Introduction
Despite the advance of therapeutic approaches,
ischemic heart disease, in particular myocardial infarction (MI),
remains a major cause of left ventricular dysfunction or heart
failure (1,2). Hypoxia and hypoperfusion disrupt
regional redox homeostasis, trigger inflammatory cascades and
subsequently facilitate excessive cell death and cardiac
remodeling, resulting in the deterioration of cardiac dysfunction.
During this process, the sympathetic stress response may also
exacerbate the imbalance between reactive oxygen species (ROS)
production and the protective antioxidant defense system, thereby
increasing the levels of oxidative stress in the heart tissue
(3).
Nicotinamide adenine dinucleotide phosphate (NADPH)
oxidase (NOX) is a prominent source of excess ROS in the
cardiovascular system (4).
Treatments targeting NOX inhibition have demonstrated therapeutic
success in a number of previous studies (5–7).
Following MI, NOX expression is significantly increased and serves
a critical role in cardiac remodeling in the infarcted myocardium.
The adverse remodeling of the left ventricle is an unfavorable
development associated with myocardial hypertrophy and increased
the sympathetic activity (8–10).
However, it is unclear whether dexmedetomidine (DEX)-mediated
sympathetic sedation contributes to curbing NOX-derived ROS,
thereby protecting MI-induced cardiac dysfunction.
DEX, with an α2 to α1 selectivity ratio of 1,600:1,
is a highly selective α adrenergic receptor agonist (11,12).
It targets the α2 adrenergic receptors in the central nervous
system to inhibit the activity of sympathetic outflow, producing
the effects of analgesia, anxiolysis and sedation (13,14).
Therefore, the application of DEX may alleviate the sympathetic
stress response induced by acute MI injury. The present study
involved an animal model of permanent coronary ligation and the
cardio-protective effects of DEX were analyzed. It was demonstrated
that DEX hindered NOX-derived oxidative stress and improved the
systolic performance of the damaged left ventricle following
MI.
Materials and methods
Induction of myocardial infarction and
drug treatment
Male C57BL/6J mice (8–10 weeks old, n=55), obtained
from the Model Animal Research Center of Nanjing University, were
divided into 4 groups (n=12/group; others failing to survive the
surgery): Sham + Normal Saline (NS) group, Sham + DEX group, MI +
NS group and MI + DEX group. The mice were maintained in
air-filtered units at 21±2°C and 50±15% relative humidity under a
12-h light/dark cycle. Mice were randomized and anesthetized using
inhaled isoflurane (2%) with the heart rate monitored
simultaneously. Surgical procedures were performed as described
previously (15). Briefly, mice in
the MI + NS and MI + DEX groups received a left thoracotomy and the
left anterior descending coronary artery was permanently ligated by
using a 7.0 polypropylene suture. The surgery in Sham + NS and Sham
+ DEX groups involved an identical procedure, with the exception of
coronary artery ligation. DEX treatment was initiated immediately
following surgery.
Mice in the Sham + DEX and MI + DEX groups were
treated with DEX (Jiangsu Nhwa Pharmaceutical Co., Ltd.; 20
µg/kg/day intraperitoneally), as previously described (16). Mice in the Sham + NS and MI + NS
groups were administered normal saline injection only. Throughout
these experiments, the therapeutic treatments were administered
once per day for 7 days, and no significant side effects were
observed in any animals following drug treatment. For ex
vivo analyses of protein expression and histological study,
animals were sacrificed using anesthesia of inhaled isoflurane,
followed by cervical dislocation, 3 or 7 days after the surgery.
All animal protocols were reviewed and approved by the Committee on
the Ethics of Animal Experiments of the Shanghai Jiao Tong
University School of Medicine.
Echocardiography analysis
A total of 1 week after surgery, cardiac function
was evaluated by transthoracic echocardiography with a
high-resolution ultrasound imaging system (Vevo 2100; FUJIFILM
VisualSonics, Inc.) equipped with a 30-MHz mechanical transducer.
M-mode tracings were used to measure percentage of ejection
fraction (EF%) and fractional shortening (FS%) as described
previously (17). M-mode
measurement data represent 3 to 6 averaged cardiac cycles from at
least 2 scans/mouse.
Histological analysis
Collagen content in the heart was analyzed using
picrosirius red staining. Briefly, the hearts were quickly removed,
weighed and then fixed in 4% buffered paraformaldehyde at room
temperature for 48 h. The hearts were embedded in paraffin, and
then cut into 6 µm serial sections using a microtome. Picrosirius
red staining (0.5% Picrosirius red at room temperature for 20 min)
was performed to evaluate the severity of fibrosis. Images were
captured with an Olympus light microscope (magnification, ×400) and
quantitatively analyzed using Image-Pro Plus v.6.0 (Media
Cybernetics, Inc.).
