Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory
disease that causes synovial hyperplasia and progressive joint
degeneration (1). The
characteristic histopathological features of arthritis include
marked proliferation of synoviocytes, infiltration of large
mononuclear cells, extensive degradation of deep cartilage and
pannus formation (2). These
features are also observed in a rat model of adjuvant-induced
arthritis (AA) (3). Angiogenesis
consists of the formation of new blood vessels from pre-existing
vasculature; this process has a critical role in the pathogenesis
of RA. Angiogenesis promotes the infiltration of inflammatory cells
into the joints and stimulates the generation of a pannus that
covers and erodes articular cartilage, subchondral bone and
peri-articular soft tissues. This eventually leads to joint
destruction and the deformities observed in chronic arthritis
(4–6).
Chemokine C-X-C motif chemokine ligand 12 (CXCL12),
also known as stromal cell-derived factor 1, is released by various
immune cells, endothelial cells and stem cells, and is widely
expressed in the whole body under physiological conditions
(7,8). C-X-C-motif chemokine receptor 4
(CXCR4), which is the specific receptor for CXCL12, is expressed on
various cell types, including tumor cells. CXCR4 serves a crucial
role in organ-specific metastasis, where it can direct the
migration of CXCR4-expressing malignant cells towards
microenvironments where its ligand is secreted (9). CXCR4 is also a co-receptor for human
immunodeficiency virus type 1 (10), and is highly expressed in synovial
tissue and endothelial cells in RA (11). It has been reported that aberrant
expression of CXCR4 causes impaired neovascularization (12) and that CXCL12 is upregulated in the
synovial fluid in RA (13).
Furthermore, it has been reported that CXCL12 and CXCR4 levels in
the synovium are associated with clinical outcome, and with bone
and joint destruction, in patients with RA treated with golimumab
(14). The present study confirmed
that the CXCL12/CXCR4 pathway may serve a pathogenic role via
angiogenesis in RA.
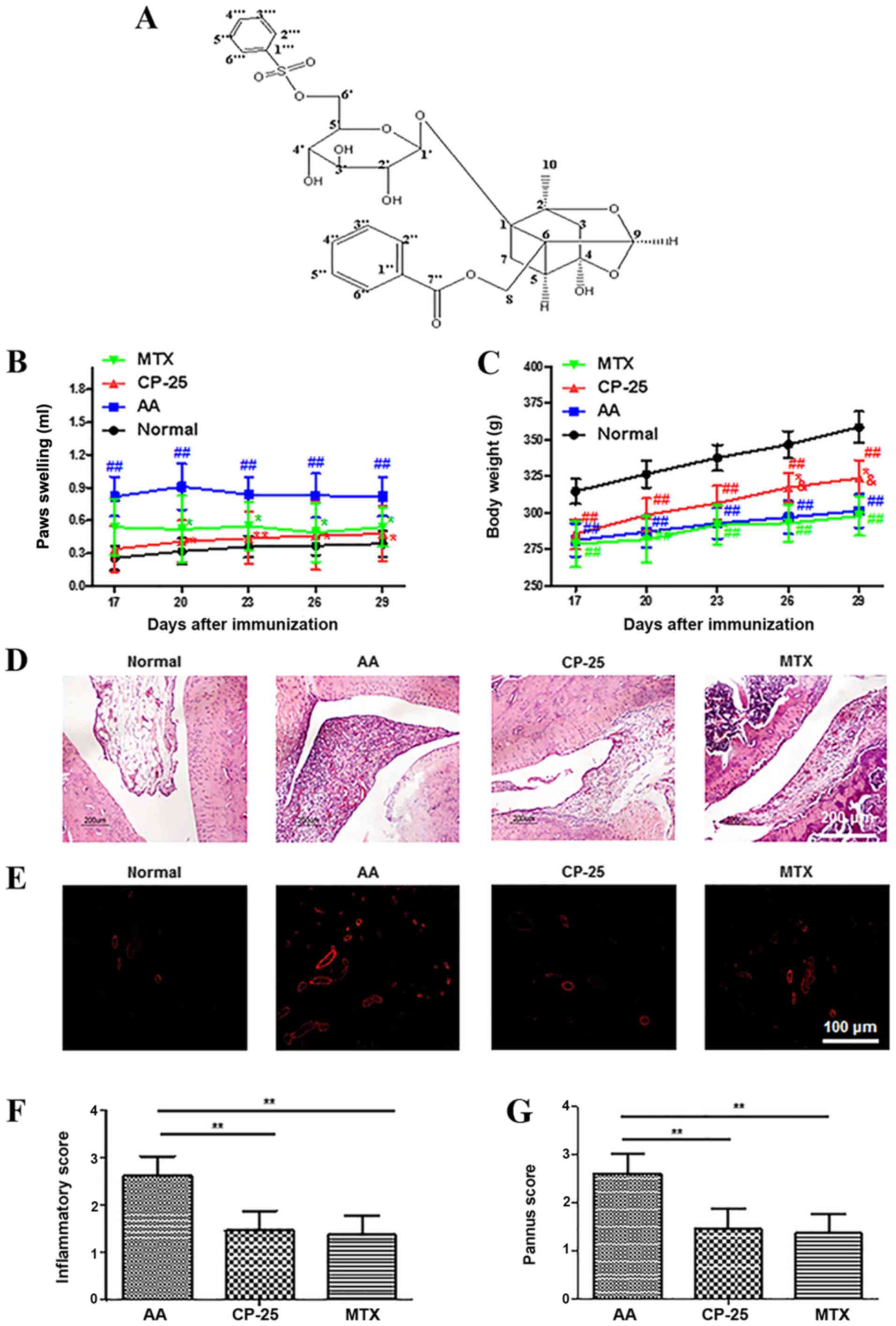
The novel compound paeoniflorin (Pae)-6′-O-benzene
sulfonate (CP-25; C29H32O13S;
molecular weight: 620 g/mol; patent no. in China: ZL20121003061
6.4; Fig. 1A) (15) is a new ester derivative of Pae. Pae
is the main effective compound in total glucosides of paeony, which
is extracted from the roots of Paeonia lactiflora Pall, and
is a traditional Chinese medicine that has anti-inflammatory and
immunoregulatory effects (16–23).
However, studies have revealed that Pae bioavailability is low
(3–4%); this phenomenon is mainly due to the poor absorption of
Pae, which is partially caused by modest permeation, efflux via
P-glycoprotein and hydrolytic degradation in the intestine
(24,25). Our previous study revealed that the
oral and venous pharmacokinetic parameters of CP-25 are better than
Pae (CP-25: t1/2=2.11 h, MRT=4.30 h, Vd=146.67 l/kg, CL/F=58.17
l/h•kg; Pae: t1/2=1.45 h, MRT=3.91 h, Vd=17.30 l/kg, CL/F=68.02
l/h•kg) (26). In addition, CP-25
has superior intestinal absorption compared with Pae (27). Consistent with another study
(28), CP-25 can attenuate the
clinical signs of AA by inhibiting the expression of
pro-inflammatory cytokines, including interleukin (IL)-1β, tumor
necrosis factor (TNF)-α, IL-6 and IL-17, and promoting the
production of the anti-inflammatory cytokine TNF-β1. In addition, a
previous in vitro study reported that CP-25 attenuates the
inflammatory response of fibroblast-like synoviocytes co-cultured
with B cell activating factor-activated CD4+ T cells
(29). However, the
anti-angiogenic effects of CP-25 and its underlying mechanisms
remain unclear.
G protein-coupled receptor kinase 2 (GRK2) is a
member of the serine/threonine protein kinases family, which has
emerged as a pivotal integrative scaffold in endothelial and
fibroblast cells (30,31). A previous study revealed the
dysfunction and overexpression of GRK2 in patients with RA and
animal models of RA, which suggests that GRK2 may be considered a
potential target in the treatment of RA (32). The present study hypothesized that
CP-25 may exert anti-angiogenic effects on AA and could suppress
the functions of human umbilical vein endothelial cells (HUVECs)
in vitro through regulation of GRK2 and the CXCL12/CXCR4
pathway. The expression levels of CXCL12 and CXCR4 were therefore
examined in the synovium of rats with AA and the effects of CP-25
on HUVEC function was investigated in vitro. To the best of
our knowledge, the present study is the first to demonstrate the
potential therapeutic effect of CP-25 on AA and to determine the
mechanism underlying its anti-angiogenic effect. The findings from
this study may provide a scientific basis for the development of
novel CP-25-containing drugs in the treatment of autoimmune
diseases.
