black snub-nosed
monkey |
ENSRBIP00000040923 | 181 |
PQYMRYVIKGGTIIILLCSVAVTVLSAINVGKSIMFAMTPVLIATSSLMPFIGFLLGYVL | 240 |
| Bolivian squirrel
monkey |
ENSSBOP00000020611 | 181 |
PQYVRYVVKGGMIIILLCSVTVIVLSAINVGKSILFAMTPLLVTTSSLMPFIGFLLGYVL | 240 |
| Bonobo |
ENSPPAP00000018811 | 181 |
PQYMRYVIKGGMIIILLCSVAVTVLSAINVGKSIMFAMTPLLIATSSLMPFIGFLLGYVL | 240 |
| Bushbaby |
ENSOGAP00000013546 | 181 |
PQYVGYVTKGGMIIILLLSVAIIALSAINVGKSIMVAMTPLLLATSSLMPFIGFLLGYIL | 240 |
| Capuchin |
ENSCCAP00000003215 | 181 |
PQYVRYVVKGGMIIILLCSVTVIVLSAINVGKSILFAMTPLLVTTSSLMPFIGFLLGYVL | 240 |
| Chimpanzee |
ENSPTRP00000010999 | 181 |
PQYMRYVIKGGMIIILLCSVAVTVLSAINVGKSIMFAMTPLLIATSSLMPFIGFLLGYVL | 240 |
| Coquerel's
sifaka |
ENSPCOP00000027513 | 181 |
PQYVSYVTKGGMLIIATFSGAIVTLSAINVGKSIVYAMTPTLLASSFLMPLIGFLLGYLL | 240 |
| Crab-eating
macaque |
ENSMFAP00000020987 | 181 |
PQYMRYVIKGGMIIILLCSVAVTVLSAINVGKSIMFAMTPLLIATSSLMPFIGFLLGYVL | 240 |
| Drill |
ENSMLEP00000033102 | 181 |
PQYMRYVIKGGMIIILLCSVAVTVLSAINVGKSIMFAMTPLLIATSSLMPFIGFLLGYVL | 240 |
| Gibbon |
ENSNLEP00000002065 | 175 |
PQYMCYVIKGGMIIILLCSVAVTVLSAINVGKSIMFAMTPLLIATSSLMPFIGFLLGYVL | 234 |
| Golden snub-nosed
monkey |
ENSRROP00000037832 | 181 |
PQYMRYVIKGGTIIILLCSVAVTVLSAINVGKSIMFAMTPVLIATSSLMPFIGFLLGYVL | 240 |
| Gorilla |
ENSGGOP00000022961 | 180 |
VDLHSLHSQGGMIIILLCSVAVTVLSAINVGKSIMFAMTPLLIATSSLMPFIGFLLGYVL | 239 |
| Macaque |
ENSMMUP00000029493 | 181 |
PQYMRYVIKGGMIIILLCSVAVTVLSAINVGKSIMFAMTPLLIATSSLMPFIGFLLGYVL | 240 |
| Marmoset |
ENSCJAP00000027394 | 179 |
PQYVRYVVKGGMIIILLCSVAVIVLSAINVGKSILLAMTPLLVTTSSLMPFIGFLLGYVL | 238 |
| Ma's night
monkey |
ENSANAP00000041565 | 181 |
PQYVRYVVKGGMIIILLCSVTVIVLSAINVGKSILFAMTPLLVTTSSLMPFIGFLLGYVL | 240 |
| Mouse Lemur |
ENSMICP00000008803 | 181 |
PQYIPYVKQGGTLIIATFSGAIVTLSAINVGKSIMFAMTPTLLATSFLMPLIGFLLGYLL | 240 |
| Olive baboon |
ENSPANP00000017171 | 181 |
PQYMRYVIKGGMIIILLCSVAVTVLSAINVGKSIMFAMTPLLIATSSLMPFIGFLLGYVL | 240 |
| Orangutan |
ENSPPYP00000006762 | 181 |
PQYMRYVIKGGMIIILLCSVAVTVLSAINVGKSIMFAMTPLLIATSSLMPFIGFLLGYVL | 240 |
| Pig-tailed
macaque |
ENSMNEP00000030827 | 181 |
PQYMRYVIKGGMIIILLCSVAVTVLSAIN
VGKSIMFAMTPLLIATSSLMPFIGFLLGYVL | 240 |
| Sooty mangabey |
ENSCATP00000043650 | 181 |
PQYMRYVIKGGMIIILLCSVAVTVLSAINVGKSIMFAMTPLLIATSSLMPFIGFLLGYVL | 240 |
| Tarsier |
ENSTSYP00000003672 | 187 |
XXXXXXXXXIGAIVGGVLLLVVAVAGVVLAKESWNADIT--LLTISFVFPLIGHVTGFLL | 244 |
| Vervet-AGM |
ENSCSAP00000009215 | 181 |
PQYMRYVIKGGMIIILLCSVAVTVLSAINVGKSIMFAMTPLLIATSSLMPFIGFLLGYVL | 240 |
In silico tools were subsequently used to
predict the pathogenicity of the novel mutation. Polyphen 2 and
DANN generated the same outcome (‘possibly damaging’), with a
prediction score of 0.977 and 0.986, respectively. MutationAssessor
yielded a result of ‘medium’ with a prediction score of 3.07, while
SIFT suggested that the mutation was ‘tolerated’, with a prediction
score of 0.08.