Western blot analysis
A total of 3 days after surgery, tissue samples of
the left ventricular myocardium were homogenized in in RIPA lysis
buffer containing 1% PMSF. Protein concentrations in supernatants
were measured with a bicinchoninic acid protein assay (Beyotime
Institute of Biotechnology). Equal amounts of prepared proteins (50
µg/lane) were subjected to 10% SDS-PAGE, separated by
electrophoresis and transferred to nitrocellulose membranes.
Following blocking in 5% non-fat milk PBS for 2 h, the membranes
were incubated overnight at 4°C with anti-Nox2 (cat. no. ab80508;
Abcam; 1:1,000), anti-Nox4 (cat. no. ab195524; Abcam; 1:1,000),
anti-Bax (cat. no. ab32503; Abcam; 1:1,000), anti-Bcl-2 (cat. no.
ab182858; Abcam; 1:1,000) or β-actin (cat. no. ab8227; Abcam;
1:1,000) primary antibodies, followed by incubation with
horseradish peroxidase-conjugated goat anti-rabbit secondary
antibodies (cat. no. 7074; Cell Signaling Technology, Inc.;
1:5,000) for 1 h at room temperature. Immunoreactive bands were
detected using an enhanced chemiluminescence system (EMD Millipore)
and quantified by Image-Pro Plus v.6.0.
Caspase-3 activity assay
Myocardial caspase-3 activity was determined by
colorimetric assay kits (Beyotime Institute of Biotechnology), as
described previously (18). The
heart tissue was collected from mice 3 days after MI and the assays
were performed according to the manufacturer's protocol.
Malondialdehyde (MDA) and superoxide
dismutase (SDS) assay
At the end of the experimental period, mice were
sacrificed, the hearts were excised and heart tissues were weighed
(wet weight) and homogenized in ice-cold PBS. The homogenates were
centrifuged at 3,000 × g for 15 min at 4°C to obtain the