Materials and methods
Animals
A total of 45 Sprague-Dawley rats (male; age, 6–8
weeks; weight, 180–200 g) were purchased from the Experimentation
Animal Center of Anhui Medical University. All rats were maintained
in a specific pathogen-free animal laboratory (20–25°C; relative
humidity, 50–60%; 12-h light/dark cycle; free access to food and
water) at the Experimentation Animal Center of Anhui Medical
University.
Reagents
RPMI-1640 medium and fetal bovine serum (FBS) were
purchased from HyClone; GE Healthcare Life Sciences. HUVECs were
purchased from All Cells BioTech Co., Ltd. Cell Counting kit-8
(CCK-8) was purchased from Dojindo Molecular Technologies, Inc. An
ELISA kit for CXCL12 (cat. no. CSB-E08729r) was purchased from
Cusabio, Inc. Antibodies against α-smooth muscle actin (α-SMA)
(cat. no. ab32575), CXCR4 (cat. no. ab124824), GRK2 (cat. no.
ab228705), ERK1/2 (cat. no. ab79853), phosphorylated (p)-ERK1/2
(cat. no. ab214362), CXCL12 (cat. no. ab9797), and β-actin (cat.
no. ab115777) were purchased from Abcam. The Na K-ATPase antibody
(cat. no. 3010S) was purchased from Cell Signaling Technology,
Inc.
Drugs
Methotrexate (MTX; 2.5 mg per tablet) was purchased
from Shanghai Xinyi Pharmaceutical Co., Ltd. CP-25 was provided by
the Chemistry Lab of the Institute of Clinical Pharmacology of
Anhui Medical University.
Arthritis induction and
treatments
To establish a rat model of AA, 100 µl (1 mg/rat)
heat-killed Mycobacterium butyricum (provided by Beijing
Biological Products Research Institute Co., Ltd.) in liquid
paraffin was intradermally injected into the right hind metatarsal
footpad, as previously described (28,33).
Rats were randomly divided into four groups (n=10/group) as
follows: i) Normal (no immunization); ii) AA model; iii) CP-25, AA
rats treated with CP-25 (50 mg/kg); and iv) MTX, AA rats treated
with MTX (0.5 mg/kg).
Our previous study demonstrated that 50 and 100
mg/kg exerted therapeutic effects on AA rats; therefore, 50 mg/kg
CP-25 was used in the present study (28). Although novel biological agents can
ameliorate inflammatory reactions and protect the joints of
patients with rheumatoid disease from progressive damage, MTX
remains one of the most effective and widely used clinical
disease-modifying anti-rheumatic drugs. Previous studies have used
MTX as a positive control in animal models including rats with AA
and mice with collagen-induced arthritis (CIA). MTX was therefore
used in the present study as a positive control drug, and the
dosage of 0.5 mg/kg was determined according to previous studies
(28,29,34).
The day of the first immunization was defined as day
0. AA joint inflammation in treated rats reached a maximum at days
13–16 following administration of the adjuvant. After the onset of
arthritis on day 16, as determined based on paw swelling of the
rats, rats were administered CP-25 (once per day) for 14 days and
MTX (once every 3 days, five times) by intragastric administration.
Normal rats and rats with AA received an equal volume of 0.9%
saline.
A blinded single observer who had no knowledge of
the treatment groups evaluated AA severity. From day 7 following
immunization, rats were examined every 3 days for paw volume and
body weight. Footpad volume was measured using a water
plethysmometer.
Histological examination
Rats were anesthetized and sacrificed on day 30
after immunization. The left ankle joints were removed, fixed in
10% neutral-buffered formalin for 2 weeks at 25°C, decalcified in
10% ethylenediaminetetracetic acid for 3 months at 25°C and
embedded in paraffin. The sections (5 µm) were stained with 0.2%
hematoxylin for 10 min and 0.5% eosin for 10 sec at 25°C and were
examined under a fluorescence microscope (80I; Nikon Corporation).
Ankle joints were histopathologically analyzed for inflammation,
synovial proliferation, cellular infiltration, pannus formation and
cartilage erosion by two blinded observers.
The expression of CXCL12 was detected by
immunohistochemical staining and was examined by a fluorescence
microscope. The 5-µm paraffin-embedded joint sections were
processed using a standard immunostaining protocol. Initially,
sections were deparaffinized in 100% xylene for 15 min and hydrated
in a descending series of alchol at room temperature. Subsequently,
sections were heated at 100°C for 15 min in 0.01 mol/l citric acid
buffer (pH 6.0) in a microwave oven for antigen retrieval and were
then treated with 3% H2O2 for 30 min at 37°C.
Sections were incubated with primary antibodies against CXCL12
(1:100) overnight at 4°C. Sections were then washed in PBS and
incubated with horseradish peroxidase (HRP)-conjugated secondary
antibodies (50 µl; cat. no. PV-9000; OriGene Technologies, Inc.) at
37°C for 30 min. Subsequently, immunostaining was observed using
3,3-diaminobenzidine (DAB) (1:20; cat. no. ZLI-9017; OriGene
Technologies, Inc.) for 2 min and counterstaining was performed
using 0.2% hematoxylin for 3 min at 25°C.
Vessel formation was observed via anti-α-SMA
immunofluorescence to determine the extent of pannus formation.
Following deparaffinization and hydration, the joint sections were
heated at 100°C for 15 min in a 0.01 mol/l citric acid buffer (pH
6.0) in a microwave oven and blocked with goat serum (cat. no.
ZLI-9022; OriGene Technologies, Inc.) at 37°C for 30 min. Sections
were incubated with rabbit anti-rat primary antibody against α-SMA
(1:100; cat. no. ab32575; Abcam) overnight at 4°C, and incubated
with TRITC-conjugated goat anti-rabbit IgG secondary antibody,
(1:100; cat. no. BA1090; Wuhan Boster Biological Technology, Ltd.)
at 37°C for 1 h in the dark. Tissue sections were washed three
times with PBS after each step.
Pathological evaluations were performed randomly by
a blinded pathologist who had no knowledge of the treatment groups.
The optical density values of CXCL12 protein staining in the
synovium and synovial vessel walls were acquired from five
different fields of view at high power magnification (×400). All
results were transformed to data using Image ProPlus 6.0 software
(Media Cybernetics, Inc.). Inflammatory scoring of synovial tissue
was assessed according to the following criteria: i) 0, normal; ii)
1, minimal inflammatory infiltration; iii) 2, moderate infiltration
with moderate edema; and iv) 3, marked infiltration with marked
edema. Pannus scores were defined as synovial tissue intimately
invading bone and/or cartilage and was scored from 0 to 3 as
follows: i) 0, none; ii) 1, minimal; iii) 2, moderate; and iv) 3,
severe.
CXCL12 detection
Synovial tissue (0.15 g) was collected, mixed with
100 µl PBS, fully ground and centrifuged at 10,956 × g for 15 min.
The supernatant was collected and reserved at 37°C. CXCL12
concentrations were subsequently measured in the synovium using
ELISA kit according to the manufacturer's protocol.
HUVEC proliferation, Transwell
migration and tube formation assays
HUVECs were cultured in RPMI-1640 containing 20%
FBS, 100 µg/ml streptomycin and 100 U/ml penicillin at 37°C in a
humidified incubator containing 5% CO2. Cells were
harvested and collected in the logarithmic phase, and were seeded
into 96-well plates at a density of 1×105 cells/well and
in triplicate for each group. Cells were cultured with fresh medium
containing CXCL12 (cat. no. ab9798; 50 ng/ml; Abcam) alone or in
combination with CP-25 at the specified concentrations
(1×10−9, 1×10−8, 1×10−7,
1×10−6 and 1×10−5 mol/l). After treatment at
37°C for 48 h, 10 µl CCK-8 reagent was added to each well, and the
plates were incubated at 37°C for 1 h. Absorbance was read using an
Infinite M1000 PRO microplate reader at 450 nm (Tecan Group,
Ltd.).
A cell migration assay was performed using Transwell
culture chambers (24 wells; 8-µm pore size; Costar; Corning Inc.).
Cells were seeded at 1×105 cells/well in 100 µl serum-free
Dulbecco's modified Eagle's medium (DMEM; cat. no. PYG0074; Wuhan
Boster Biological Technology, Ltd.) containing CXCL12 alone or in
combination with CP-25 at the specified concentrations. After
incubation at 37°C for 24 h, cells that migrated to the bottom
chamber were stained with 0.1% crystal violet at 37°C for 15 min.