SWISS-MODEL software was used for structural
prediction of the NTCP protein. The c.595A>C(p.Ser199Arg)
variant resulted in the replacement of a serine by an arginine at
the amino acid position 199 of the NTCP molecule. Arginine produced
a new hydrogen bond with the proline at position 286. Additionally,
the hydrogen bond distances between specific amino acids in the
NTCP protein were altered (Fig.
4), distorting the molecular structure of the NTCP protein.
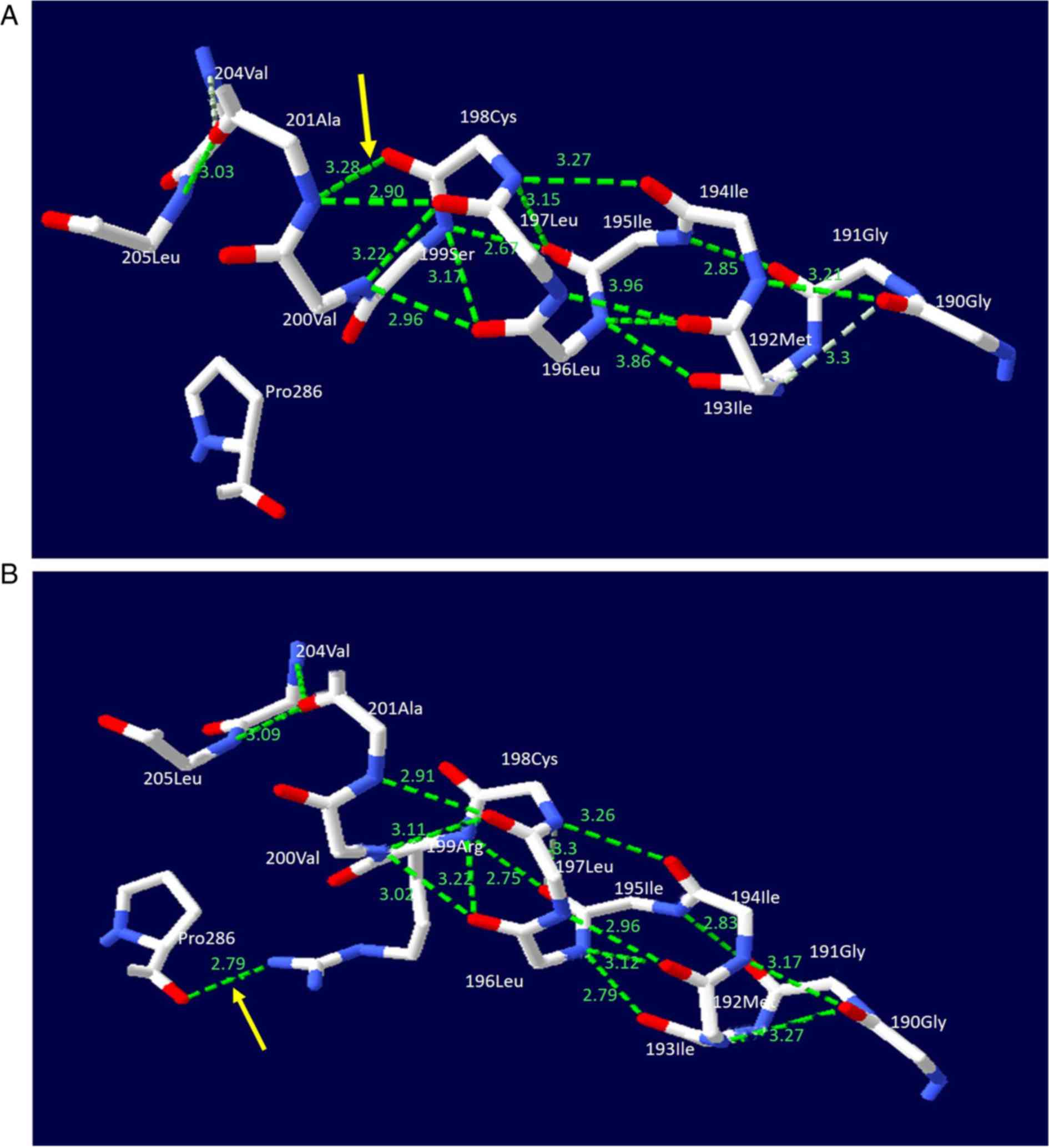 | Figure 4.NTCP molecular alteration caused by
the solute carrier family 10 member 1 variant
c.595A>C(p.Ser199Arg). The figure illustrates a ball-and-stick
model of the human NTCP protein. The solid lines in white, red and
blue represent carbon, oxygen and nitrogen atoms, respectively,
while the dashed lines in green represent hydrogen bonds. (A) In
the wild-type model, the serine at position 199 in NTCP was not
associated with the proline at position 286. (B) In the mutant NTCP
model, the serine at position 199 was changed to an arginine. This
change led to the formation of a new hydrogen bond between the
arginine and proline (denoted by the yellow arrow). Meanwhile, the
hydrogen bond lengths between the Gly at 190 and the Ile at 194,
the Gly at 190 and Ile at 193, the Gly at 191 and Ile at 195, the
Met at 192 and Leu at 196 as well as the Ile at 195 and Cys at 198,
were changed from 3.17, 3.3, 2.85, 2.86 and 3.15 Å to 3.21, 3.27,
2.83, 2.79 and 3.3 Å, respectively. The changes in the hydrogen
bonds resulted in structural distortion of the normal NTCP
molecule. NTCP, sodium taurocholate cotransporting polypeptide. |
Discussion
The present study identified 4 patients with NTCP
deficiency, including 2 pediatric and 2 adult individuals. Analysis
revealed that the patients were compound heterozygous for the