supernatant. The MDA content, a reliable index of ROS-induced lipid
peroxidation, and SOD activity were measured by commercially
available kits according to the manufacturer's protocol (Nanjing
Jiancheng Bioengineering Institute Co., Ltd.).
Statistical analysis
Continuous data are presented as means ± standard
error of the mean and were analyzed by paired or unpaired Student's
t-test unless otherwise stated. Differences between multiple groups
were determined with one way analysis of variance followed by a
Bonferroni post hoc analysis. P<0.05 was considered to indicate
a statistically significant difference. Data were analyzed with the
use of GraphPad Prism v.5 software (GraphPad Software, Inc.).
Results
DEX treatment leads to an improvement
in LV dysfunction following MI
As demonstrated in Fig.
1, echocardiographic parameters were measured in each group at
day 7 post-MI. For sham mice, the results revealed no significant
changes in ejection fraction or fractional shortening regardless of
whether or not DEX treatment was given. LAD ligation resulted in
the dilatation of LV and a serious impairment of cardiac function.
Notably, the MI-induced acceleration of cardiac dilatation and
deterioration of LV function were inhibited in MI + DEX mice, in
comparison with those in the MI + NS mice.
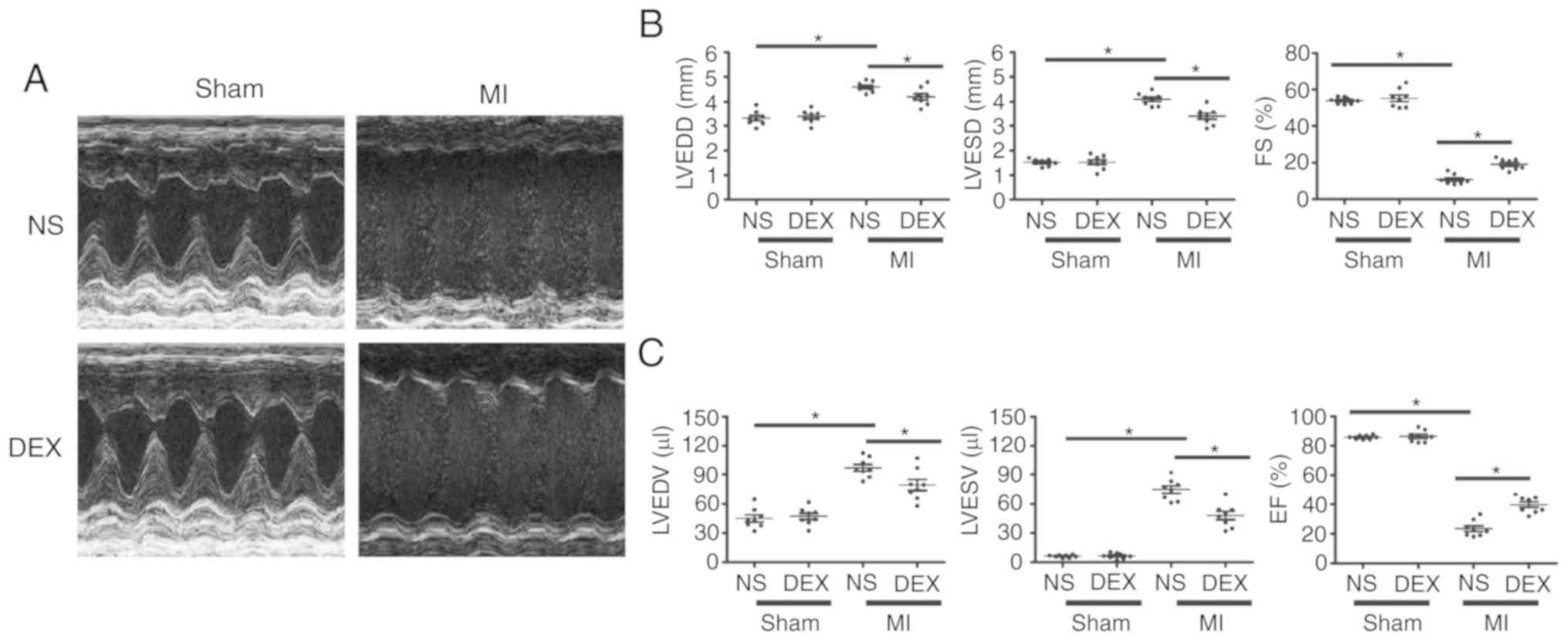 | Figure 1.Functional analysis of the left
ventricle with echocardiography. (A) Representative M-mode
echocardiograms from the Sham + NS, Sham + DEX, MI + NS, and MI +
DEX mice. (B) The mean left ventricular LVEDD, LVESD and FS in the
Sham + NS, Sham + DEX, MI + NS, and MI + DEX-treated mice. (C) The
mean left ventricular LVEDV, LVESV and EF in the Sham + NS, Sham +
DEX, MI + NS, and MI + DEX mice. Data are presented as mean ±
