Introduction
Cardiovascular disease (CVD) is the predominant
complication in patients with chronic kidney disease (CKD), and is
also the leading cause of mortality among patients with end stage
renal failure (1–3). Left ventricular hypertrophy (LVH) is
a typical pathological feature of cardiovascular lesions observed
in these patients, which was previously thought to be caused by
long-term hypertension and volume overload. However, even when
controlling blood pressure and volume load, LVH still exists
(4), suggesting that other
pro-hypertrophy factors must be present.
There is a large body of evidence to suggest that
the uremic milieu itself serves a critical function in the
development and progression of CVD (5). Although CVD prevalence and mortality
rates may vary amongst different studies, the prevalence of CVD is
estimated to be between 14.4–34.0% across a range of different
ethnic groups (6,7). One previous study has revealed that
indoxyl sulfate (IS), a typical protein-bound uremic toxin that is
produced by intestinal bacteria and accumulates in the blood with
the progression of CKD, participates in cardiac hypertrophy
(8). Another previous study has
suggested that the serum levels of IS were independently associated
with LVH, and IS-induced cardiomyocyte hypertrophy was observed
in vitro, and was further confirmed by the intraperitoneal
injection of IS in mice (9). In
addition, it has also been demonstrated that IS may induce reactive
oxygen species (ROS) production by inhibiting the protein kinase
AMP-activated catalytic subunit α2 (AMPK)/uncoupling protein 2
(UCP2) signaling pathway (10).
However, the ROS production induced by IS cannot be completely
reversed by an AMPK activator. This suggests that another signaling
pathway may also be involved in its biological functions. On the
other hand, oral charcoal adsorbents that are able to lower serum
IS levels were reported to prevent the progression of cardiac
hypertrophy in CKD (11,12). These studies reveal the deleterious
vascular effects of IS under uremic conditions.
Previously, IS was demonstrated to be a potent
endogenous agonist of the aryl hydrocarbon receptor (AhR), which is
a ligand-activated helix-loop-helix transcription factor involved
in the regulation of biological responses to planar aromatic
hydrocarbons (13,14). Prior to ligand binding, AhR is
sequestered in the cytoplasm; following activation by ligands, AhR
is translocated into the nucleus where it complexes with the AhR
nuclear translocator (ARNT), and binds to a specific DNA promoter
sequence including the DNA replication-related element (DRE) or the
xenobiotic responsive element (XRE), and modulates the subsequent
transcription of its downstream target genes, including
xenobiotic-metabolizing enzymes (15). It is generally accepted that
induction of cytochrome P450 family 1 subfamily A member 1,
cytochrome P450 family 1 subfamily A member 2 and cytochrome P450
family 1 subfamily B member 1 (CYP1B1) is considered cardiotoxic
through generating ROS and endogenous arachidonic acid (AA)
metabolites (16). Among these
targets, CYP1B1 has an important function in CVD.
CYP1B1, a heme-thiolate monooxygenase that catalyzes
numerous reactions involved in drug metabolism and synthesis of
cholesterol, steroids and other lipids, is able to metabolize AA
into hydroxyeicosatetraenoic acids (HETEs), which are thought to
serve a central function in the pathophysiology of the
cardiovascular system (17).
CYP1B1 was reported to mediate angiotensin (Ang) II-induced
vascular smooth muscle cell migration, proliferation and
hypertrophy (18). In a rat model
of isoprenaline-induced cardiac hypertrophy, expression of the
CYP1B1 gene was substantially increased during pressure overloads
(19). Furthermore, the selective
inhibition of CYP1B1 with 2,4,3′,5′-tetramethoxystilbene (TMS)
protected against Ang II or isoproterenol induced cardiac
hypertrophy (20,21). Overall, these results suggest that
CYP1B1 is associated with cardiac hypertrophy.
However, whether CYP1B1 is involved in cardiac
hypertrophy induced by uremic toxins remains unknown. Gondouin
et al (22) revealed that
IS is able to induce the upregulation of CYP1B1 in endothelial
cells. Therefore, the present study hypothesized that IS may also
possess the same function in cardiomyocytes. In order to
investigate this theory, rat embryonic cardiomyoblast H9c2 cells
were treated with various doses of IS or uremic toxin in the
present study, and the gene expression of CYP1B1 was detected. The
data demonstrated that CYP1B1 was involved in cardiac hypertrophy
induced by uremic toxins, while suppression of CYP1B1 ameliorated
cardiac hypertrophy.
Materials and methods
Materials
IS was purchased from Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany). Primary antibodies against CYP1B1 (cat no.
ab185954) and AhR (cat no. ab2769) were obtained from Abcam
(Cambridge, MA, USA). The antibodies anti-Histone H3 (cat no. 4499)
and anti-β-actin (cat no. 3700) were obtained from Cell Signaling
Technology, Inc. Small interfering RNA (siRNA) of CYP1B1 and AhR
were obtained from Shanghai GenePharma Co., Ltd. and the sequences
of these siRNA are presented in Table
SI. Transfection reagent Lipofectamine® 2000 was
obtained from Invitrogen (Thermo Fisher Scientific, Inc.). The
chromatin immunoprecipitation (ChIP) kit EZ-ChIP was obtained from
EMD Millipore. All primers were synthesized by Shanghai Sangong
Pharmaceutical Co., Ltd.
Human serum
A total of 15 patients with primary CKD at stage V
(age range 55–64 years, mean age 59.4, 8 male and 7 female, Chinese
and ethnically Han, and 15 sex- and age-matched controls were
enrolled from the Department of Nephrology of Xinqiao Hospital
(Chongqing, China) between July and October 2018. The protocol of
the present study was ethically approved by the Ethics Committee of
Xinqiao Hospital (approval no. 2018-006-01), and was performed in
accordance with the Declaration of Helsinki. All patients and
healthy controls provided written informed consent as a form
provided by the Ethical Committee of Xinqiao Hospital (Third
Military Medical University) prior to the start of the study to
confirm the collection and use of the samples in the present
study.
Animals
Male C57BL/6J mice were obtained from Beijing HFK
Bioscience Co., Ltd. Mice (n=18) at 8 weeks of age were housed
under controlled temperature (25°C), humidity (50–70%) and 12-h
light/dark cycle with free access to food and water. Mice were
divided into 3 groups. The first group (n=6) received dimethyl
sulfoxide (DMSO; 50% in saline, v/v) intraperitoneally. The second
group (n=6) was treated with a single daily dose of 100 mg/kg IS
for 8 weeks intraperitoneally. The third group (n=6) was
administered 300 µg/kg TMS (MedChemExpress LLC) in DMSO every third
day plus a daily dose of 100 mg/kg IS for 8 weeks
intraperitoneally. Subsequently, the mice were euthanized by
cervical dislocation under isoflurane (2%)-inhaled anesthesia 24 h
following the final injection. Hearts were immediately frozen in
liquid nitrogen and stored at −80°C. The parameters of heart
function were monitored by micro-computed tomography. All animal
studies were ethically approved by the Institutional Animal Care
and Use Committee of Third Military Medical University, and were
performed in accordance with the animal care guidelines of Third
Military Medical University.
Cell culture and treatment
Rat embryonic cardiomyoblast H9c2 cell line was
purchased from Shanghai Cell Bank (http://www.cellbank.org.cn). Cells were cultured in
Dulbecco's modified Eagle's medium with 10% fetal bovine serum, at
37°C in a humidified incubator containing 5% CO2. For
the IS treatment, increasing concentrations of IS (0, 0.1, 0.25 and
0.5 mM) were added to the medium. Cells were treated with IS for
the indicated periods (0, 24, 48 and 72 h). For treatment with
human serum, individual serum samples were diluted at room
temperature with the complete culture media (1:4, v/v), and the
cells were treated with diluted serum for 48 h.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from the cell culture and
heart tissues using TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc.). The RNA was reverse transcribed into cDNA and
RT-qPCR was performed as previously described (9). Briefly, a reaction volume of 10 µl
containing primers (1.0 µM), cDNA (100 ng) and 1XPCR mixture with
SYBR I Green (Takara Biotechnology Co., Ltd.) was amplified on an
iCycler iQ (Bio-Rad Laboratories, Inc.), with the thermocycling
conditions of one cycle of 95°C for 5 min, 36 cycles of 95°C for 30
sec, 60°C for 30 sec and 72°C for 60 sec, then one cycle of 72°C
for 5 min. Expression levels of CYP1B1, AhR, atrial natriuretic
factor (ANF), brain natriuretic peptide (BNP) and β-myosin heavy
chain (β-MHC) were calculated relative to GAPDH expression, using
the 2−ΔΔCq method (23). All primers are listed in Table SII.
Western blot analysis
Protein samples were prepared from the cell culture
(H9c2, 1×106) and heart tissues using RIPA buffer
containing protease inhibitors (Beyotime Institute of
Biotechnology, Haimen, China). Protein expression levels were
measured by assessing band thickness in the gel-phase of the
western blot analysis as previously described (9). The following antibodies were used for
the western blot analysis: Anti-CYP1B1 (1:1,000), anti-AhR
(1:1,000), anti-Histone H3 (1:1,000) and anti-β-Actin (1:1,000).