SLC10A1 variants c.800C>T(p.Ser267Phe) and c.595A>C
(p.Ser199Arg). The former variant has previously been reported to
be pathogenic by functional and bioinformatics approaches and
clinical studies (6,9,12–16).
The latter unreported variant demonstrated a frequency of <1% in
50 healthy individuals in the present study and was absent from
controls in the 1000 Genomes Project, the Exome Sequencing Project
and the Human Gene Mutation Database. Exome Aggregation Consortium
database has recorded the variant c.595A>C (p.Ser199Arg);
however, this database is not based on findings on patients but
general populations and the low allele frequencies of this variant
in East Asian (0.3%) and all humans (0.025%) indicated a mutation,
constituting supporting evidence of pathogenicity. The
c.595A>C(p.Ser199Arg) mutation exhibited co-segregation with
hypercholanemia and was detected in trans with the
c.800C>T(p.Ser267Phe) variant in 4 patients from two unrelated
families. Moreover, this novel mutation exhibited a conserved amino
acid and distorted the conformation of the NTCP molecule. While
functional analysis of the novel SLC10A1 variant was not
performed in the current study due to technical limitations, the
results obtained supported the diagnosis of NTCP deficiency in the
patients investigated. Particularly, the functional study
constituted a work plan for the authors' future investigation,
since it provides much stronger evidence of pathogenicity.
Moreover, the present group has been committed to the clinical
diagnosis of NTCP deficiency since the first pediatric patient in
China and the first adult worldwide with such a condition were
reported in 2016 (12), but
unfortunately no additional patients harboring the novel
SLC10A1 variant c.595A>C (p.Ser199Arg) were diagnosed,
suggesting that this variant was not so prevalent in Chinese
populations.