standard error of the mean. n=6. *P<0.05. NS, normal saline;
DEX, dexmedetomidine; MI, myocardial infarction; LVEDD, left
ventricle end-diastolic dimension; LVESD, LV end-systolic
dimension; FS, fractional shortening. |
DEX treatment mitigates cardiac
fibrosis following MI
Picrosirius red staining was performed to determine
the severity of fibrotic changes in each group. No significant
difference was observed in the positive picrosirius red stained
areas between the Sham + NS and Sham + DEX mice. In the MI mice,
DEX treatment resulted in significantly decreased collagen content
in the peri-infarct zone (Fig.
2).
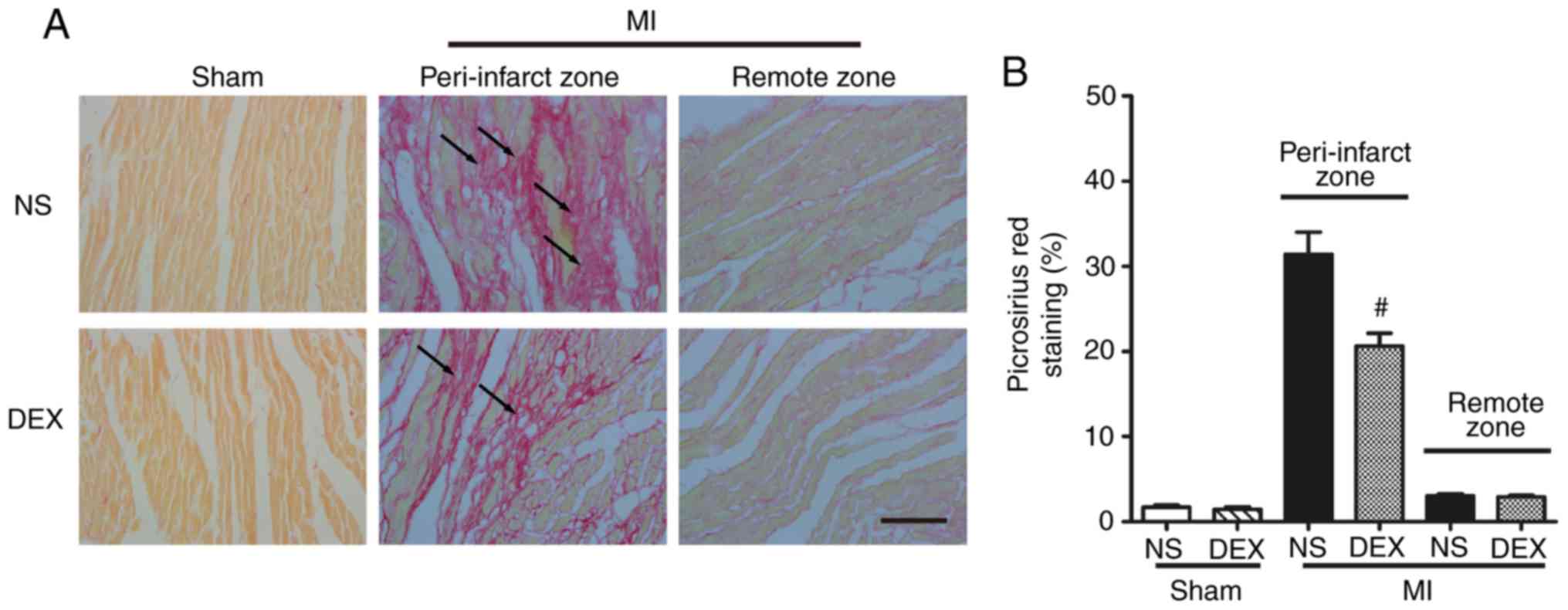 | Figure 2.Analysis of pathological cardiac
remodeling with picrosirius red staining. (A) Representative images
of picrosirius red-stained sections in the Sham + NS, Sham + DEX,
MI + NS, and MI + DEX mice. Scale bar, 50 µm. (B) Quantitative
analysis of picrosirius red-positive area in the Sham + NS, Sham +
DEX, MI + NS, and MI + DEX mice. Data are presented as mean ±
standard error of the mean (n=6. #P<0.05 vs. MI + NS
group. NS, normal saline; DEX, dexmedetomidine; MI, myocardial
infarction. |
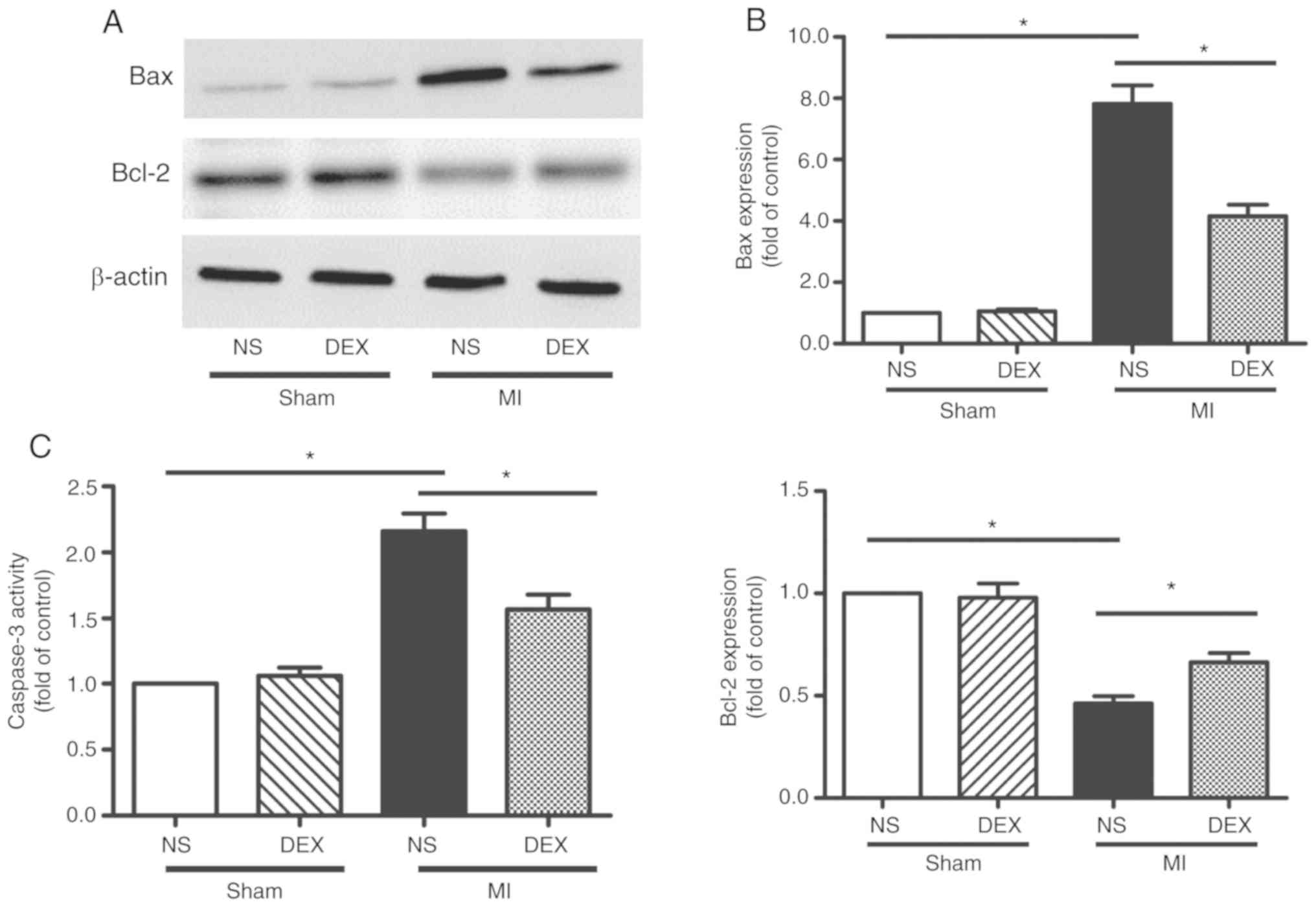
DEX treatment protects the myocardium
against apoptosis after MI
The levels apoptosis of myocardium was analyzed by
western blot analysis and the detection of caspase-3 activity. The
data from the western blot analysis performed in the present study
revealed significant upregulation of Bax and downregulation of
Bcl-2 expression in myocardium following MI, indicating the
occurrence of enhanced cardiomyocyte apoptosis. MI-induced
apoptosis was decreased following the administration of DEX
(Fig. 3A and B).