The blots were then incubated with the corresponding secondary
antibodies (1:2,000) conjugated with horseradish peroxidase (cat
nos. ZDR-5306 or ZDR-5307, OriGene Technologies, Inc.). The
immunoreactive proteins were detected using a ECL Detection System
(Thermo Fisher Scientific, Inc.). Finally, the membranes were
scanned and the densitometry analysis was performed using an image
analysis system (Image Lab, v.3.0; Bio-Rad Laboratories Inc.). To
validate the data, the western blot analysis was repeated at least
3 times.
Immunofluorescence assay
The immunofluorescence assay was performed on H9c2
cells (2×105) fixed in 4% paraformaldehyde for 15 min at
room temperature as previously described (9). The following antibodies were used:
Anti-AhR (1:100) and anti-α-actinin (1:200, ab137346; Abcam). The
nucleus was labeled with 1 mg/ml 4′,6-Diamidino-2-phenylindole,
dihydrochloride in phosphate buffered saline (PBS; Roche
Diagnostics) for 5 min at room temperature. Images were captured
using a Leica TCS-SP5 laser-scanning confocal microscope
(magnification, ×400; Leica Microsystems, Inc.).
Heart histology
The hearts were fixed in buffered formalin for 24 h
at room temperature and dehydrated with increasing concentrations
of ethanol (70, 80, 95 and 100% for 2 h, respectively) and
subsequently with xylene (30 min, 3 times). Finally, the hearts
were embedded in paraffin. The embedded tissues were cut into 3 µm
sections and were processed with xylene (10 min, twice) and
rehydrated with decreasing concentrations of ethanol (100, 95, 90,
80 and 70% for 5 min, respectively). Then the sections were stained
with hematoxylin-eosin (HE) with a commercial kit (C0105, Beyotime
Institute of Biotechnology) in accordance with the manufacturer's
protocol and were observed under a light microscope (magnification,
×200, Olympus CX22; Olympus Corporation).
Immunohistochemistry assay
The paraffin sections (3 µm) of the hearts were
analyzed via immunohistochemistry using antibodies against CYP1B1
or AhR using standard methods. Following rehydration, nonenzymatic
antigen retrieval was performed by heating the sections to 95–100°C
in citrate buffer (10 mM, pH 6.0) for 15 min and washed three times
with phosphate buffered saline (PBS) for 5 min. The sections were
immersed in 3% (v/v) H2O2 for 5 min and
washed three times with PBS for 5 min prior to using QuickBlock
Blocking Buffer (P0231, Beyotime Institute of Biotechnology) for 10
min. Following incubation with primary antibodies (CYP1B1 or AhR,
1:100) at 4°C overnight, the slides were then incubated with
enzyme-conjugated secondary antibodies (PV9001 or PV9002, 1:200,
OriGene Technologies, Inc.) and visualized using
3,3′-diaminobenzidine. Slides were then observed using an inverted
microscope (magnification, ×400, Olympus BX63; Olympus
Corporation). The negative controls used PBS in place of the
primary antibody. Images were randomly captured using the
microscope.
ChIP analysis
ChIP was performed using a commercial EZ-ChIP kit as
previously described (24).
Briefly, IS-treated H9c2 cells or normal controls
(1×107) were harvested and washed in cold PBS. Cells
were then immediately fixed in 1% formaldehyde for 10 min at room
temperature and halted using 0.125 M glycine. Cell lysates were
sonicated, and the sheared cross-linked chromatin was
immunoprecipitated with antibodies against CYP1B1 (target), histone
H3 (positive control) or normal immunoglobulin G (IgG; negative
control) overnight. Chromatin was then collected, washed and
decross-linked at 65°C. DNA was purified using spin columns and
detected with regular and quantitative PCR. Primers are listed in
Table SIII.
Knockdown of CYP1B1 and AhR in H9c2
cells
H9c2 cells (2×104) in 6-well plate were
transfected with 50 µM of siRNA against CYP1B1 or AhR or negative
control siRNA using Lipofectamine® 2,000 according to
the manufacturer's protocol. After incubating with siRNA for 4 h,
the media were refreshed. Following 2 days, the efficiency of the
knockdown of the target was evaluated by analyzing the protein
level of CYP1B1 or AhR. In order to analyze the biological
functions of the knockdown, H9c2 cells were further treated with IS
as aforementioned.
Statistical analysis
All data are expressed as the mean ± standard error
of the mean. Unpaired, two-tailed t-tests and one-way analysis of
variance followed by Tukey's multiple comparison test were used.
The statistical analysis was performed using SPSS software (version
13.0; SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to
indicate a statistically significant result.
Results
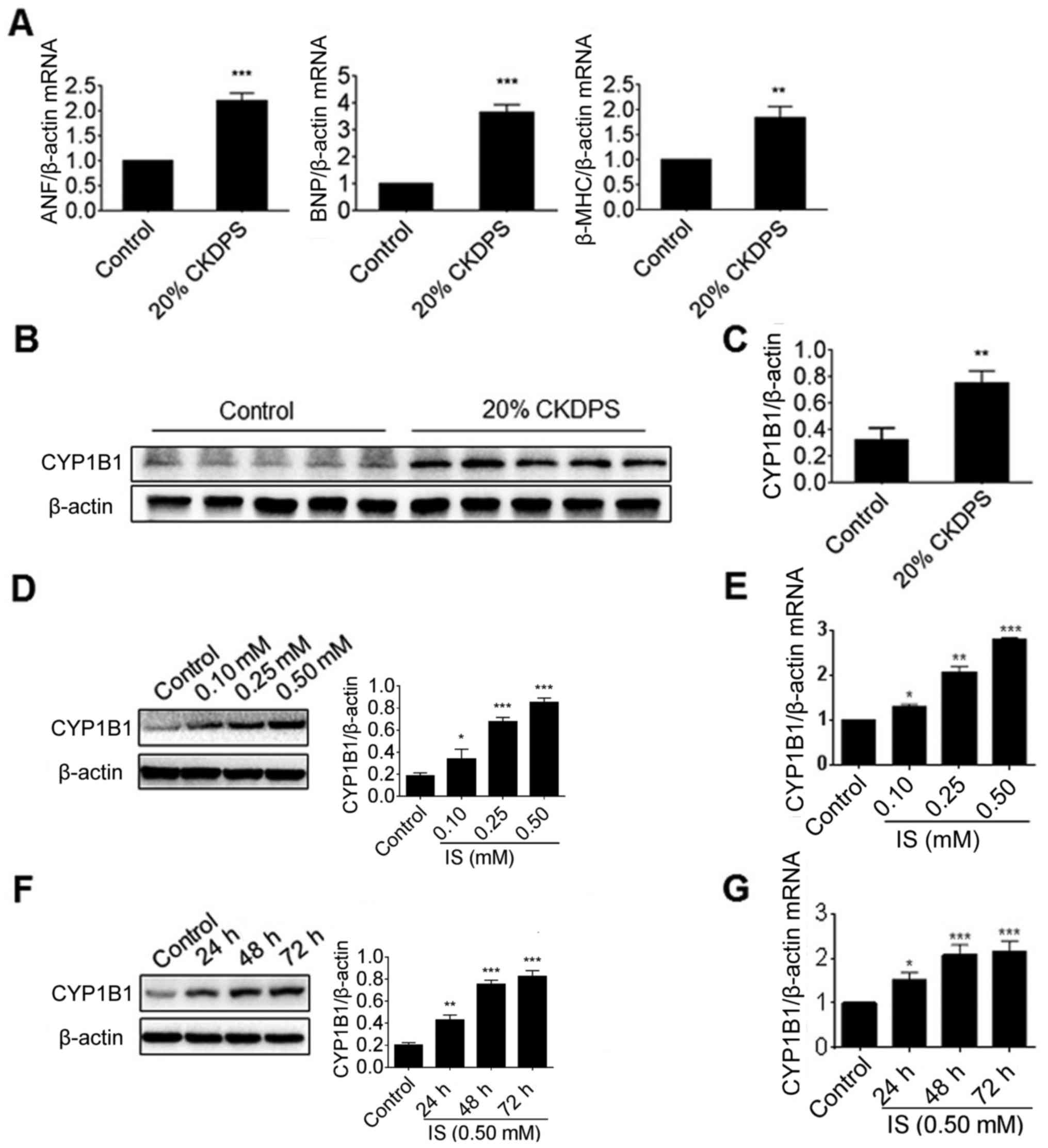
Induction of CYP1B1 by CKD serum and
IS in H9c2 cells
H9c2 cells were treated with serum from patients
with CKD in the present study. The effect of the serum on cardiac
hypertrophy markers, including ANF, BNP and β-MHC, was detected
using RT-qPCR. As presented in Fig.
1A, all these markers were significantly induced by CKD serum
when compared with that of the healthy controls (P<0.01). The
protein levels of CYP1B1 were then detected via western blot
analysis, and the results revealed that the CYP1B1 protein was
substantially upregulated by CKD serum compared with the control
(Fig. 1B). In order to further
determine whether the induction of CYP1B1 was at the translational
or transcriptional level, the mRNA level of CYP1B1 was detected via
RT-PCR. As presented in Fig. 1C,
the CYP1B1 mRNA levels in H9c2 cells treated with CKD serum were
significantly increased compared with the controls (P<0.01).