In silico tools were used to predict the
pathogenicity of the SLC10A1 variant
c.595A>C(p.Ser199Arg). Polyphen 2 and DANN predicted pathogenic
outcomes, and this might be considered as computational evidence
supporting the deleterious effect on the SLC10A1 gene,
according to the American College of Medical Genetics and Genomics
standards and guidelines for the interpretation of sequence
variants (22). However,
MutationAssessor and SIFT generated inconsistent results. This
discrepancy therefore requires further investigation as the
accuracy of prediction algorithms is affected by a number of
variables, including the gene examined, the number of sequences in
the alignment, the evolutionary distances among species and the
importance of absolute amino acid conservation vs. relatively
conservative missense changes (23). In general, the accuracy of the
majority of algorithms used for missense variant prediction is
~65–80%, even when applied to established disease-causing variants
(22). As clinical data describe
human disease more directly, clinical observations should be
considered more persuasive when a discrepancy or conflict arises
between clinical and functional observations (24).
The 4 patients in the current study presented with
persistent hypercholanemia and supported the primary role of NTCP
in the uptake of bile acids from plasma. However, the plasma TBA
levels tended to decrease over time in the two pediatric patients
and the father of patient 1 exhibited an occasionally normal TBA
level. The aforementio-ned trends suggested that mechanisms other
than NTCP are involved in the hepatic uptake of bile acids. Solute
carrier organic anion transporter family member 1B1 and 3
(OATP1B1/3), two members of the organic anion transporting
polypeptides family, are heterodimers expressed in the basal
membrane of hepatocytes and clear bile acid salts such as cholic
acid, glycolic acid and taurocholic acid from the plasma (25). OATP1B1 and OATP1B3
demonstrate an age-dependent maturation process in humans and it
has been reported that their hepatic expression in all pediatric
age groups was significantly lower compared with adults (26). Furthermore, the expression level of
OATP1 at birth is 15% of adult levels (25). Additionally, the organic solute
transporter α/β is a heterodimer expressed in the membrane of
hepatocytes, where it functions as a bidirectional organic solute
transport system that transports bile acids (27). The farnesoid X receptor is
activated by bile acids (28) and
subsequently inhibits the synthesis of bile acids in hepatocytes
and upregulates the expression of organic solute transporters α and
β (OSTα/β) to remove bile acids from the plasma (29–31).
The presence of the aforementioned maturation and regulation
mechanisms suggests that the bile acid levels in patients with NTCP
deficiency may not increase uncontrollably, may decline with age
and may approach normal as observed in the father of patient 1 in
the current study.
The hypoglobulinemia observed in patients 1 and 2
and the hypoalbuminemia seen in patient 2 may be a transient
alteration associated with their age and not related to NTCP
deficiency. Globulin is closely associated with immune function
(32), which gradually develops
and matures in children (33).
Moreover, hypoalbuminemia is associated with immature liver
synthesis function in children, particularly preterm infants
(34) and may result in the
gradually-corrected hypoalbuminemia observed in patient 2.
The patients with NTCP deficiency reported in
previous studies were homozygous or compound heterozygous for
SLC10A1 variants, indicating an autosomal recessive disorder
(8–14). In the current study, however, the
maternal grandfather of patient 1 and the sister of patient 2, were
carriers of the variant c.800C>T(p.Ser267Phe) and c.595A>C
(p.Ser199Arg) respectively and exhibited hypercholanemia. In the
former case, the specific cause for the elevated TBA level was
unknown but may be attributed at least in part to hepatic
hemangioma. In the latter case, a reasonable explanation may be
physiological cholestasis, which results in temporarily high TBA
levels in normal newborns and infants due to immature liver
function (35–37). Moreover, her hyperbilirubinemia and
high γ-glutamyl transpeptidase level may also reflect her immature
liver function as a preterm infant. The NTCP protein is gradually
expressed on the plasma membrane of hepatocytes in an age-dependent
manner and its glycosylation is not completed until ~1 year after
birth (38).