Caspase-3 is involved in a number of important
events in the apoptosis process (19). LAD ligation led to the upregulation
of caspase-3 activity compared with sham mice. The data from the
present study also revealed that this pro-apoptotic change was
markedly hindered by the administration of DEX (Fig. 3C).
DEX treatment inhibits NOX-derived
oxidative stress in myocardium following MI
Whether NOX served an important role in DEX-induced
cardiac protection was examined. The protein expression levels of
NOX2 and NOX4 were evaluated by western blot analysis. The results
revealed that DEX significantly decreased the expression levels of
both NOX2 and NOX4, which were upregulated following MI (Fig. 4A and B).
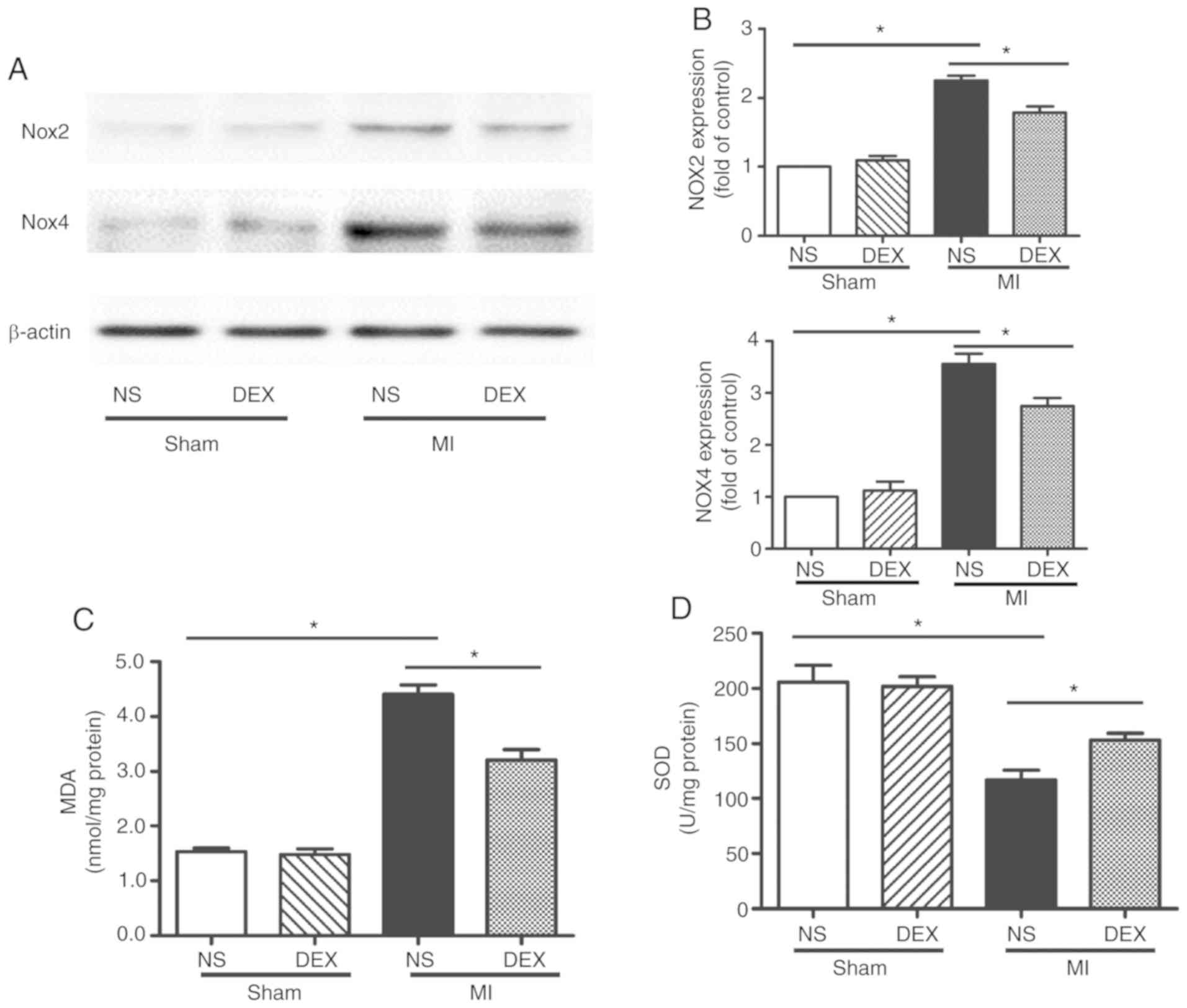 | Figure 4.Western blot analysis of NOX2 and NOX4
expression and SOD and MDA assays. (A) Representative gel images
and (B) quantitative densitometric analysis of western blot
analysis data demonstrating the expression levels of NOX2 and NOX4
proteins in the Sham + NS, Sham + DEX, MI + NS, and MI + DEX mice.