IS is the most abundant uremic toxin in CKD serum.
The present study further investigated its effect on gene
expression of CYP1B1. As the IS concentration in patients with CKD
was reported to vary from 7.6–656.0 µM (25), 500 µM IS was selected as the
maximal dose in the present study. Consistent with previous
results, all markers of myocardial hypertrophy were significantly
increased at an IS concentration of 0.25 or 0.50 mM compared with
the control (P<0.05), as determined by RT-PCR (Fig. S1A-B). In order to assess whether
IS may induce the gene expression of CYP1B1, H9c2 cells were also
treated with an increasing dose of IS. The results from the present
study revealed that the protein levels of CYP1B1 were significantly
upregulated by IS in a dose- and time-dependent manner (P<0.05;
Fig. 1D and E). In addition, the
mRNA levels of CYP1B1 were detected using RT-PCR. As presented in
Fig. 1F and G, the CYP1B1 mRNA
levels in IS-treated H9c2 cells were significantly increased
compared with the control (P<0.05), while also revealing a
similar dose- and time-dependent manner. These data indicated that
the gene expression of CYP1B1 in H9c2 cells was induced by the
uremic toxin.
Gene expression of CYP1B1 in CKD or
IS-treated mice
In order to further investigate the effect of the
uremic toxin on the gene expression of CYP1B1 in vivo, two
mouse models were applied. As the IS concentration in CKD mice has
previously been reported to be increased (26), a CKD model was established by 5/6
electrocoagulation and was applied in the present study. The other
model was established by direct intraperitoneal injections of IS.
The pathological changes of myocardial hypertrophy of mouse models
were confirmed by HE staining (Fig.
2A). In order to investigate the CYP1B1 mRNA level, total RNA
was isolated from the heart tissues and detected using RT-PCR. As
presented in Fig. 2B, the CYP1B1
mRNA levels were significantly increased in the CKD and IS groups
compared with the control (P<0.05). The heart tissues from these
groups were then assessed via western blot analysis and the results
revealed that the CYP1B1 protein levels were significantly
increased in the CKD and IS groups when compared with those in the
normal control group (P<0.05; Fig.
2C). Similarly, the mRNA levels of ANF, BNF and β-MHC were also
significantly increased in these two groups, compared with that in
controls (P<0.05; Fig. 2D). To
further confirm the gene expression of CYP1B1 in vivo,
sections from the heart tissues were detected using
immunochemistry. As presented in Fig.
2E, CYP1B1 was substantially increased in the CKD and IS groups
compared with the control, which was consistent with the results of
the western blot analysis. These data clearly demonstrated that the
expression of CYP1B1 was induced by IS in vivo.
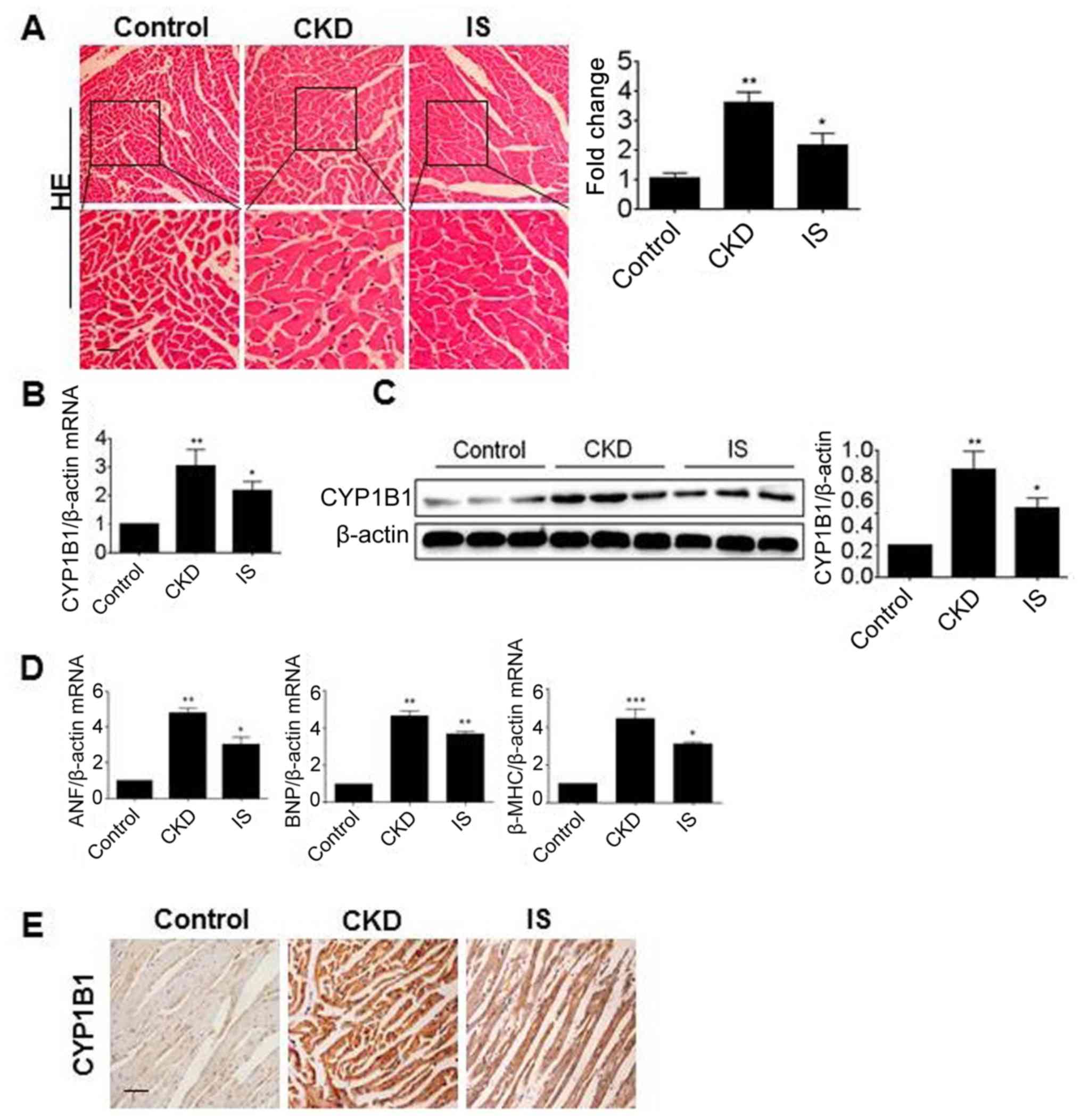 | Figure 2.Gene expression of CYP1B1 in CKD or
IS-treated mice. (A) Heart tissues from control, CKD and IS-treated
mouse were stained using HE. Scale bar, 200 µm. RNA and protein
samples were obtained from the heart tissues of control, CKD and
IS-treated mice, and CYP1B1 expression was detected using (B)
RT-PCR and (C) western blot analysis. (D) RNA samples were detected
using RT-PCR for ANF, BNP and β-MHC. (E) Paraffin sections from the
heart tissues were detected using immunohistochemistry with
antibodies against CYP1B1. *P<0.05, **P<0.01 and
***P<0.001 vs. the control. Scale bar, 100 µm. CKD, chronic
kidney disease; IS, indoxyl sulfate; CYP1B1, cytochrome P450 family
1 subfamily B member 1; HE, hematoxylin and eosin; RT-PCR, reverse
transcription-polymerase chain reaction; ANF, atrial natriuretic
factor; BNP, brain natriuretic peptide; β-MHC, β-myosin heavy
chain. |
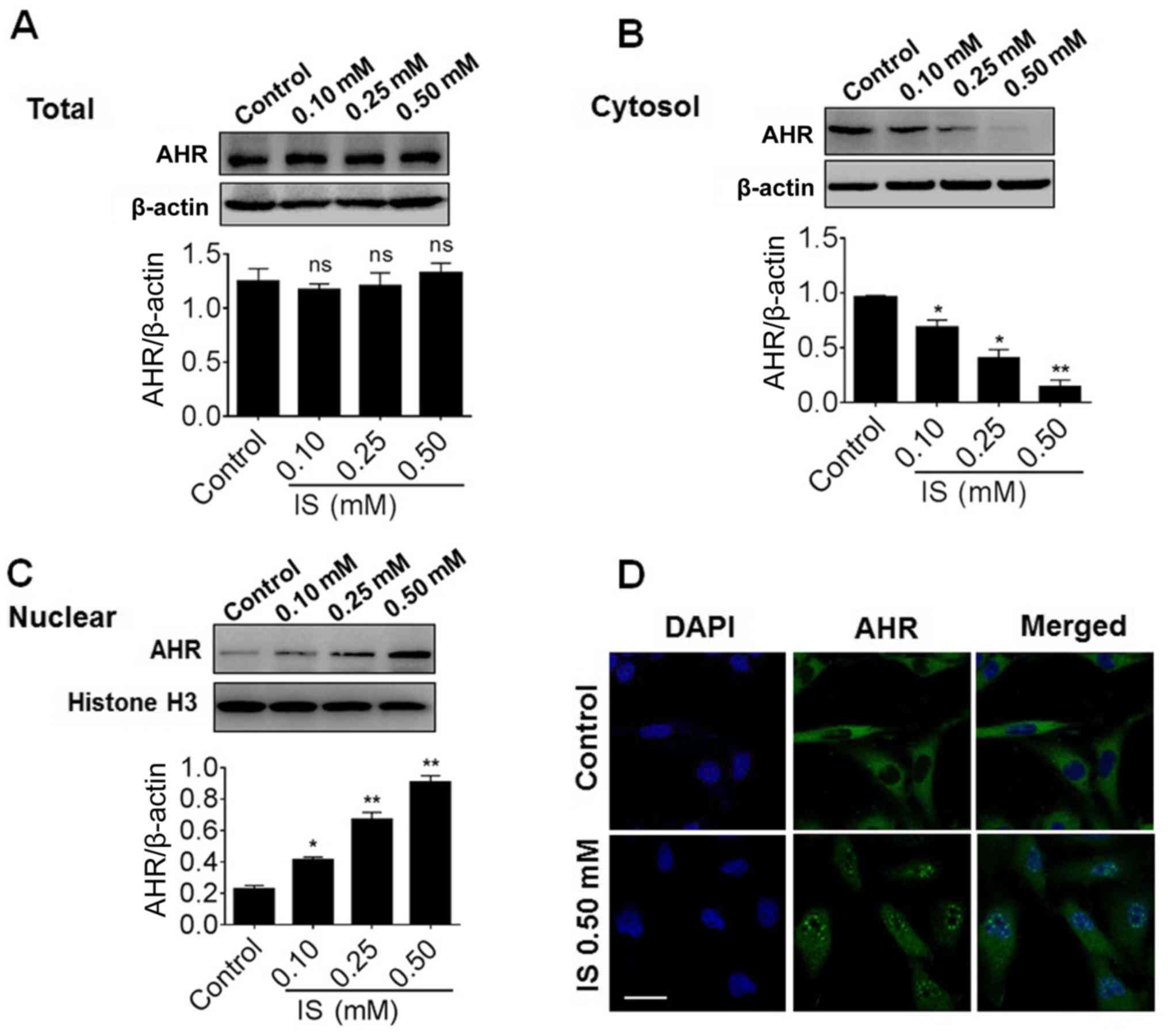
Nuclear translocation of AhR induced
by IS
The present study then investigated whether the
induction of CYP1B1 by IS is dependent on AhR. Thus, the effect of
IS on the protein expression of AhR in H9c2 cells was examined.