After air-drying, migrated cells in each chamber were counted under
a light microscope (magnification, ×200; BX53; Olympus
Corporation). These experiments were performed independently three
times.
Prior to the tube formation assay, HUVECs were
treated with CXCL12 with or without CP-25 for 24 h. Cells were then
washed with PBS, harvested and seeded in 96-well plates coated with
Matrigel (BD Bioscience) at a density of 2×104
cells/well in DMEM containing 20% FBS for 6 h at 37°C. Images of
tube formation were captured using a 1X71 Olympus microscope
(Olympus Corporation), and branch point quantifications were
assessed using Image Pro Plus 6.0 software (Media Cybernetics,
Inc.).
Western blotting
Synovium was homogenized in ice-cold RIPA lysis
buffer (cat. no. P0013C; Beyotime Institute of Biotechnology) with
a homogenizer. Samples were centrifuged at 10,956 × g for 20 min at
4°C and supernatants were collected. Furthermore, proteins were
extracted from HUVECs using RIPA lysis buffer. Protein
concentration was determined using the Bradford assay. Equal
amounts of protein samples were mixed with 5X sample buffer (4:1)
(Bio-Rad Laboratories, Inc.) and heated in boiling water for 10
min. Proteins (20 µl; 10 µg/µl) were then separated by 12% SDS-PAGE
and transferred onto polyvinylidene fluoride membranes (EMD
Millipore). Membranes were blocked for 1 h at room temperature in
5% skimmed milk and incubated overnight at 4°C with primary
antibodies against CXCR4 (1:1,000; cat. no. ab124824; Abcam) and
GRK2 (1:1000; cat. no. ab228705; Abcam). Membranes were washed with
Tris-buffered saline containing 0.05% Tween-20 (TBST) three times
for 10 min and were then incubated with HRP-conjugated secondary
antibodies (1:10,000; cat. no. ab205718; Abcam). for 2 h at room
temperature. Membranes were washed three times with TBST and bands
were detected using enhanced chemiluminescence substrate (Pierce;
Thermo Fisher Scientific, Inc.). Autoradiographs were scanned using
an ImageQuant LAS 4000 mini-system (GE Healthcare Life Sciences).
Relative expression levels of the specific bands were quantified
using ImageJ software 1.8.0 (National Institutes of Health).
In addition, HUVECs were treated with CXCL12 for 30
min and with CP-25 at the specified concentrations for 24 h. Cells
were then lysed in RIPA lysis buffer and the total proteins were
collected following centrifugation at 10,956 × g for 10 min at 4°C.
Membrane and cytoplasmic proteins were separated from total
proteins using a cell membrane protein and cytoplasmic protein
extraction kit (cat. no. P0033; Beyotime Institute of
Biotechnology) by ultracentrifugation at 559,000 × g for 60 min at
4°C. The protein fractions then underwent western blotting as
aforementioned. The primary antibodies used included ant-CXCR4,
anti-GRK2, anti-ERK1/2 and anti-p-ERK1/2 (1:1,000). The secondary
antibody used was a HRP-conjugated secondary antibody (1:10,000;
cat. no. ab205718; Abcam).
Co-immunoprecipitation
The co-expression of GRK2 with CXCR4 and p-ERK1/2
was analyzed by co-immunoprecipitation. HUVECs were divided into
three groups as follows: i) Control group (untreated HUVECs); ii)
CXCL12 group (HUVECs treated with 100 ng/ml CXCL12 for 30 min at
37°C); and iii) CP-25 group (HUVECs treated with 100 ng/ml CXCL12
for 30 min and with 1×10−6 mol/l CP-25 for 24 h at
37°C). Subsequently, cells were lysed in RIPA lysis buffer and
total proteins were collected following centrifugation at 10,956 ×
g for 10 min at 4°C. A supernatant (20 µl) was taken as the input
fraction and the remainder was frozen at −80°C prior to use. The
GRK2 protein in the control group served as control for
co-immunoprecipitation. Briefly, 10 µl antibodies (anti-CXCR4,
anti-GRK2, anti-p-ERK; 1:1,000) were conjugated to 50 µl protein G
agarose (cat. no. P2053; Beyotime Institute of Biotechnology) and
diluted with RIPA lysis buffer to a volume of 500 µl. The mixture
was shaken slowly for 2 h at 4°C, and was incubated overnight at
4°C following centrifugation at 56 × g for 5 min at 4°C.
Subsequently, the sample was centrifuged at 56 × g for 2 min at
4°C. The supernatant was discarded and the pellet at the bottom of
the tube was centrifuged again with 500 µl RIPA lysis buffer at 56
× g for 2 min at 4°C. Proteins were eventually evaluated by western
blotting as aforementioned. The expression of GRK2 was used as a
loading control and the relative expression levels of the specific
bands were semi-quantified using ImageJ software 1.8.0 (National
Institutes of Health).
Statistical analysis
Data are expressed as the means ± standard
deviation. Each experiment was repeated at least three times
independently. Statistical analysis was performed using SPSS 16.0
for Windows (SPSS, Inc.). Statistical differences among groups were
analyzed by one-way analysis of variance and pairwise comparisons
were performed using the Bonferroni test. Spearman correlation was
used for correlation analysis of two variables. P<0.05 was
considered to indicate a statistically significant difference.
Results
CP-25 attenuates the clinical signs
and pannus formation in the ankle joint of rats with AA
The effect of CP-25 was evaluated
using a murine AA model
Arthritis developed rapidly in rats following a
single injection of Mycobacterium butyricum. By day 17–29
following immunization, paw swelling in the AA group had increased
compared with that in the normal group. By day 20–29 following
immunization, paw swelling was significantly reduced in the MTX and
CP-25 groups compared with that in the AA group (Fig. 1B). CP-25 and MTX treatments
therefore resulted in a significant decrease in paw volume. By day
17–29 following immunization, body weight in the AA, CP-25 and MTX
groups was lower than that in the normal group (AA group,
12.5±2.5%; CP-25 group, 8.8±1.0%; MTX group, 12.1±1.8%; average of
five time points). By day 26–29 following immunization, body weight
in the CP-25 group was increased compared with in the AA group,
whereas MTX caused a substantial reduction in body weight (Fig. 1C).
The typical characteristics of RA include synovial
hyperplasia and progressive joint destruction. Rats with AA
developed severe arthritis, which was characterized by marked
synovial proliferation, pannus formation and infiltration of
inflammatory cells. Histopathological examination of the ankle
joints revealed that there were marked differences in cell
proliferation and inflammatory infiltration at the synovial margin
among the four groups, and the inflammatory scores of synovial
tissue in the CP-25 or MTX groups were significantly lower than in
the AA group (Fig. 1D and F).
Compared with in the AA group, the number of blood vessels and
pannus score of synovial tissue was significantly reduced in rats
with AA following administration of CP-25 and MTX (Fig. 1E and G).