(C) Analysis of MDA content in the Sham + NS, Sham + DEX, MI + NS,
and MI + mice. (D) Analysis of SOD activity in the Sham + NS, Sham
+ DEX, MI + NS, and MI + DEX mice. Data are presented as mean ±
standard error of the mean. n=6. *P<0.05. NOX, nicotinamide
adenine dinucleotide phosphate oxidase; NS, normal saline; DEX,
dexmedetomidine; MI, myocardial infarction. |
MDA is an oxidative stress marker, the expression
levels of which are considered the index of ROS-induced lipid
peroxidation in cardiac tissue (20). In the present study, in the cardiac
tissue, MI significantly increased the MDA level in comparison with
the sham mice. In the MI mice, treatment with DEX resulted in a
significant decrease in MDA level in comparison with NS-treated
mice.
In addition, SOD, a component of several myocardial
endogenous antioxidants, was decreased significantly in the MI
groups as compared with the sham groups, whereas SOD levels were
increased significantly in MI + DEX mice compared with MI + NS
mice.
Discussion
Ischemic injury leads to the loss of viable
myocardium, which is followed by adverse cardiac remodeling.
Eventually, progressive changes in the molecular and structural
components of the myocardium result in cardiac dysfunction. In the
present study, the administration of DEX, a highly selective α2
adrenergic receptor agonist, was identified as a protective factor
for the recovery of heart tissue post-MI. DEX treatment contributed
to the successful healing process and limited the level of
oxidative stress, through the over-activation of NOXs.
Clinically, DEX is frequently prescribed during
perioperative periods, and contributes to an enhancement of vagus
nerve excitability, hemodynamic stability and permits the use of a
lower dosage of anesthetic with sedation and analgesia (21). As an anesthetic adjuvant, DEX
produces sedative and analgesic effects, primarily through its
agonistic action on the α2 adrenergic receptors in the locus
coeruleus of the pons and the spinal cord (22). Without depressing respiration, DEX
is also a potential regulator of inflammatory and immune responses
(23,24). These properties suggest that DEX
may have an effect on post-MI remodeling and cardiac dysfunction,
in spite of the fact that DEX is not a component of conventional
therapies for patients who have suffered MI. In the present study,
DEX treatment was identified to markedly decrease the levels of
fibrotic changes and improve the cardiac performance after MI.
While the deposition of non-contractile scar tissue is important
for a successful healing process, excess fibrogenesis is a key
component of adverse cardiac remodeling.
The fibrosis and dysfunction of MI hearts may be
attributed to the loss of viable myocardium. Bax, Bcl-2 and
caspase-3 are all closely associated with apoptosis (17). The present study demonstrated that
DEX administration restored the Bax and Bcl-2 ratio and
simultaneously decreased the activity of caspase-3, implying that
DEX decreased the levels of apoptosis of the myocardium. The
underlying mechanism for the anti-apoptosis properties of DEX
requires further investigation.
Oxidative stress serves a vital role in the
development of post-MI remodeling and cardiomyocytic apoptosis
following MI (25,26). SOD and MDA are two common indexes
used for evaluating the ability to eliminate oxygen free radicals
in cells (27). SOD, engaged in
scavenging free radicals, protects cells from damage elicited by
ROS, while MDA, together with excessive oxyradicals, attacks the
cell membrane, leading to cell death. The present study revealed
that ischemic injury resulted in an increased MDA level and a
decreased SOD level; furthermore, the homeostasis between MDA and
SOD was partially restored following the administration of DEX,
suggesting that DEX may participate in the inhibition of the
activated oxidative stress response.
Previous studies have confirmed the importance of
ROS-generating NOX family in redox signaling following ischemic
injury (28,29). Of the 5 NOX family members, Nox2
and Nox4 are expressed in the murine heart (17,30).