However, the total protein of AhR was not significantly altered by
IS treatment, even at the maximal dose of IS (Fig. 3A). Similarly, the total protein and
mRNA levels of AhR remained unchanged when the cells were treated
with CKD serum (Fig. S2). The
protein samples were further assessed to isolate the nuclear and
cytosol proportions. Notably, the cytosol AhR level was
significantly decreased compared with the control (P<0.05) while
the nuclear AhR level was significantly increased compared with the
control (P<0.05), indicating that nuclear translocation of AhR
occurs in a dose-dependent manner (Fig. 3B and C). In order to further
confirm this result, the cellular distribution of AhR was detected
using immunofluorescence. As presented in Fig. 3D, AhR was detected as smeared in
the cytosol in control cells. However, following the treatment of
IS, AhR was visible as dotted foci in the nucleus of the H9c2
cells, which indicated the marked nuclear translocation of AhR
induced by IS.
ChIP analysis of the binding of AhR in
the promoter region of CYP1B1
AhR is able to bind with the ARNT to form a
heterodimeric transcription factor, which is responsible for the
induction of a number of different targets (15). It was further investigated whether
AhR may directly bind to the promoter region of CYP1B1. Therefore,
ChIP-PCR was performed in order to analyze the location of AhR in
the promoter region (P1) and the intron 2 (I2) of CYP1B1 (Fig. 4A). Following IS treatment, H9c2
cells were immunoprecipitated with antibodies against AhR, IgG
(negative control) and histone H3 (positive control). The
immunoprecipitated DNA was detected via PCR and positive bands of
primer pair P1 were detected in the immunoprecipitated samples of
AhR and histone H3, but not in IgG (Fig. 4B, lower panel). Of note, the band
of primer pair I2 was only visible in immunoprecipitated samples of
histone H3, indicating the specific binding of AhR in the promoter
region (Fig. 4B, upper panel).
RT-qPCR was further applied to detect the enrichment of AhR. As
presented in Fig. 4C and D,
significantly increased amounts of AhR-binding DNA were detected in
IS-treated cells compared with the negative control (P<0.001).
Overall, the data in the present study clearly demonstrated that
AhR was able to bind with the promoter region of CYP1B1 and this
binding was enhanced by IS treatment.
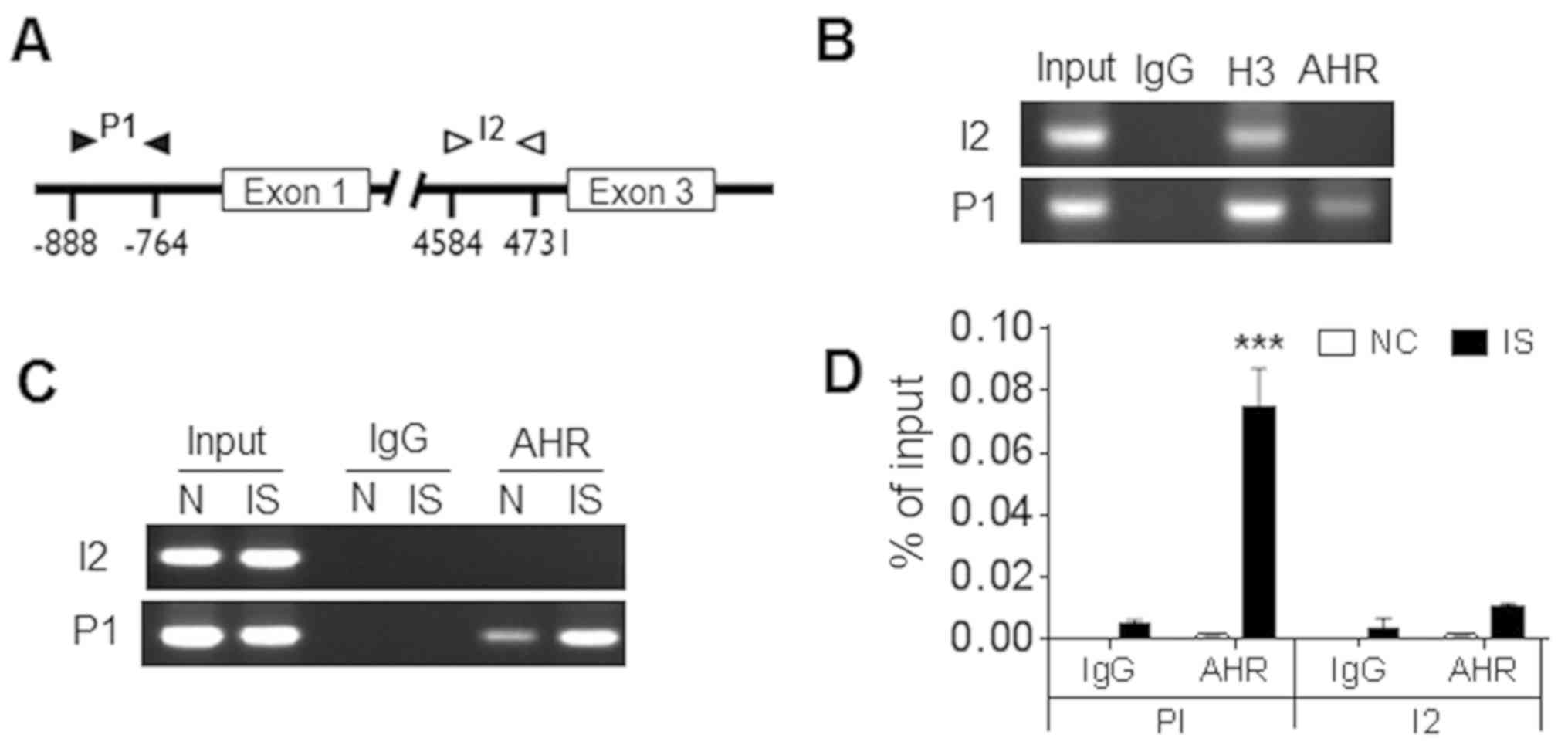 | Figure 4.ChIP analysis of the binding of AhR
in the promoter region of CYP1B1. (A) Diagram of the CYP1B1
promoter and the intron region. Black and white arrows indicate the
locations of the ChIP-PCR primers. The transcriptional start site
in the promoter region was considered to be +1. (B) IS-treated H9c2
cells and controls were harvested for ChIP analysis with antibodies
against AhR, histone H3 (positive control) or normal rabbit IgG
(negative control). The immunoprecipitated DNA and input DNA were
detected by routine PCR. The immunoprecipitated DNA and input DNA
were detected by (C) semi-quantitative PCR or reverse
transcription-PCR. (D) Data were displayed as the percentage of
input DNA. ***P<0.001 vs. the negative control. N, control;
ChIP, chromatin immunoprecipitation; AhR, aryl hydrocarbon
receptor; CYP1B1, cytochrome P450 family 1 subfamily B member 1;
PCR, polymerase chain reaction; IgG, immunoglobulin G; IS, indoxyl
sulfate; P1, promoter region; I2, intron 2. |
Effect of AhR knockdown on induction
of CYP1B1 by IS
To further prove that AhR is crucial for the
induction of CYP1B1 by IS, siRNAs of AhR were used to treat H9c2
cells. Compared with NC-siRNA, si-AhR-2 had the greatest
significant efficiency when inhibiting the expression of AhR and
was used in the subsequent experiments (P<0.001; Fig. 5A). H9c2 cells were pre-treated with
si-AhR or its control prior to IS treatment, and the gene
expression of CYP1B1 was detected. It was revealed that the
pretreatment of si-AhR significantly reversed the induction of
CYP1B1 protein and mRNA induced by IS (P<0.01; Fig. 5B and C). Accordingly, the
transcription of cardiac hypertrophy markers including ANF, BNF and
β-MHC was also reversed by si-AhR (P<0.05; Fig. 5D). Thus, AhR knockdown attenuated
the induction of CYP1B1 by IS, and prevented cardiac
hypertrophy.