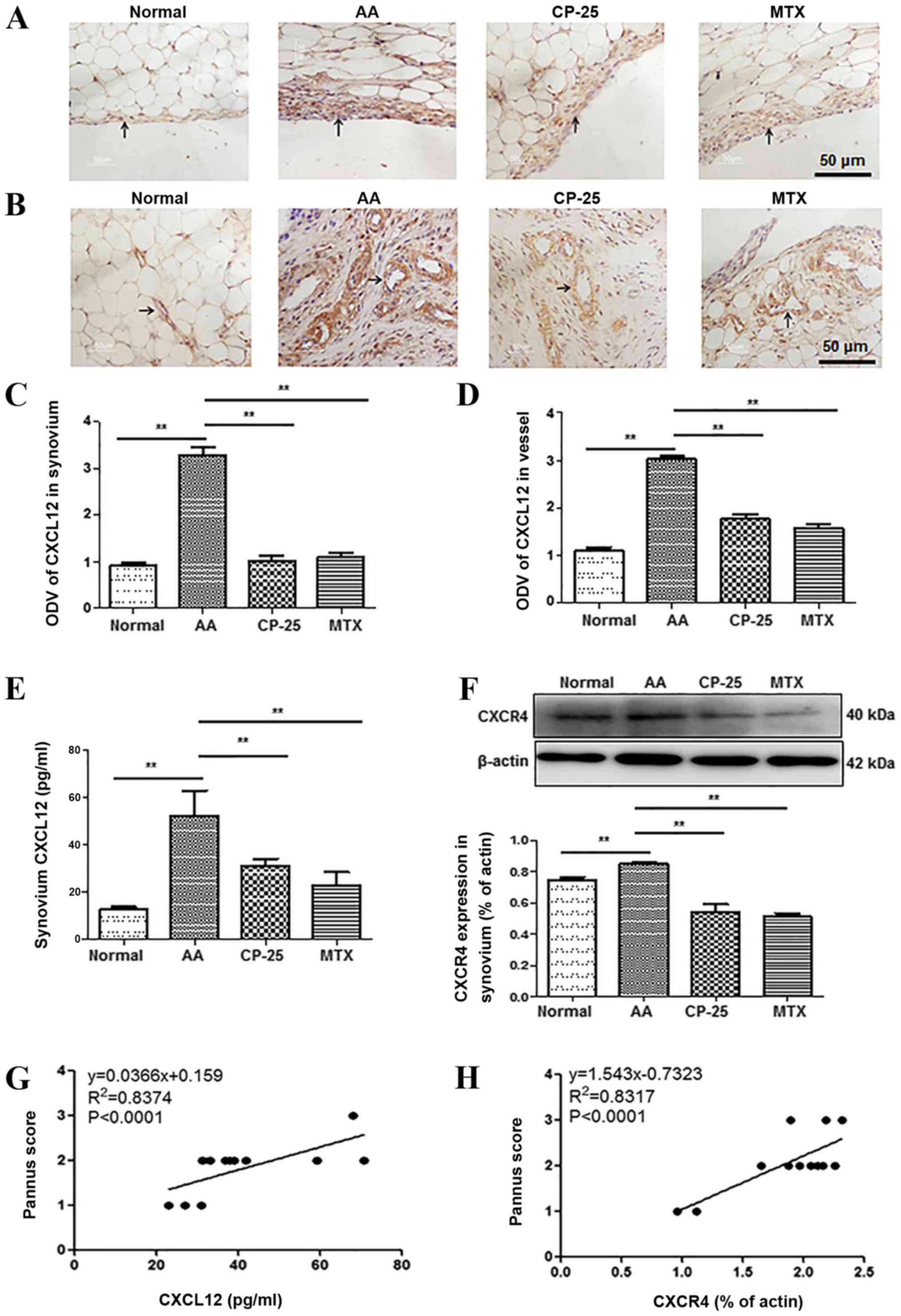
CP-25 alters the expression of
CXCL12/CXCR4 in the synovium of rats with AA
Immunohistochemistry was performed to detect the
expression of CXCL12 in synovium and in the wall of blood vessels
in the synovium. The results demonstrated that the expression of
CXCL12 in the AA group was increased compared with in rats in the
normal group. Compared with in the AA group, CXCL12 was markedly
decreased in rats treated with CP-25 (Fig. 2A and B). Furthermore, the optical
density value of CXCL12 was consistent with the immunohistochemical
results (Fig. 2C and D). As
presented in Fig. 2, CXCL12 and
CXCR4 expression was increased in synovial tissues from the AA
group compared with in those from the normal group. Compared with
in the AA group, CXCL12 and CXCR4 expression was significantly
decreased in CP-25 and MTX-treated rats (Fig. 2E and F). In addition, there was a
positive correlation between pannus score and CXCL12 and CXCR4
expression (Fig. 2G and H). These
results indicated that CP-25 inhibited pannus formation in the
synovium of rats with AA, which may be associated with
downregulation of CXCL12/CXCR4 expression in the synovium.
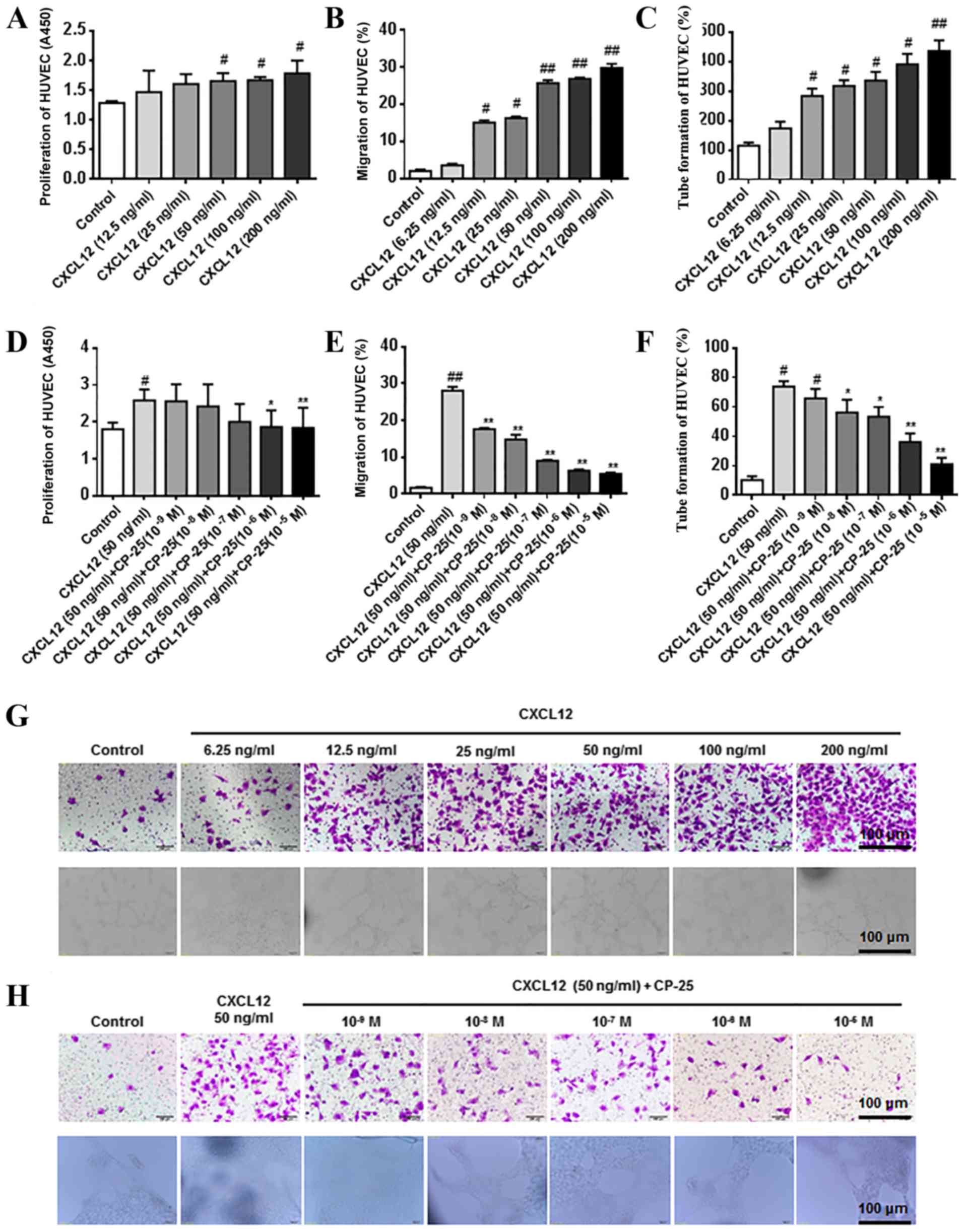
Effects of CP-25 on proliferation,
migration and tube formation of HUVECs treated with CXCL12
CCK-8, Transwell and tube formation assays were used
to examine HUVEC proliferation, migration and tube formation,
respectively. Cells were incubated with various concentrations of
CXCL12 for 24 h. The results demonstrated that treatment with high
concentrations of CXCL12 (50, 100 and 200 ng/ml) significantly
promoted HUVEC proliferation (Fig.
3A). In addition, 12.5, 25, 50, 100 and 200 ng/ml CXCL12
significantly promoted the migration and tube formation of HUVECs
(Fig. 3B, C and G). HUVECs were
then treated with 50 ng/ml CXCL12 alone or in combination with
increasing concentrations of CP-25 (1×10−9,
1×10−8, 1×10−7, 1×10−6 and
1×10−5 mol/l) for 24 h. The results revealed that CP-25
significantly inhibited CXCL12-induced cell proliferation (at
1×10−6 and 1×10−5 mol/l), migration (at
1×10−9, 1×10−8, 1×10−7, 1×10-6 and
1×10−5 mol/l) and tube formation (at 1×10−8,
1×10−7, 1×10−6 and 1×10−5 mol/l)
(Fig. 3D-F and H).