In the present study, the expression levels of these two molecules
were analyzed by western blot analysis. The results indicated that
their expression levels were both decreased following the
administration of DEX in MI mice. Understanding of the mechanisms
of DEX-induced cardioprotective signaling is complex, as α2
adrenergic receptors are also present in the myocardial tissue
(31,32). In addition to the action of
DEX-induced neurohumoral systemic modulation, the direct
stimulation of cardiac α2 adrenergic receptors may also facilitate
the benefits of DEX administration.
Taken together, the results of the present study
demonstrated that the administration of DEX promoted recovery from
ischemic injury and improved the performance of damaged left
ventricles following MI. The therapeutic effect may be associated
with the inhibition of excess NOX-derived ROS, thereby decelerating
apoptosis in the myocardium and the subsequent adverse cardiac
remodeling.
Acknowledgements
Not applicable.
Funding
The study was supported by the National Natural
Science Foundation of China (grant nos. 81570316, 81670389,
81770249 and 81800375).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
GT and RZ provided the concept, administration,
supervision, resources and funding for the present study, and
validated the data. HH, DD, JZ and JH collected the data. HH, DD
and LL analyzed the data. DD and JZ analyzed the data with the
software. HH, DD and JH prepared the figures. HH and DD wrote the
manuscript. All authors reviewed and edited the final
manuscript.
Ethics approval and consent to
participate
All animal protocols were reviewed and approved by
the Committee on the Ethics of Animal Experiments of the Shanghai
Jiao Tong University School of Medicine.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cahill TJ, Choudhury RP and Riley PR:
Heart regeneration and repair after myocardial infarction:
Translational opportunities for novel therapeutics. Nat Rev Drug
Discov. 16:699–717. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lefer DJ and Marban E: Is cardioprotection
dead? Circulation. 136:98–109. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Malfitano C, Barboza CA, Mostarda C, da
Palma RK, dos Santos CP, Rodrigues B, Freitas SC, Belló-Klein A,
Llesuy S, Irigoyen MC and De Angelis K: Diabetic hyperglycemia
attenuates sympathetic dysfunction and oxidative stress after
myocardial infarction in rats. Cardiovasc Diabetol. 13:1312014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li B, Tian J, Sun Y, Xu TR, Chi RF, Zhang
XL, Hu XL, Zhang YA, Qin FZ and Zhang WF: Activation of NADPH
oxidase mediates increased endoplasmic reticulum stress and left
ventricular remodeling after myocardial infarction in rabbits.
Biochim Biophys Acta. 1852:805–815. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yu L, Yang G, Zhang X, Wang P, Weng X,
Yang Y, Li Z, Fang M, Xu Y, Sun A and Ge J: Megakaryocytic leukemia
1 (MKL1) bridges epigenetic activation of NADPH oxidase in
macrophages to cardiac ischemia-reperfusion injury. Circulation.
138:2820–2836. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cadenas S: ROS and redox signaling in
myocardial ischemia-reperfusion injury and cardioprotection. Free
Radic Biol Med. 117:76–89. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Asensio-Lopez MDC, Lax A, Fernandez Del
Palacio MJ, Sassi Y, Hajjar RJ and Pascual-Figal DA:
Pharmacological inhibition of the mitochondrial NADPH oxidase
4/PKCalpha/Gal-3 pathway reduces left ventricular fibrosis
following myocardial infarction. Transl Res. 199:4–23. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shi S, Liang J, Liu T, Yuan X, Ruan B, Sun
L, Tang Y, Yang B, Hu D and Huang C: Depression increases
sympathetic activity and exacerbates myocardial remodeling after
myocardial infarction: Evidence from an animal experiment. PLoS
One. 9:e1017342014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang BS and Leenen FH: The brain
renin-angiotensin-aldosterone system: A major mechanism for
sympathetic hyperactivity and left ventricular remodeling and
dysfunction after myocardial infarction. Curr Heart Fail Rep.
6:81–88. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xiong L, Liu Y, Zhou M, Wang G, Quan D,
Shuai W, Shen C, Kong B, Huang C and Huang H: Targeted ablation of
cardiac sympathetic neurons attenuates adverse post-infarction
remodeling and left ventricle dysfunction. Exp Physiol.