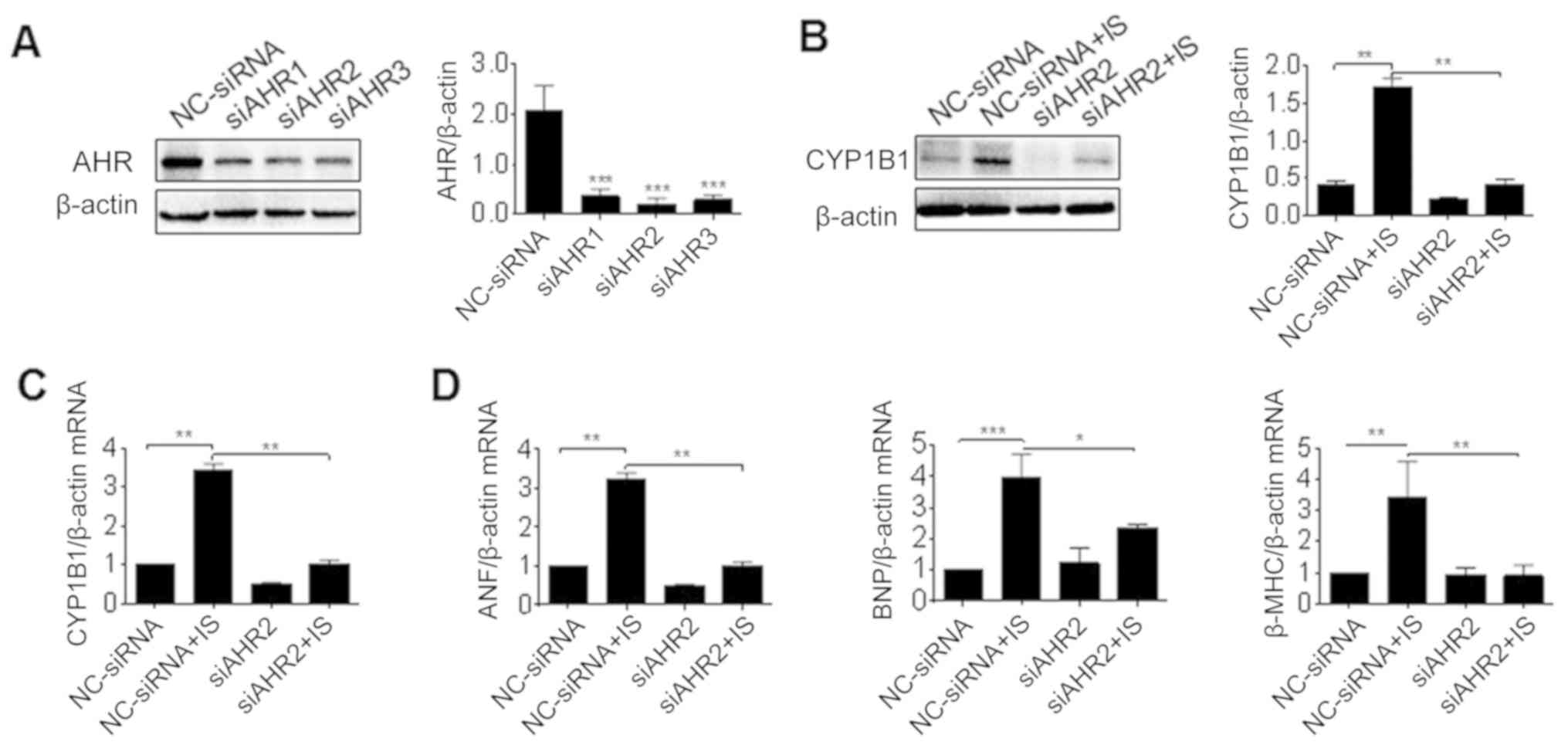 | Figure 5.Effect of AhR knockdown on the
induction of CYP1B1 by IS. (A) H9c2 cells were transfected with
siRNA against AhR (siAHR1-3) or control (NC-siRNA) for 2 days, and
protein samples were prepared for western blot analysis. H9c2 cells
were pretreated with siRNA and further incubated with IS media for
3 days, then total proteins and RNA were isolated for (B) western
blot analysis and (C) RT-PCR analysis of AhR expression. (D) RNA
samples were detected using RT-PCR analysis of ANF, BNP and β-MHC
expression. *P<0.05, **P<0.01 and ***P<0.001 with
comparisons shown by lines. AhR, aryl hydrocarbon receptor; CYP1B1,
cytochrome P450 family 1 subfamily B member 1; IS, indoxyl sulfate;
siRNA, small interfering RNA; NC, negative control; RT-PCR, reverse
transcription-polymerase chain reaction; ANF, atrial natriuretic
factor; BNP, brain natriuretic peptide; β-MHC, β-myosin heavy
chain. |
Effect of CYP1B1 knockdown on cardiac
hypertrophy induced by IS
In order to elucidate the association between CYP1B1
expression and cardiac hypertrophy, siRNAs of CYP1B1 were
synthesized and used to knockdown the expression of CYP1B1 in H9c2
cells. It was revealed that si-CYP1B1-2 was the optimum siRNA for
inhibiting the expression of CYP1B1, and was therefore used in
subsequent experiments (Fig. 6A).
Furthermore, the pretreatment of si-CYP1B1 significantly reversed
the induction of CYP1B1 protein induced by IS (P<0.01; Fig. 6B). In order to gain an improved
understanding of this association, H9c2 cells were pretreated with
si-CYP1B1 prior to IS treatment, and the gene expression of ANF,
BNF and β-MHC was detected via RT-PCR. As presented in Fig. 6C, the induction of all these
cardiac hypertrophy markers was significantly reversed by
pretreatment with si-CYP1B1 (P<0.01; Fig. 6C). In addition, the cell size was
determined by immunofluorescence staining using α-actinin
antibodies. The results revealed that the cell size induced by IS
treatment was significantly inhibited by si-CYP1B1 (P<0.01;
Fig. 6D). This suggested that
CYP1B1 knockdown inhibited cardiac hypertrophy.