Effects of CP-25 on GRK2 and CXCR4
expression in HUVECs treated with CXCL12
Western blotting was performed to measure the
expression of GRK2 and CXCR4 in HUVECs treated with 100 ng/ml
CXCL12. The results demonstrated that GRK2 expression in cells
treated with CXCL12 was significantly upregulated compared with
that in the control group. Compared with in CXCL12-treated cells,
GRK2 expression in cells treated with CP-25 (1×10−7,
1×10−6 and 1×10−5 mol/l) and CXCL12 was
significantly downregulated. Notably, CXCR4 expression was not
significantly altered by CXCL12 or CP-25 treatments (Fig. 4A-C). Furthermore, the cytoplasmic
expression of GRK2 in CXCL12-treated cells was significantly
downregulated compared with that in the control group. Compared
with in the CXCL12-treated group, the cytoplasmic expression of
GRK2 was significantly upregulated in cells treated with CP-25
(1×10−6 and 1×10−5 mol/l). Conversely, CXCR4
expression was significantly downregulated in the cytoplasm in
response to CP-25 (1×10−6 and 1×10−5 mol/l)
(Fig. 4D-F). The membrane
expression of GRK2 in cells treated with CXCL12 was significantly
upregulated compared with that in the control cells. Compared with
CXCL12 treatment, the membrane expression of GRK2 in cells treated
with CP-25 (1×10−6 and 1×10−5 mol/l) was
significantly reduced. The expression of CXCR4 was significantly
upregulated in cells treated with CP-25 (1×10−5 mol/l)
(Fig. 4G-I).
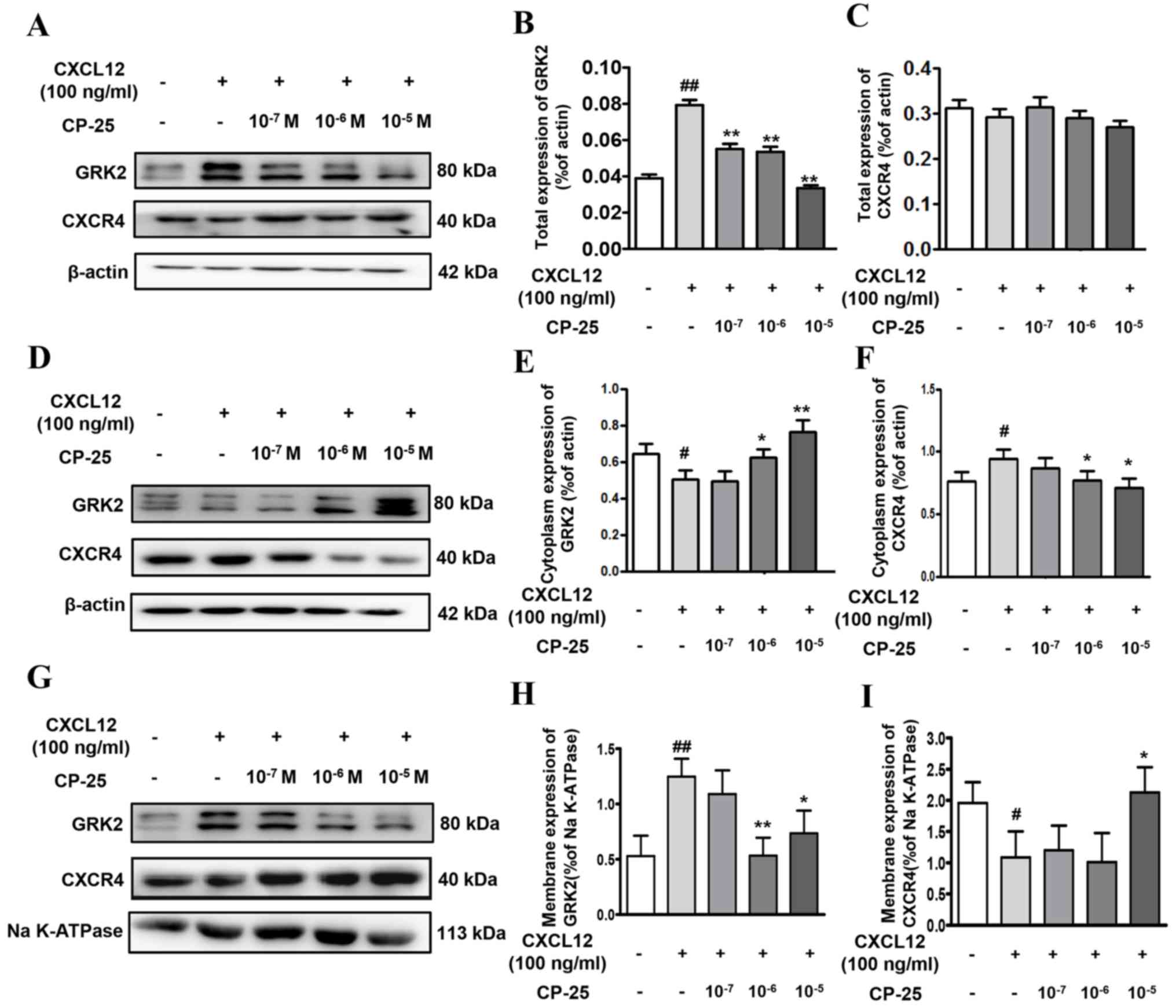 | Figure 4.Effects of CP-25 on GRK2 and CXCR4
expression in HUVECs treated with CXCL12. Representative images of
western blotting of (A) total, (D) cytoplasmic and (G) membrane
expression of GRK2 and CXCR4. (B, C, E, F, H and I) Western
blotting semi-quantification of GRK2 and CXCR4 expression. Data are
expressed as the means ± standard deviation of three independent
experiments. #P<0.05, ##P<0.01 vs.
control; *P<0.05, **P<0.01 vs. CXCL12. CP-25,
paeoniflorin-6′-O-benzene sulfonate; CXCL12, C-X-C motif chemokine
ligand 12; CXCR4, C-X-C chemokine receptor type 4; GRK2; G
protein-coupled receptor kinase 2. |
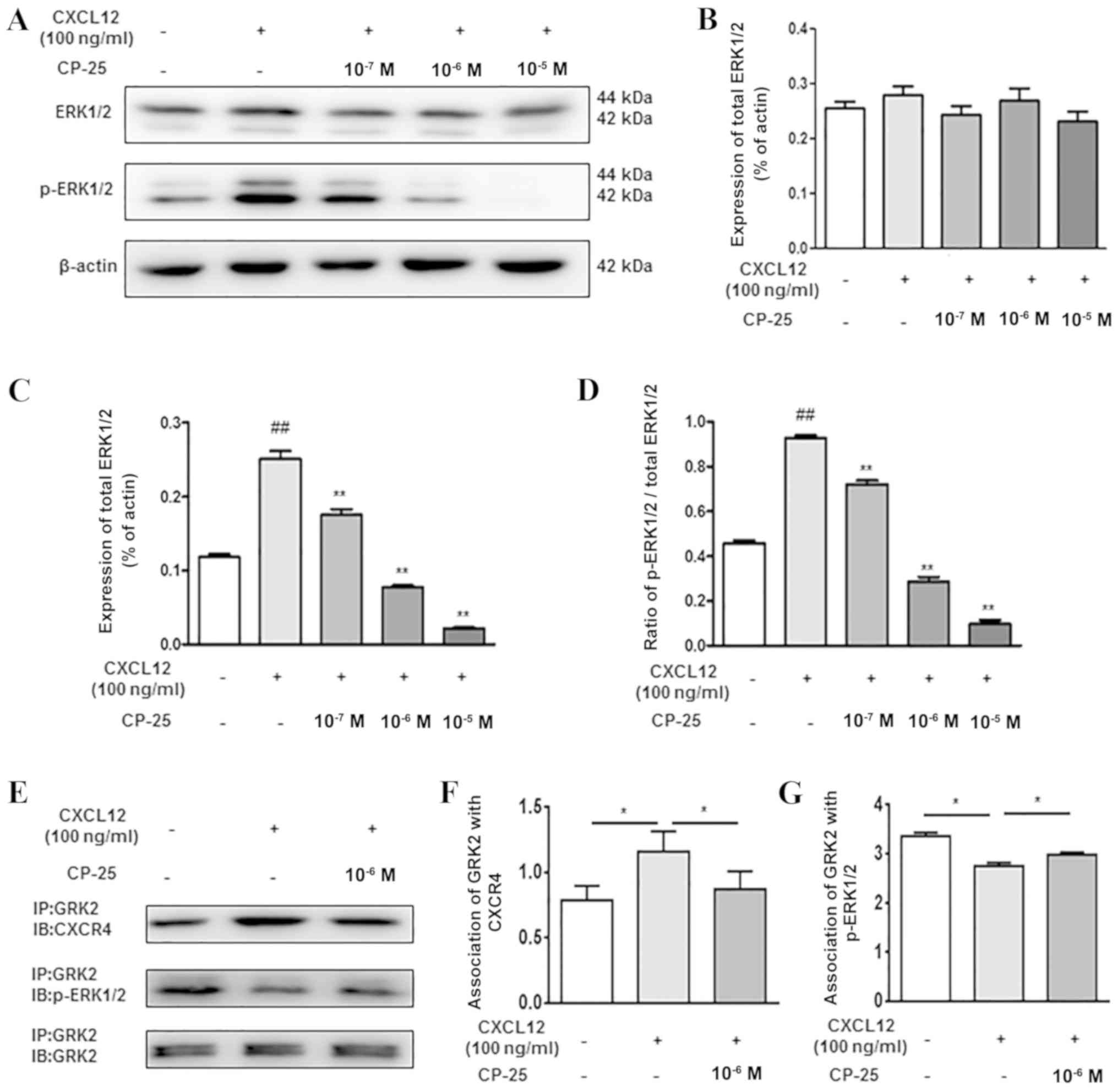
Effects of CP-25 on ERK1/2 expression
in HUVECs treated with CXCL12
As presented in Fig.