103:1221–1229. 2018. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ji F, Li Z, Nguyen H, Young N, Shi P,
Fleming N and Liu H: Perioperative dexmedetomidine improves
outcomes of cardiac surgery. Circulation. 127:1576–1584. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Menon DV, Wang Z, Fadel PJ, Arbique D,
Leonard D, Li JL, Victor RG and Vongpatanasin W: Central
sympatholysis as a novel countermeasure for cocaine-induced
sympathetic activation and vasoconstriction in humans. J Am Coll
Cardiol. 50:626–633. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Parati G and Esler M: The human
sympathetic nervous system: Its relevance in hypertension and heart
failure. Eur Heart J. 33:1058–1066. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sun Z, Zhao T, Lv S, Gao Y, Masters J and
Weng H: Dexmedetomidine attenuates spinal cord ischemia-reperfusion
injury through both anti-inflammation and anti-apoptosis mechanisms
in rabbits. J Transl Med. 16:2092018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yan X, Zhang H, Fan Q, Hu J, Tao R, Chen
Q, Iwakura Y, Shen W, Lu L, Zhang Q and Zhang R: Dectin-2
deficiency modulates Th1 differentiation and improves wound healing
after myocardial infarction. Circ Res. 120:1116–1129. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sun Y, Jiang C, Jiang J and Qiu L:
Dexmedetomidine protects mice against myocardium
ischaemic/reperfusion injury by activating an AMPK/PI3K/Akt/eNOS
pathway. Clin Exp Pharmacol Physiol. 44:946–953. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Han H, Zhu J, Zhu Z, Ni J, Du R, Dai Y,
Chen Y, Wu Z, Lu L and Zhang R: p-Cresyl sulfate aggravates cardiac
dysfunction associated with chronic kidney disease by enhancing
apoptosis of cardiomyocytes. J Am Heart Assoc. 4:e0018522015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu J, Deng G, Tian Y, Pu Y, Cao P and Yuan
W: An in vitro investigation into the role of bone
marrowderived mesenchymal stem cells in the control of disc
degeneration. Mol Med Rep. 12:5701–5708. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang B, Ye D and Wang Y: Caspase-3 as a
therapeutic target for heart failure. Expert Opin Ther Targets.
17:255–263. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang J, Wang H, Hao P, Xue L, Wei S, Zhang
Y and Chen Y: Inhibition of aldehyde dehydrogenase 2 by oxidative
stress is associated with cardiac dysfunction in diabetic rats. Mol
Med. 17:172–179. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dong J, Guo X, Yang S and Li L: The
effects of dexmedetomidine preconditioning on aged rat heart of
ischaemia reperfusion injury. Res Vet Sci. 114:489–492. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nguyen V, Tiemann D, Park E and Salehi A:
Alpha-2 agonists. Anesthesiol Clin. 35:233–245. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu W, Yu W, Weng Y, Wang Y and Sheng M:
Dexmedetomidine ameliorates the inflammatory immune response in
rats with acute kidney damage. Exp Ther Med. 14:3602–3608. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang X, Wang J, Qian W, Zhao J, Sun L,
Qian Y and Xiao H: Dexmedetomidine inhibits tumor necrosis
factor-alpha and interleukin 6 in lipopolysaccharide-stimulated
astrocytes by suppression of c-Jun N-terminal kinases.
Inflammation. 37:942–949. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hou L, Guo J, Xu F, Weng X, Yue W and Ge
J: Cardiomyocyte dimethylarginine dimethylaminohydrolase1
attenuates left-ventricular remodeling after acute myocardial
infarction: Involvement in oxidative stress and apoptosis. Basic
Res Cardiol. 113:282018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Becher UM, Ghanem A, Tiyerili V, Furst DO,
Nickenig G and Mueller CF: Inhibition of leukotriene C4 action
reduces oxidative stress and apoptosis in cardiomyocytes and
impedes remodeling after myocardial injury. J Mol Cell Cardiol.
50:570–577. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jimenez-Fernandez S, Gurpegui M,
Díaz-Atienza F, Perez-Costillas L, Gerstenberg M and Correll CU:
Oxidative stress and antioxidant parameters in patients with major
depressive disorder compared to healthy controls before and after
antidepressant treatment: Results from a meta-analysis. J Clin
Psychiatry. 76:1658–1667. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yu Q, Lee CF, Wang W, Karamanlidis G,
Kuroda J, Matsushima S, Sadoshima J and Tian R: Elimination of
NADPH oxidase activity promotes reductive stress and sensitizes the
heart to ischemic injury. J Am Heart Assoc. 3:e0005552014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cave AC, Brewer AC, Narayanapanicker A,
Ray R, Grieve DJ, Walker S and Shah AM: NADPH oxidases in
cardiovascular health and disease. Antioxid Redox Signal.
8:691–728. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Matsushima S, Tsutsui H and Sadoshima J:
Physiological and pathological functions of NADPH oxidases during
myocardial ischemia-reperfusion. Trends Cardiovasc Med. 24:202–205.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Imamura M, Lander HM and Levi R:
Activation of histamine H3-receptors inhibits carrier-mediated
norepinephrine release during protracted myocardial ischemia.
Comparison with adenosine A1-receptors and alpha2-adrenoceptors.
Circ Res. 78:475–481. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zefirov TL, Khisamieva LI, Ziyatdinova NI
and Zefirov AL: Selective blockade of α2-adrenoceptor
subtypes modulates contractility of rat myocardium. Bull Exp Biol
Med. 162:177–179. 2016. View Article : Google Scholar : PubMed/NCBI
|