Patient 1 in the current study was delivered by
cesarean section due to early fetal heart decelerations that were
associated with uterine contractions and vaginal examination during
labor. NTCP deficiency itself in the affected fetus may not be
associated with the heart decelerations. Bile acids are synthesized
in the fetus from 12 weeks of gestation (39) and are exported into the maternal
circulation via the placenta (40,41).
Trophoblast cells in the placental barrier express bile acid
carrier proteins, which are involved in the one-way transport of
bile acids from the fetus to the mother (42). The mother of patient 1 had normal
serum TBA levels during pregnancy, therefore fetal NTCP deficiency
was unlikely to affect the fetus to cause early fetal heart
decelerations during labor. Moreover, to the best of our knowledge,
early fetal heart decelerations were not observed in 8 pediatric
patients with NTCP deficiency with uncomplicated deliveries
(8,10,12–15),
suggesting that early fetal heart decelerations are not associated
with NTCP deficiency.
In conclusion, by way of clinical and SLC10A1
genetic analysis, a total of 4 new patients with NTCP deficiency
from two unrelated families were diagnosed in the current study.
The results obtained in the current study suggested an autosomal
recessive trait for NTCP deficiency, supported the primary role of
NTCP in the uptake of bile acids from plasma and suggested the
presence of mechanisms of bile acid hepatic uptake other than NTCP.
Moreover, the novel missense variant c.595A>C (p.Ser199Arg)
enriched the SLC10A1 mutation spectrum and constituted a new
genetic marker for the molecular diagnosis and genetic counseling
of NTCP deficiency.
Acknowledgements
The authors would like to thank Dr Shu Chen from the
Department of Gynecology and Obstetrics of the First Affiliated
Hospital of Jinan University for assisting with the analysis of the
birth history of patient 1.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81570793, 81741080
and 81974057).
Availability of data and materials
The datasets used or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
YZS, MD, JQ and LG conceived and designed the
experiments. MD, JQ and LG performed the experiments. HL and XL
collected the clinical data. YS, HL and XX analyzed the data. YS
and HL wrote the manuscript.
Ethics approval and consent to
participate
The current study was approved by the Committee for
Medical Ethics, The First Affiliated Hospital, Jinan University,
Guangzhou, China and adhered to the World Medical Association
Declaration of Helsinki (2008). Written informed consent was
obtained from the parents of the patients and all the healthy
controls.
Patient consent for publication
Written informed consent was obtained from the
parents of the patients and all the volunteers.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hagenbuch B and Meier PJ: Molecular
cloning, chromosomal localization, and functional characterization
of a human liver Na+/bile acid cotransporter. J Clin Invest.
93:1326–1331. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shiao T, Iwahashi M, Fortune J, Quattrochi
L, Bowman S, Wick M, Qadri I and Simon FR: Structural and
functional characterization of liver cell-specific activity of the
human sodium/taurocholate cotransporter. Genomics. 69:203–213.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Anwer MS and Stieger B: Sodium-dependent
bile salt transporters of the SLC10A transporter family: More than
solute transporters. Pflugers Arch. 466:77–89. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hagenbuch B and Dawson P: The sodium bile
salt cotransport family SLC10. Pflugers Arch. 447:566–570. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ho RH, Leake BF, Roberts RL, Lee W and Kim
RB: Ethnicity-dependent polymorphism in Na+-taurocholate
cotransporting polypeptide (SLC10A1) reveals a domain critical for
bile acid substrate recognition. J Biol Chem. 279:7213–7222. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pan W, Song IS, Shin HJ, Kim MH, Choi YL,
Lim SJ, Kim WY, Lee SS and Shin JG: Genetic polymorphisms in
Na+-taurocholate co-transporting polypeptide (NTCP) and
ileal apical sodium-dependent bile acid transporter (ASBT) and
ethnic comparisons of functional variants of NTCP among Asian
populations. Xenobiotica. 41:501–510. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shneider BL, Fox VL, Schwarz KB, Watson
CL, Ananthanarayanan M, Thevananther S, Christie DM, Hardikar W,
Setchell KD, Mieli-Vergani G, et al: Hepatic basolateral
sodium-dependent-bile acid transporter expression in two unusual
cases of hypercholanemia and in extrahepatic biliary atresia.