5, there was no difference in ERK1/2 expression among the
treatment groups. However, the expression of p-ERK1/2 and the ratio
of p-ERK/ERK in CXCL12-treated cells were significantly upregulated
compared with those in the control group. Compared with in the
CXCL12 treatment group, the expression of p-ERK1/2 and the ratio of
p-ERK/ERK in CP-25-treated cells (1×10−7,
1×10−6 and 1×10−5 mol/l) was significantly
downregulated (Fig. 5A-D).
Co-immunoprecipitation and western blotting was also performed to
investigate the interaction between GRK2 and CXCR4, and between
GRK2 and p-ERK1/2 in HUVECs treated with CXCL12. Following CXCL12
(100 ng/ml) cell stimulation for 30 min and treatment with CP-25
(1×10−6 mol/l) for 24 h, the results demonstrated that
CXCL12 increased the interaction between GRK2 and CXCR4, and
downregulated the interaction between GRK2 and p-ERK1/2.
Conversely, CP-25 could downregulate the interaction between GRK2
and CXCR4, and upregulate the interaction between GRK2 and p-ERK1/2
(Fig. 5E-G).
Discussion
The present study investigated the anti-angiogenic
activity of CP-25, which is a novel ester derivative of Pae. The
results from this study suggested that treatment with CP-25 may
inhibit the biological function of endothelial cells that
contribute to the progression of autoimmune arthritis by reducing
synovial angiogenesis.
The rat model of AA has been used in numerous
studies to elucidate the pathogenesis of RA and to determine
potential therapeutic targets. The arthritic etiology of AA and RA
exhibits similar pathological and immunological features, including
immune dysfunction, synoviocyte proliferation and pannus formation
(3,32). In RA, pannus formation corresponds
to an invasive and destructive front of inflammatory vascular
tissue that covers and erodes articular cartilage, subchondral bone
and peri-articular soft tissues. Therapeutic options aiming to
suppress angiogenesis may therefore represent a useful strategy for
preventing RA progression. CP-25 has a clear therapeutic effect in
AA, and our previous studies demonstrated that it inhibits synovial
and immune cell functions (26,35).
However, the anti-angiogenic effects and underlying mechanism of
CP-25 remain unclear. In the present study, CP-25 treatment
significantly attenuated the clinical features of AA, synovial
proliferation, and inflammatory and pannus scores in vivo.
Furthermore, there was a positive correlation between pannus score
and the expression of CXCL12 and CXCR4 in the synovium.
Endothelial cells serve a crucial role in
angiogenesis. The effect of CP-25 on endothelial cell function and
CXCL12/CXCR4 signaling was therefore examined using HUVECs.
Synovial angiogenesis and pannus formation may represent specific
features that make RA difficult to cure. Circulating pro-angiogenic
factors are commonly increased in patients with RA, and the joint
microenvironment is frequently characterized by low oxygen levels
and numerous inflammatory factors and active pro-angiogenic
molecules (36). A previous study
reported that CXCL12 levels in the synovium of patients with RA
treated with golimumab are correlated with disease activity, and
that CXCR4 is associated with joint degeneration (14). Notably, small molecules acting as
CXCR4 antagonists have exhibited promising outcomes in animal
models of arthritis (37); for
example, AMD3100, which is a CXCR4 antagonist, inhibits CIA in
interferon-γ-deficient mice (38).
In the present study, HUVECs were used to explore
the effects and underlying mechanisms of CP-25 on angiogenesis.
Currently, CXCL12 is considered to have significant effects on
promoting angiogenesis in RA and certain types of cancer (39,40).
As demonstrated in the present study, high levels of CXCL12 (50,
100 and 200 ng/ml) significantly promoted the proliferation,
migration and tube formation of HUVECs. Furthermore, CP-25
significantly inhibited CXCL12-induced cell proliferation,
migration and tube formation, in a dose-dependent manner, which
suggested that CXCL12 may have a crucial role in HUVEC functions,
and that CP-25 may inhibit these functions.
GRK2, a member of the serine/threonine protein
kinases family, can specifically recognize and phosphorylate
agonist-activated G protein-coupled receptors (GPCRs), and is
emerging as a key integrative node in various signaling pathways
(41). The expression and activity
of GRK2 may be important in the pathophysiology of RA (20). A previous study reported that GRK2
exerts some effects on cell chemotaxis due to its ability to
trigger desensitization or internalization of specific chemokine
receptors (42). Our previous
study also demonstrated that paroxetine, which is a GRK2
antagonist, can attenuate the symptoms of rats with CIA (43). GRK2, a labile protein ordinarily
degraded via the ubiquitin/proteasome pathway, can be
phosphorylated by numerous kinases, including ERK1/2 and MAPK
(43,44). CXCL12 binds to CXCR4 to activate
downstream signaling pathways, including phosphoinositide
3-kinase-protein kinase B, phospholipase C-protein kinase C and
Raf-MAPK kinase-ERK1/2, which increase cell chemotaxis and
proliferation ability (45).