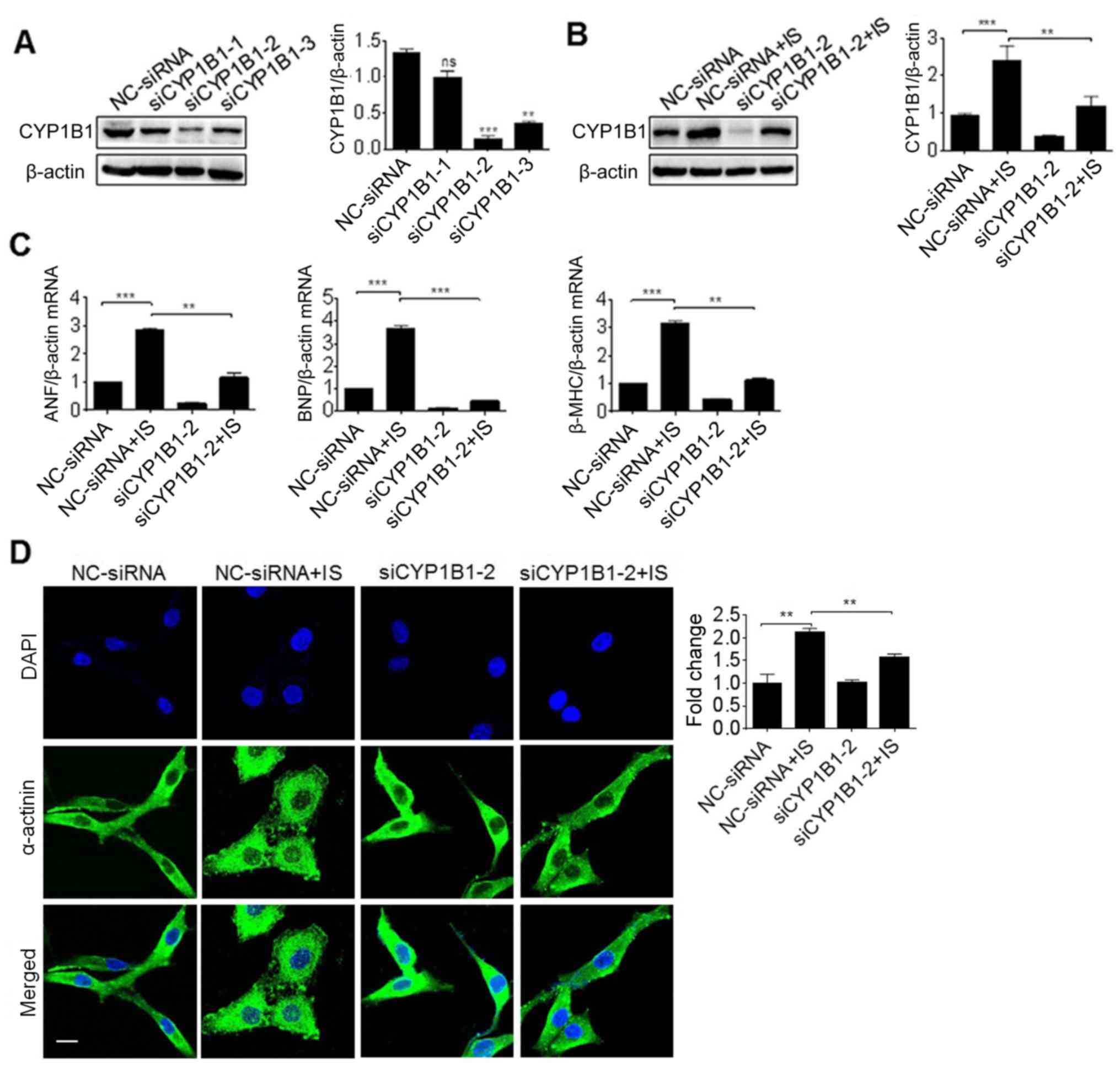 | Figure 6.Effect of CYP1B1 knockdown on cardiac
hypertrophy induced by IS. (A) H9c2 cells were transfected with
siRNA against CYP1B1 (siCYP1B1-1~3) or control (NC-siRNA) for 2
days, and protein samples were then prepared for the western blot
analysis. H9c2 cells were pretreated with siRNA and further
incubated with IS media for 3 days, then total proteins and RNA
were subjected to (B) the western blot analysis and (C) reverse
transcription-polymerase chain reaction analysis of ANF, BNP and
β-MHC expression. (D) Similar treated H9c2 cells on slides were
detected by immunofluorescence with antibodies against of
α-actinin. **P<0.01 and ***P<0.001 with comparisons shown by
lines. Scale bar, 50 µm. ns, not significant; CYP1B1, cytochrome
P450 family 1 subfamily B member 1; IS, indoxyl sulfate; siRNA,
small interfering RNA; NC, negative control; ANF, atrial
natriuretic factor; BNP, brain natriuretic peptide; β-MHC, β-myosin
heavy chain. |
Effect of CYP1B1 inhibition in
vivo
Finally, in order to investigate the function of
CYP1B1 inhibition in vivo, a specific inhibitor of CYP1B1
was combined with IS for use in the mouse model. Previous studies
(21,27) have reported TMS as a selective
inhibitor of CYP1B1, and the dosage in vivo was selected
according to these studies. Following 2 months of treatment, the
thickness of the ventricular wall and the relative heart weight in
the IS group were significantly increased, compared with that in
the controls (P<0.01). However, these parameters were
significantly ameliorated by TMS treatment (P<0.05; Fig. 7A and B). Furthermore, the results
of the HE staining revealed that the decreased length of the left
ventricular midchamber and the increased cell size induced by IS
were also substantially ameliorated by TMS treatment (Fig. 7C). In addition, the increased mRNA
levels of cardiac hypertrophy markers induced by IS were
significantly reversed by TMS treatment (P<0.001; Fig. 7D). These results clearly indicate
that the inhibition of CYP1B1 ameliorated cardiac hypertrophy
induced by IS.
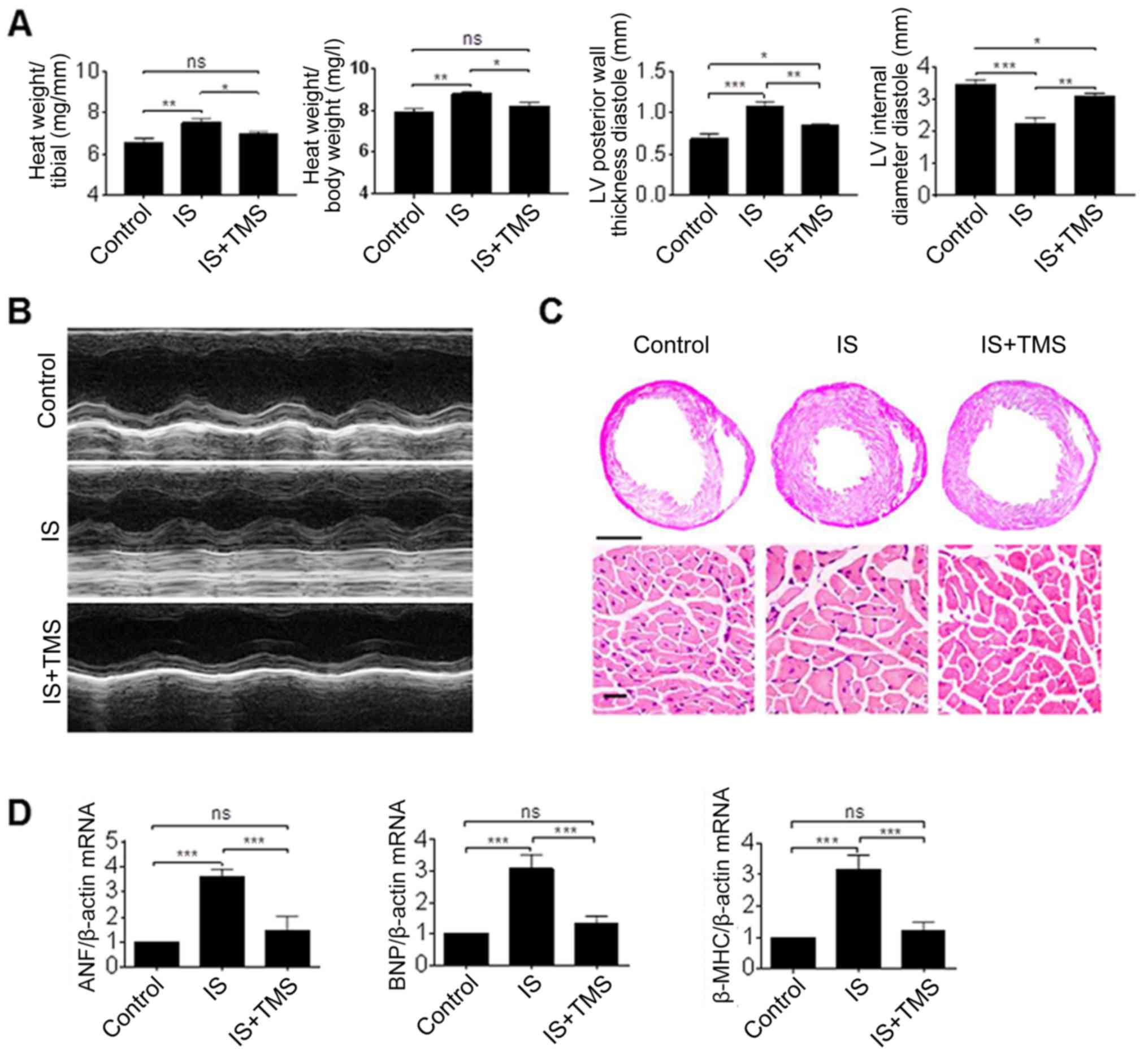 | Figure 7.Effect of CYP1B1 inhibitor on cardiac
hypertrophy in vivo. (A) Mice were treated with IS or/and
CYP1B1 inhibitor (TMS) for 8 weeks and (B) the LV hypertrophy was
determined by echocardiography. (C) Gross pathology of the
midchamber sections of the heart was stained using hematoxylin and
eosin. Scale bar, 200 mm in the upper panel and 50 µm in the lower
panel. (D) Gene expression of the cardiac hypertrophy markers in
mouse heart tissues were detected using a reverse
transcription-polymerase chain reaction. *P<0.05, **P<0.01
and ***P<0.001 with comparisons shown by lines. ns, not
significant; CYP1B1, cytochrome P450 family 1 subfamily B member 1;
IS, indoxyl sulfate; ANF, atrial natriuretic factor; BNP, brain
natriuretic peptide; β-MHC, β-myosin heavy chain; LV, left
ventricular; TMS, 2,4,3′,5′-tetramethoxystilbene. |
Discussion
CYP1B1 is a member of the CYP1 subfamily, and also
the most abundantly expressed CYP gene in human hearts (28). It has been reported that CYP1B1
contributes to the development of hypertension and renal
dysfunction in male mice (29).