Hepatology. 25:1176–1183. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vaz FM, Paulusma CC, Huidekoper H, de Ru
M, Lim C, Koster J, Ho-Mok K, Bootsma AH, Groen AK, Schaap FG, et
al: Sodium taurocholate cotransporting polypeptide (SLC10A1)
deficiency: Conjugated hypercholanemia without a clear clinical
phenotype. Hepatology. 61:260–267. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Van Herpe F, Waterham HR, Adams CJ,
Mannens M, Bikker H, Vaz FM and Cassiman D: NTCP deficiency and
persistently raised bile salts: An adult case. J Inherit Metab Dis.
40:313–315. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Qiu JW, Deng M, Cheng Y, Atif RM, Lin WX,
Guo L, Li H and Song YZ: Sodium taurocholate cotransporting
polypeptide (NTCP) deficiency: Identification of a novel SLC10A1
mutation in two unrelated infants presenting with neonatal indirect
hyperbilirubinemia and remarkable hypercholanemia. Oncotarget.
8:106598–106607. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu R, Chen C, Xia X, Liao Q, Wang Q,
Newcombe PJ, Xu S, Chen M, Ding Y, Li X, et al: Homozygous
p.Ser267Phe in SLC10A1 is associated with a new type of
hypercholanemia and implications for personalized medicine. Sci
Rep. 7:92142017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Deng M, Mao M, Guo L, Chen FP, Wen WR and
Song YZ: Clinical and molecular study of a pediatric patient with
sodium taurocholate cotransporting polypeptide deficiency. Exp Ther
Med. 12:3294–3300. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Song YZ and Deng M: Sodium taurocholate
cotransporting polypeptide deficiency manifesting as cholestatic
jaundice in early infancy: A complicated case study. Zhongguo Dang
Dai Er Ke Za Zhi. 19:350–354. 2017.(In Chinese). PubMed/NCBI
|
|
14
|
Li H, Qiu JW, Lin GZ, Deng M, Lin WX,
Cheng Y and Song YZ: Clinical and genetic analysis of a pediatric
patient with sodium taurocholate cotransporting polypeptide
deficiency. Zhongguo Dang Dai Er Ke Za Zhi. 20:279–284. 2018.(In
Chinese). PubMed/NCBI
|
|
15
|
Tan HJ, Deng M, Qiu JW, Wu JF and Song YZ:
Monozygotic twins suffering from sodium taurocholate cotransporting
polypeptide deficiency: A case report. Front Pediatr. 6:3542018.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen R, Deng M, Rauf YM, Lin GZ, Qiu JW,
Zhu SY, Xiao XM and Song YZ: Intrahepatic cholestasis of pregnancy
as a clinical manifestation of sodium-taurocholate cotransporting
polypeptide deficiency. Tohoku J Exp Med. 248:57–61. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mao F, Liu T, Hou X, Zhao H, He W, Li C,
Jing Z, Sui J, Wang F, Liu X, et al: Increased sulfation of bile
acids in mice and human subjects with sodium taurocholate
cotransporting polypeptide deficiency. J Biol Chem.
294:11853–11862. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bacalhau M, Pratas J, Simoes M, Mendes C,
Ribeiro C, Santos MJ, Diogo L, Macario MC and Grazina M: In silico
analysis for predicting pathogenicity of five unclassified
mitochondrial DNA mutations associated with mitochondrial
cytopathies' phenotypes. Eur J Med Genet. 60:172–177. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kumar P, Henikoff S and Ng PC: Predicting
the effects of coding non-synonymous variants on protein function
using the SIFT algorithm. Nat Protoc. 4:1073–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Quang D, Chen Y and Xie X: DANN: A deep
learning approach for annotating the pathogenicity of genetic
variants. Bioinformatics. 31:761–763. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hicks S, Wheeler DA, Plon SE and Kimmel M:
Prediction of missense mutation functionality depends on both the
algorithm and sequence alignment employed. Hum Mutat. 32:661–668.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nykamp K, Anderson M, Powers M, Garcia J,
Herrera B, Ho YY, Kobayashi Y, Patil N, Thusberg J, Westbrook M, et
al: Sherloc: A comprehensive refinement of the ACMG-AMP variant
classification criteria. Genet Med. 19:1105–1117. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hagenbuch B and Meier PJ: The superfamily
of organic anion transporting polypeptides. Biochim Biophys Acta.