Arrestin proteins can bind and regulate GPCR cell-surface
expression, often functioning together with GRK2. Furthermore, GRK2
overexpression enhances CXCR4 internalization and inhibits the
function of CXCL12 (46). In
addition, ERK1/2 activation is reduced by GRK2 in the cytoplasm,
which may inhibit the proliferation of endothelial cells induced by
CXCL12 (47). In the present
study, the interaction between CXCR4 and GRK2 in HUVECs treated
with CXCL12 and CP-25 was examined in order to investigate the
mechanisms underlying the effects of CP-25 on HUVECs. The results
demonstrated that CXCL12 significantly promoted the proliferation,
migration and tube formation of HUVECs. In addition, CXCL12 (100
ng/ml) upregulated the expression of total GRK2, membrane GRK2,
cytoplasmic CXCR4 and p-ERK1/2, downregulated the expression of
cytoplasmic GRK2 and membrane CXCR4, but had no effect on the
expression of total CXCR4 and ERK1/2. Furthermore, CXCL12 (100
ng/ml) upregulated the interaction between GRK2 and CXCR4, and
downregulated GRK2 and p-ERK1/2 interaction. Following CP-25
administration, this trend induced by CXCL12 was reversed, which
indicated that CXCL12 could improve HUVECs function by increasing
the membrane localization of GRK2, weakening the inhibitory effect
of GRK2 on ERK1/2 in the cytoplasm, and stimulating ERK1/2
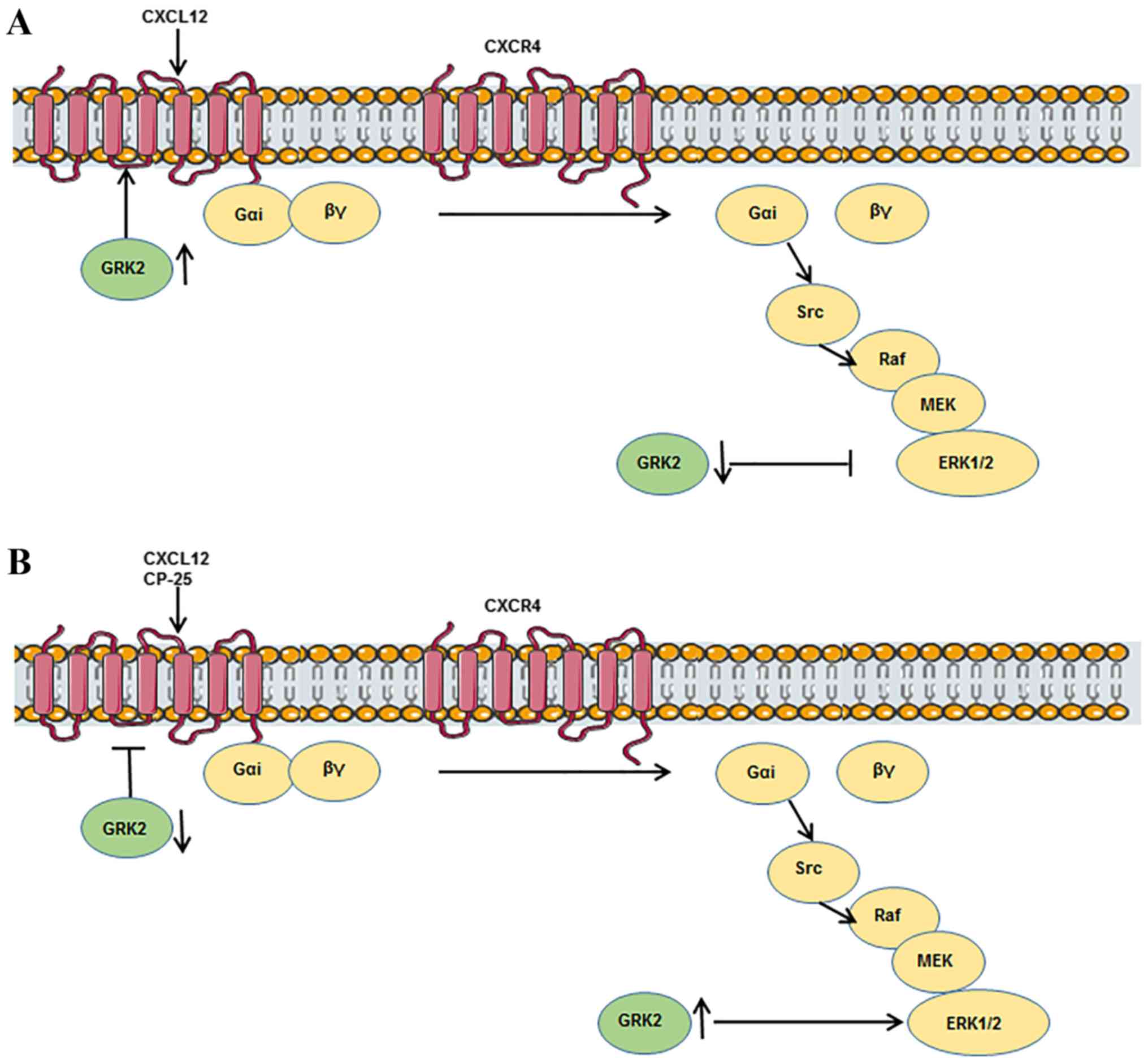
phosphorylation (Fig. 6A). In
addition, CP-25 regulated HUVECs function by reducing the membrane
localization of GRK2, enhancing the inhibitory effect of GRK2 on
ERK1/2 in the cytoplasm, and reducing ERK1/2 activation to alter
HUVECs function (Fig. 6B).
Unfortunately, this study did not analyze the β-arrestins, which
may affect the internalization of CXCR4 (48). This was one limitation in the
present study; however, our group is currently studying the
translocation of β-arrestins and their role in regulating CXCR4
function.
In conclusion, CP-25 markedly inhibited pannus
formation in a rat model of AA. The results suggested that
upregulation of CXCL12 and its receptor CXCR4 may be associated
with synovial inflammatory lesions and angiogenesis in AA rats.
Furthermore, the therapeutic effects of CP-25 on AA may be related
to inhibition of angiogenesis. CP-25 inhibited GRK2 transfer from
the cytoplasm to the membrane, thus restoring cytoplasmic GRK2
expression, which may inhibit the activation of ERK; this is the
potential mechanism underlying the inhibitory effects of CP-25 on
proliferation, migration and tube-forming ability of HUVECs. The
findings from the present study provided a potential novel
mechanism for the anti-angiogenic activity of CP-25 in the
treatment of RA. However, in order to increase our understanding of
the anti-inflammatory and immunomodulatory effects of CP-25 in RA,
further investigation is required at clinical and molecular
levels.
Acknowledgements
The authors would like to thank Dr Li Gui and Dr
Dake Huang (Synthetic Laboratory of Anhui Medical University) for
their technical assistance.
Funding
This study was supported by the National Natural
Science Foundation of China (grant nos. 81330081 and 81503084).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
MZ, LS and WW conceived the original idea and
planned the experiments. MZ, MG and JC participated in the
experiments. All authors analyzed and interpreted the data. MZ
wrote the manuscript with the support of LS and WW. All authors
approved the final version of the manuscript.
Ethics approval and consent to
participate
All experiments were approved by the Ethics Review
Committee for Animal Experimentation of the Institute of Clinical
Pharmacology, Anhui Medical University. All experiments were
conducted according to the animal care and use committee
guidelines.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bottini N and Firestein GS: Duality of
fibroblast-like synoviocytes in RA: Passive responders and
imprinted aggressors. Nat Rev Rheumatol. 9:24–33. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
McInnes IB and Schett G: The pathogenesis
of rheumatoid arthritis. N Engl J Med. 365:2205–2219. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Choudhary N, Bhatt LK and Prabhavalkar KS:
Experimental animal models for rheumatoid arthritis.
Immunopharmacol Immunotoxicol. 40:193–200. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Elshabrawy HA, Chen Z, Volin MV, Ravella
S, Virupannavar S and Shahrara S: The pathogenic role of
angiogenesis in rheumatoid arthritis. Angiogenesis. 18:433–448.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Szekanecz Z and Koch AE: Vascular
involvement in rheumatic diseases: ‘Vascular rheumatology’.
Arthritis Res Ther. 10:2242008. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lainer-Carr D and Brahn E: Angiogenesis
inhibition as a therapeutic approach for inflammatory synovitis.
Nat Clin Pract Rheumatol. 3:434–442. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Komatsu N and Takayanagi H: Bone and
cartilage destruction in rheumatoid arthritis. Clin Calcium.
22:179–185. 2012.(In Japanese). PubMed/NCBI
|
|
8
|
Kawaguchi N, Zhang TT and Nakanishi T:
Involvement of CXCR4 in normal and abnormal development. Cells.
8:E1852019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Orimo A, Gupta PB, Sgroi DC,
Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL
and Weinberg RA: Stromal fibroblasts present in invasive human
breast carcinomas promote tumor growth and angiogenesis through
elevated SDF-1/CXCL12 secretion. Cell. 121:335–348. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Patrussi L and Baldari CT: The
CXCL12/CXCR4 axis as a therapeutic target in cancer and HIV-1
infection. Curr Med Chem. 18:497–512. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pablos JL, Santiago B, Galindo M, Torres
C, Brehmer MT, Blanco FJ and García-Lázaro FJ: Synoviocyte-derived
CXCL12 is displayed on endothelium and induces angiogenesis in
rheumatoid arthritis. J Immunol. 170:2147–2152. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tachibana K, Hirota S, Iizasa H, Yoshida
H, Kawabata K, Kataoka Y, Kitamura Y, Matsushima K, Yoshida N,
Nishikawa S, et al: The chemokine receptor CXCR4 is essential for
vascularization of the gastrointestinal tract. Nature. 393:591–594.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Blades MC, Ingegnoli F, Wheller SK, Manzo
A, Wahid S, Panayi GS, Perretti M and Pitzalis C: Stromal
cell-derived factor 1 (CXCL12) induces monocyte migration into
human synovium transplanted onto SCID Mice. Arthritis Rheum.