Furthermore, mutations in this gene have been associated with
primary congenital glaucoma (30).
As a monooxygenase, it catalyzes a number of reactions involved in
drug metabolism and the synthesis of cholesterol, steroids and
other lipids (31). This enzyme
has been reported to metabolize procarcinogens, including
polycyclic aromatic hydrocarbons and 17β-estradiol (21). The functions of CYP1B1 have been
extensively studied in cancer, due to its bioactivity and
overexpression in a variety of different types of human tumor
(32). Furthermore, the
association between CYP1B1 polymorphisms and cancer has been
extensively studied. For example, an association between the
CYP1B1-rs1056836 genetic polymorphism and clinical features of
esophageal squamous cell carcinoma was identified in an Iranian
Mashhad cohort study (33). In
addition, a single nucleotide polymorphism in CYP1B1 resulted in
differential prostate cancer risk and telomere length (34). As it metabolizes AA to mid-chain
HETEs with vascular deleterious effects, studies have also
demonstrated that there is an association between CYP1B1 and
cardiac diseases. Previous studies have demonstrated that CYP1B1
was significantly induced in the left ventricular tissue of
spontaneously hypertensive rats and in drug-induced hypertrophied
hearts (35–37). In the present study, it was
revealed that CYP1B1 expression may be efficiently induced by IS
and CKD serum via the AhR pathway. In addition, it was demonstrated
that CYP1B1 knockdown or inhibition with TMS may reverse cardiac
hypertrophy induced by IS in in vitro and in vivo
studies. These results further confirmed the association between
CYP1B1 and cardiac diseases.
Lower estimated glomerular filtration rate (eGFR)
has been accepted as an independent risk factor for CVD (38). It is well known that various
metabolites, or so-called uremic toxins, are detained in the body
as the GFR declines. A number of these have been confirmed as
having cardiovascular toxicity, including IS. As a typical
protein-bound uremic toxin, IS is a tryptophan-derived uremic toxin
(8,39,40).
Tryptophan, an essential amino acid in the diet, may be metabolized
by tryptophanase to indole in the gut where IS is absorbed, and
further converted into IS in the liver (41). A gradual rise in serum IS was
observed in CKD development from the earliest stages of the
disease, and higher serum IS levels predicted higher overall and
cardiovascular mortality risk, which was independent of age, sex,
diabetes mellitus, phosphate, albumin and hemoglobin levels,
vascular stiffness and aortic calcification (42). In the present study, the
concentration of serum IS from patients with CDK varied from
125.8–356.1 µM (data not shown), which was markedly higher compared
with the normal value. Previous studies indicated that the serum
levels of IS were independently associated with LVH, and IS-induced
cardiomyocyte hypertrophy was identified in vitro and was
further confirmed by the intraperitoneal injection of IS in mice
(9), which is consistent with
other research results (43,44).
Furthermore, IS was demonstrated to induce increased ROS
production, which inhibited the p38, extracellular signal-regulated
kinase 1/2 and AMPK/UCP2 signaling pathway and then promote LVH
development (10). But the
signaling pathways associated with IS exposure have not yet been
fully understood.
Previous studies have demonstrated that IS is a
human AhR ligand and a potent activator of its transcriptional
activity, and that there is an association between the deleterious
effects of IS and AhR activation (45–47).
Expression of AhR is relatively high in the lungs, placenta,
spleen, pancreas and liver, and relatively low in the heart, brain
and skeletal muscles in adults (48). Despite the low expression level of
AhR in the heart, AhR does have noticeable effects on the
physiological functioning of the heart. It exists as an AhR
molecular chaperone complex comprising an AhR, two heat shock
protein 90 and X-associated protein 2 and 23 in the cytosol
(49,50). Following binding with ligands, AhR
translocates from the cytosol to the nucleus where it disassociates
from the complex. In the present study, the nuclear translocation
of AhR induced by IS was demonstrated via western blot analysis and
an immunofluorescence assay (Fig.
3), which was consistent with previous studies. Subsequently,
the ligand-AhR complex combines with ARNT and binds to DRE or XRE,
promoting the transcription of a large number of target genes
activating and triggering a number of biological or toxicological
effects. In fact, other uremic toxins, including indole-3-acetic
acid (IAA) and p-cresyl sulfate, are also thought to be potent
activators of AhR transcriptional activity under uremic conditions
(51). Likewise, IAA was reported
to induce endothelial inflammation and oxidative stress, and
activate the inflammatory AhR/p38 mitogen-activated protein
kinase/nuclear factor-κB pathway (52). In using the ChIP analysis in the
present study, it was revealed that AhR may bind with the promoter
region of CYP1B1, but not with the intron (Fig. 4). This suggested that AhR
activation may occur in patients with CKD.
In addition to the classical signaling pathway, AhR
may also interact with other pathways by competing for
transcriptional co-activators or co-repressors. The present study
focused on the AhR-CYP1B1 pathway. Other non-classical AhR
signaling pathways may also be associated with cardiac hypertrophy
induced by IS. Notably, the function of AhR in cardiac hypertrophy
is controversial, as marked cardiac hypertrophy was observed in
AhR-deficient mice and the underlying molecular mechanism may be
associated with an elevated level of vascular endothelial growth
factor in AhR-deficient mice (53,54).
These studies suggest that the effects of AhR on the cardiovascular
system are greater than expected. One explanation may be that the
AhR deficiency model is not suitable for analyzing its
pathophysiological function, as AhR is vital for the development of
the heart (55). Although there
are contradictions among currently available studies, the functions
of AhR signaling in cardiac hypertrophy are important.
A previous study has indicated that CYP1B1
polymorphisms are closely associated with cardiac disease; the
association between two common CYP1B1 polymorphisms, CYP1B1*3 and
CYP1B1*4, and the risk of CVD was reported in a large population of
50,000 individuals (56). However,
to the best of our knowledge, there are currently no reports
regarding the effects of these polymorphisms on AA metabolism. One
of the reasons that the induction of CYP1B1 results in cardiac
hypertrophy is that it may give rise to a higher level of
deleterious metabolites. Previous studies have demonstrated that
CYP1B1 is able to metabolize AA to mid-chain HETEs, of which the
vascular deleterious effects have been extensively investigated
(17,57,58).
For example, the increased levels of mid-chain HETE metabolism were
associated with increased CYP1B1 expression in a rat model of
pressure overload cardiac hypertrophy (59). Although the present study did not
assess HETE metabolism, it is rational to speculate that these
deleterious metabolisms are involved in cardiac hypertrophy induced
by IS, which will be investigated in future studies. Another reason
may be that CYP1B1 may generate a higher level of superoxide
radicals. It has previously been reported that CYP1B1 served an
important function in the activation of NADPH oxidase, which was
thought to generate ROS. This was evident by the result that CYP1B1
siRNA inhibited AA-induced increases in NADPH oxidase activity
(60). A previous study also
demonstrated that IS may induce ROS production. Thus it is
believable that CYP1B1 contributes to the generation of ROS.
One limitation of present study is that cardiac
hypertrophy was not measured in these patients. The main purpose of
the present study was to assess the biological effects of CKD serum
on cell culture, including cell proliferation, apoptosis and its
associated gene expression. However, CKD is recognized as a strong
and independent risk factor for developing CVD, which has been
confirmed in numerous studies (3,5,61).
Therefore, the present study did not measure cardiac
hypertrophy.
In summary, the present study revealed that the
expression of the CYP1B1 gene was induced by CKD serum and IS,
which was associated with the AhR pathway. CYP1B1 was involved in
cardiac hypertrophy under uremic conditions, which may be a
potential therapy target for cardiovascular disease among patients
with CKD. Thus, the present study provided evidence to support
another molecular mechanism underlying cardiac hypertrophy induced
by the uremic toxin.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Key
R&D Program of China (grant nos. 2018YFC1312700 and
2017YFA0106600), the Natural Science Foundation of China (grant
nos. 81873605, 81800468, 81700379, 81400747 and 81800616), the
Personal Training Program for Clinical Medicine Research of Army
Medical University (grant no. 2018XLC1007) and the Frontier
specific projects of Xinqiao Hospital (grant no. 2018YQYLY004).
Availability of data and materials
All data generated or analyzed during the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
LN, JZ and BZ designed the study. YZ, SW, YH, KY and
YL performed the experiments. XB, CL and JX analyzed and
interpreted the data. JZ made substantial contributions to the
design and supervision of the present study. LN wrote the
manuscript. All authors reviewed the results and approved the final
version of the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Xinqiao Hospital, Army Medical University, and written
informed consent was obtained from the patients. The animal
protocol was approved by the Animal Care and Use Committee of Army
Medical University.
Patient consent for publication
Written informed consent for publication was
obtained from enrolled patients.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chen SC, Huang JC, Su HM, Chiu YW, Chang
JM, Hwang SJ and Chen HC: Prognostic cardiovascular markers in
chronic kidney disease. Kidney Blood Press Res. 43:1388–1407. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cunningham MW Jr and LaMarca B: Risk of
cardiovascular disease, end-stage renal disease, and stroke in
postpartum women and their fetuses after a hypertensive pregnancy.