1609:1–18. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mooij MG, Schwarz UI, de Koning BA, Leeder
JS, Gaedigk R, Samsom JN, Spaans E, van Goudoever JB, Tibboel D,
Kim RB and de Wildt SN: Ontogeny of human hepatic and intestinal
transporter gene expression during childhood: Age matters. Drug
Metab Dispos. 42:1268–1274. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ballatori N, Christian WV, Lee JY, Dawson
PA, Soroka CJ, Boyer JL, Madejczyk MS and Li N: OSTalpha-OSTbeta: A
major basolateral bile acid and steroid transporter in human
intestinal, renal and biliary epithelia. Hepatology. 42:1270–1279.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Goodwin B, Jones SA, Price RR, Watson MA,
McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME, et al:
A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1
represses bile acid biosynthesis. Mol Cell. 6:517–526. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Boyer JL, Trauner M, Mennone A, Soroka CJ,
Cai SY, Moustafa T, Zollner G, Lee JY and Ballatori N: Upregulation
of a basolateral FXR-dependent bile acid efflux transporter
OSTalpha-OSTbeta in cholestasis in humans and rodents. Am J Physiol
Gastrointest Liver Physiol. 290:G1124–G1130. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Frankenberg T, Rao A, Chen F, Haywood J,
Shneider BL and Dawson PA: Regulation of the mouse organic solute
transporter alpha-beta, Ostalpha-Ostbeta, by bile acids. Am J
Physiol Gastrointest Liver Physiol. 290:G912–G922. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Landrier JF, Eloranta JJ, Vavricka SR and
Kullak-Ublick GA: The nuclear receptor for bile acids, FXR,
transactivates human organic solute transporter-alpha and -beta
genes. Am J Physiol Gastrointest Liver Physiol. 290:G476–G485.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tom Lissauer and Will Carroll: Illustrated
textbook of paediatricsElsevier; Amsterdam: pp. 2572018
|
|
33
|
Ignjatovic V, Lai C, Summerhayes R,
Mathesius U, Tawfilis S, Perugini MA and Monagle P: Age-related
differences in plasma proteins: How plasma proteins change from
neonates to adults. PLoS One. 6:e172132011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Robert M, Kliegman, Behrman, Jenson and
Stanton: Nelson textbook of pediatricsSaunders; Philadelphia: pp.
16602007
|
|
35
|
Suchy FJ, Balistreri WF, Heubi JE, Searcy
JE and Levin RS: Physiologic cholestasis: Elevation of the primary
serum bile acid concentrations in normal infants. Gastroenterology.
80:1037–1041. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Balistreri WF, Suchy FJ, Farrell MK and
Heubi JE: Pathologic versus physiologic cholestasis: Elevated serum
concentration of a secondary bile acid in the presence of
hepatobiliary disease. J Pediatr. 98:399–402. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kawasaki H, Yamanishi Y, Miyake M, Mura T
and Ikawa S: Age- and sex-related profiles of serum primary and
total bile acids in infants, children and adults. Tohoku J Exp Med.
150:353–357. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sargiacomo C, El-Kehdy H, Pourcher G,
Stieger B, Najimi M and Sokal E: Age-dependent glycosylation of the
sodium taurocholate cotransporter polypeptide: From fetal to adult
human livers. Hepatol Commun. 2:693–702. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Colombo C, Zuliani G, Ronchi M,
Breidenstein J and Setchell KD: Biliary bile acid composition of
the human fetus in early gestation. Pediatr Res. 21:197–200. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Colombo C, Roda A, Roda E, Buscaglia M,
dell'Agnola CA, Filippetti P, Ronchi M and Sereni F: Correlation
between fetal and maternal serum bile acid concentrations. Pediatr
Res. 19:227–231. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Itoh S, Onishi S, Isobe K, Manabe M and
Inukai K: Foetomaternal relationships of serum bile acid pattern
estimated by high-pressure liquid chromatography. Biochem J.
204:141–145. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Monte MJ, Rodriguez-Bravo T, Macias RI,
Bravo P, el-Mir MY, Serrano MA, Lopez-Salva A and Marin JJ:
Relationship between bile acid transplacental gradients and
transport across the fetal-facing plasma membrane of the human
trophoblast. Pediatr Res. 38:156–163. 1995. View Article : Google Scholar : PubMed/NCBI
|