46:824–836. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kanbe K, Chiba J, Inoue Y, Taguchi M and
Yabuki A: SDF-1 and CXCR4 in synovium are associated with disease
activity and bone and joint destruction in patients with rheumatoid
arthritis treated with golimumab. Mod Rheumatol. 26:46–50. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang F, Shu JL, Li Y, Wu YJ, Zhang XZ,
Han L, Tang XY, Wang C, Wang QT, Chen JY, et al: CP-25, a novel
anti-inflammatory and immunomodulatory drug, inhibits the functions
of activated human B cells through regulating BAFF and TNF-α
signaling and comparative efficacy with biological agents. Front
Pharmacol. 8:9332017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jia XY, Chang Y, Sun XJ, Wu HX, Wang C, Xu
HM, Zhang L, Zhang LL, Zheng YQ, Song LH, et al: Total glucosides
of paeony inhibit the proliferation of fibroblast-like synoviocytes
through the regulation of G proteins in rats with collagen-induced
arthritis. Int Immunopharmacol. 18:1–6. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu H, Wei W, Song L, Zhang L, Chen Y and
Hu X: Paeoniflorin induced immune tolerance of mesenteric lymph
node lymphocytes via enhancing beta 2-adrenergic receptor
desensitization in rats with adjuvant arthritis. Int
Immunopharmacol. 7:662–673. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang QT, Zhang LL, Wu HX and Wei W: The
expression change of β-arrestins in fibroblast-like synoviocytes
from rats with collagen-induced arthritis and the effect of total
glucosides of paeony. J Ethnopharmacol. 133:511–516. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang C, Yuan J, Wu HX, Chang Y, Wang QT,
Wu YJ, Liu LH and Wei W: Paeoniflorin inhibits inflammatory
responses in mice with allergic contact dermatitis by regulating
the balance between inflammatory and anti-inflammatory cytokines.
Inflamm Res. 62:1035–1044. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang LL, Wei W, Wang NP, Wang QT, Chen
JY, Chen Y, Wu H and Hu XY: Paeoniflorin suppresses inflammatory
mediator production and regulates G protein-coupled signaling in
fibroblast-like synoviocytes of collagen induced arthritic rats.
Inflamm Res. 57:388–395. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen JY, Wu HX, Chen Y, Zhang LL, Wang QT,
Sun WY and Wei W: Paeoniflorin inhibits proliferation of
fibroblast-like synoviocytes through suppressing G-protein-coupled
receptor kinase 2. Planta Med. 78:665–671. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chang Y, Zhang L, Wang C, Jia XY and Wei
W: Paeoniflorin inhibits function of synoviocytes pretreated by
rIL-1α and regulates EP4 receptor expression. J Ethnopharmacol.
137:1275–1282. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chang Y, Wei W, Zhang L and Xu HM: Effects
and mechanisms of total glucosides of paeony on synoviocytes
activities in rat collagen-induced arthritis. J Ethnopharmacol.
121:43–48. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu ZQ, Jiang ZH, Liu L and Hu M:
Mechanisms responsible for poor oral bioavailability of
paeoniflorin: Role of intestinal disposition and interactions with
sinomenine. Pharm Res. 23:2768–2780. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang C, Yuan J, Yang ZY, Nie XX, Song LH
and Wei W: Pharmacokinetics of paeoniflorin microemulsion after
repeated dosing in rats with adjuvant arthritis. Pharmazie.
67:997–1001. 2012.PubMed/NCBI
|
|
26
|
Wang C, Yuan J, Zhang LL and Wei W:
Pharmacokinetic comparisons of Paeoniflorin and
Paeoniflorin-6′O-benzene sulfonate in rats via different routes of
administration. Xenobiotica. 46:1142–1150. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang XD, Wang C, Zhou P, Yu J, Asenso J,
Ma Y and Wei W: Absorption characteristic of
paeoniflorin-6-O-benzene sulfonate (CP-25) in in situ single-pass
intestinal perfusion in rats. Xenobiotica. 46:775–783. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chang Y, Jia X, Wei F, Wang C, Sun X, Xu
S, Yang X, Zhao Y, Chen J, Wu H, et al: CP-25, a novel compound,
protects against autoimmune arthritis by modulating immune
mediators of inflammation and bone damage. Sci Rep. 6:262392016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jia X, Wei F, Sun X, Chang Y, Xu S, Yang
X, Wang C and Wei W: CP-25 attenuates the inflammatory response of
fibroblast-like synoviocytes co-cultured with BAFF-activated CD4(+)
T cells. J Ethnopharmacol. 189:194–201. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sun WY, Wu JJ, Peng WT, Sun JC and Wei W:
The role of G protein-coupled receptor kinases in the pathology of
malignant tumors. Acta Pharmacol Sin. 39:1699–1705. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Premont RT and Gainetdinov RR:
Physiological roles of G protein-coupled receptor kinases and
arrestins. Annu Rev Physiol. 69:511–534. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Métayé T, Gibelin H, Perdrisot R and
Kraimps JL: Pathophysiological roles of G-protein-coupled receptor
kinases. Cell Signal. 17:917–928. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yu H, Yang YH, Rajaiah R and Moudgil KD:
Nicotine-induced differential modulation of autoimmune arthritis in
the Lewis rat involves changes in interleukin-17 and anti-cyclic
citrullinated peptide antibodies. Arthritis Rheum. 63:981–991.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen J, Wang Q, Wu H, Liu K, Wu Y, Chang Y
and Wei W: The ginsenoside metabolite compound K exerts its
anti-inflammatory activity by downregulating memory B cell in
adjuvant-induced arthritis. Pharm Biol. 54:1280–1288. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang Y, Han CC, Cui D, Luo TT, Li Y, Zhang
Y, Ma Y and Wei W: Immunomodulatory Effects of CP-25 on splenic T
cells of rats with adjuvant arthritis. Inflammation. 41:1049–1063.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Semerano L, Clavel G, Assier E, Denys A
and Boissier MC: Blood vessels, a potential therapeutic target in
rheumatoid arthritis? Joint Bone Spine. 78:118–123. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Khan A, Greenman J and Archibald SJ: Small
molecule CXCR4 chemokine receptor antagonists: Developing drug
candidates. Curr Med Chem. 14:2257–2277. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hatse S, Princen K, Bridger G, De Clercq E
and Schols D: Chemokine receptor inhibition by AMD3100 is strictly
confined to CXCR4. FEBS Lett. 527:255–262. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Matsuo Y, Ochi N, Sawai H, Yasuda A,
Takahashi H, Funahashi H, Takeyama H, Tong Z and Guha S: CXCL8/IL-8
and CXCL12/SDF-1alpha co-operatively promote invasiveness and
angiogenesis in pancreatic cancer. Int J Cancer. 124:853–861. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Salcedo R, Wasserman K, Young HA, Grimm
MC, Howard OM, Anver MR, Kleinman HK, Murphy WJ and Oppenheim JJ:
Vascular endothelial growth factor and basic fibroblast growth
factor induce expression of CXCR4 on human endothelial cells: In
vivo neovascularization induced by stromal-derived factor-1alpha.
Am J Pathol. 154:1125–1135. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gurevich EV, Tesmer JJ, Mushegian A and
Gurevich VV: G protein-coupled receptor kinases: More than just
kinases and not only for GPCRs. Pharmacol Ther. 133:40–69. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Brondello JM, Pouysségur J and McKenzie
FR: Reduced MAP kinase phosphatase-1 degradation after
p42/p44MAPK-dependent phosphorylation. Science. 286:2514–2517.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang Q, Wang L, Wu L, Zhang M, Hu S, Wang
R, Han Y, Wu Y, Zhang L, Wang X, et al: Paroxetine alleviates T
lymphocyte activation and infiltration to joints of
collagen-induced arthritis. Sci Rep. 7:453642017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Penela P, Ruiz-Gómez A, Castaño JG and
Mayor F Jr: Degradation of the G protein-coupled receptor kinase 2
by the proteasome pathway. J Biol Chem. 273:35238–35244. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Woodard LE and Nimmagadda S: CXCR4-based
imaging agents. J Nucl Med. 52:1665–1669. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Clift IC, Bamidele AO, Rodriguez-Ramirez
C, Kremer KN and Hedin KE: β-Arrestin1 and distinct CXCR4
structures are required for stromal derived factor-1 to
downregulate CXCR4 cell-surface levels in neuroblastoma. Mol
Pharmacol. 85:542–552. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Busillo JM, Armando S, Sengupta R, Meucci
O, Bouvier M and Benovic JL: Site-specific phosphorylation of CXCR4
is dynamically regulated by multiple kinases and results in
differential modulation of CXCR4 signaling. J Biol Chem.
285:7805–7817. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cheng ZJ, Zhao J, Sun Y, Hu W, Wu YL, Cen
B, Wu GX and Pei G: β-arrestin differentially regulates the
chemokine receptor CXCR4-mediated signaling and receptor
internalization, and this implicates multiple interaction sites
between β-arrestin and CXCR4. J Biol Chem. 275:2479–2485. 2000.
View Article : Google Scholar : PubMed/NCBI
|