Am J Physiol Regul Integr Comp Physiol. 315:R521–R528. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shah SR and Winchester DE: The impact of
chronic kidney disease on medication choice and pharmacologic
management in patients with heart failure. Expert Rev Clin
Pharmacol. 11:571–579. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Siedlecki AM, Jin X and Muslin AJ: Uremic
cardiac hypertrophy is reversed by rapamycin but not by lowering of
blood pressure. Kidney Int. 75:800–808. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sethna CB, Merchant K and Reyes A:
Cardiovascular disease risk in children with kidney disease. Semin
Nephrol. 38:298–313. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim H, Yoo TH, Choi KH, Oh KH, Lee J, Kim
SW, Kim TH, Sung S and Han SH; KNOW-CKD Group, : Baseline
cardiovascular characteristics of adult patients with chronic
kidney disease from the korean cohort study for outcomes in
patients with chronic kidney disease (KNOW-CKD). J Korean Med Sci.
32:231–239. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lash JP, Go AS, Appel LJ, He J, Ojo A,
Rahman M, Townsend RR, Xie D, Cifelli D, Cohan J, et al: Chronic
Renal Insufficiency Cohort (CRIC) Study: Baseline characteristics
and associations with kidney function. Clin J Am Soc Nephrol.
4:1302–1311. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gao H and Liu S: Role of uremic toxin
indoxyl sulfate in the progression of cardiovascular disease. Life
Sci. 185:23–29. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang K, Wang C, Nie L, Zhao X, Gu J, Guan
X, Wang S, Xiao T, Xu X, He T, et al: Klotho protects against
indoxyl sulphate-induced myocardial hypertrophy. J Am Soc Nephrol.
26:2434–2446. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang K, Xu X, Nie L, Xiao T, Guan X, He T,
Yu Y, Liu L, Huang Y, Zhang J and Zhao J: Indoxyl sulfate induces
oxidative stress and hypertrophy in cardiomyocytes by inhibiting
the AMPK/UCP2 signaling pathway. Toxicol Lett. 234:110–119. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Six I, Gross P, Rémond MC, Chillon JM,
Poirot S, Drueke TB and Massy ZA: Deleterious vascular effects of
indoxyl sulfate and reversal by oral adsorbent AST-120.
Atherosclerosis. 243:248–256. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yoshifuji A, Wakino S, Irie J, Matsui A,
Hasegawa K, Tokuyama H, Hayashi K and Itoh H: Oral adsorbent
AST-120 ameliorates gut environment and protects against the
progression of renal impairment in CKD rats. Clin Exp Nephrol.
22:1069–1078. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kamiński T, Michałowska M and Pawlak D:
Aryl hydrocarbon receptor (AhR) and its endogenous agonist-indoxyl
sulfate in chronic kidney disease. Postepy Hig Med Dosw (Online).
71:624–632. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wheeler MA, Rothhammer V and Quintana FJ:
Control of immune-mediated pathology via the aryl hydrocarbon
receptor. J Biol Chem. 292:12383–12389. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Al-Dhfyan A, Alhoshani A and Korashy HM:
Aryl hydrocarbon receptor/cytochrome P450 1A1 pathway mediates
breast cancer stem cells expansion through PTEN inhibition and
β-Catenin and Akt activation. Mol Cancer. 16:142017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Korashy HM and El-Kadi AO: The role of
aryl hydrocarbon receptor in the pathogenesis of cardiovascular
diseases. Drug Metab Rev. 38:411–450. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
El-Sherbeni AA and El-Kadi AO: Repurposing
resveratrol and Fluconazole to modulate human cytochrome
P450-mediated arachidonic acid metabolism. Mol Pharm. 13:1278–1288.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yaghini FA, Song CY, Lavrentyev EN,
Ghafoor HU, Fang XR, Estes AM, Campbell WB and Malik KU:
Angiotensin II-induced vascular smooth muscle cell migration and
growth are mediated by cytochrome P450 1B1-dependent superoxide
generation. Hypertension. 55:1461–1467. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Althurwi HN, Tse MM, Abdelhamid G, Zordoky
BN, Hammock BD and El-Kadi AO: Soluble epoxide hydrolase inhibitor,
TUPS, protects against isoprenaline-induced cardiac hypertrophy. Br
J Pharmacol. 168:1794–1807. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Elkhatali S, Maayah Z, El-Sherbeni AA,
Elshenawy OH, Abdelhamid G, Shoieb SM and El-Kadi AOS: Inhibition
of Mid-chain HETEs protects against angiotensin II-induced cardiac
hypertrophy. J Cardiovasc Pharmacol. 70:16–24. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Maayah ZH, Althurwi HN, El-Sherbeni AA,
Abdelhamid G, Siraki AG and El-Kadi AO: The role of cytochrome P450
1B1 and its associated mid-chain hydroxyeicosatetraenoic acid
metabolites in the development of cardiac hypertrophy induced by
isoproterenol. Mol Cell Biochem. 429:151–165. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gondouin B, Cerini C, Dou L, Sallée M,
Duval-Sabatier A, Pletinck A, Calaf R, Lacroix R, Jourde-Chiche N,
Poitevin S, et al: Indolic uremic solutes increase tissue factor
production in endothelial cells by the aryl hydrocarbon receptor
pathway. Kidney Int. 84:733–744. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pang X, He G, Luo C, Wang Y and Zhang B:
Knockdown of Rad9A enhanced DNA damage induced by trichostatin A in
esophageal cancer cells. Tumour Biol. 37:963–970. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Deltombe O, Van Biesen W, Glorieux G,
Massy Z, Dhondt A and Eloot S: Exploring protein binding of uremic
toxins in patients with different stages of chronic kidney disease
and during hemodialysis. Toxins (Basel). 7:3933–3946. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Adesso S, Popolo A, Bianco G, Sorrentino
R, Pinto A, Autore G and Marzocco S: The uremic toxin indoxyl
sulphate enhances macrophage response to LPS. PLoS One.
8:e767782013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jennings BL, Montanez DE, May ME Jr, Estes
AM, Fang XR, Yaghini FA, Kanu A and Malik KU: Cytochrome P450 1B1
contributes to increased blood pressure and cardiovascular and
renal dysfunction in spontaneously hypertensive rats. Cardiovasc
Drugs Ther. 28:145–161. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jennings BL, George LW, Pingili AK, Khan
NS, Estes AM, Fang XR, Gonzalez FJ and Malik KU: Estrogen
metabolism by cytochrome P450 1B1 modulates the hypertensive effect
of angiotensin II in female mice. Hypertension. 64:134–140. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pingili AK, Thirunavukkarasu S, Kara M,
Brand DD, Katsurada A, Majid DS, Navar LG, Gonzalez FJ and Malik
KU: 6β-hydroxytestosterone, a cytochrome P450
1B1-testosterone-metabolite, mediates angiotensin II-induced renal
dysfunction in male mice. Hypertension. 67:916–926. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jennings BL, Anderson LJ, Estes AM, Fang
XR, Song CY, Campbell WB and Malik KU: Involvement of cytochrome
P-450 1B1 in renal dysfunction, injury, and inflammation associated
with angiotensin II-induced hypertension in rats. Am J Physiol
Renal Physiol. 302:F408–F420. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li F, Zhu W and Gonzalez FJ: Potential
role of CYP1B1 in the development and treatment of metabolic
diseases. Pharmacol Ther. 178:18–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
D'Uva G, Baci D, Albini A and Noonan DM:
Cancer chemoprevention revisited: Cytochrome P450 family 1B1 as a
target in the tumor and the microenvironment. Cancer Treat Rev.
63:1–18. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Moghadam AR, Mehramiz M, Entezari M,
Aboutalebi H, Kohansal F, Dadjoo P, Fiuji H, Nasiri M, Aledavood
SA, Anvari K, et al: A genetic polymorphism in the CYP1B1 gene in
patients with squamous cell carcinoma of the esophagus: An Iranian
Mashhad cohort study recruited over 10 years. Pharmacogenomics.
19:539–546. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gu CY, Li GX, Zhu Y, Xu H, Zhu Y, Qin XJ,
Bo D and Ye DW: A single nucleotide polymorphism in CYP1B1 leads to
differential prostate cancer risk and telomere length. J Cancer.
9:269–274. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tse MM, Aboutabl ME, Althurwi HN,
Elshenawy OH, Abdelhamid G and El-Kadi AO: Cytochrome P450
epoxygenase metabolite, 14,15-EET, protects against
soproterenol-induced cellular hypertrophy in H9c2 rat cell line.
Vasc Pharmacol. 58:363–373. 2013. View Article : Google Scholar
|
|
36
|
Thum T and Borlak J: Testosterone,
cytochrome P450, and cardiac hypertrophy. FASEB J. 16:1537–1549.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Maayah ZH and El-Kadi AO: The role of
mid-chain hydroxyeicosatetraenoic acids in the pathogenesis of
hypertension and cardiac hypertrophy. Arch Toxicol. 90:119–136.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Evans M, Grams ME, Sang Y, Astor BC,
Blankestijn PJ, Brunskill NJ, Collins JF, Kalra PA, Kovesdy CP,
Levin A, et al: Risk Factors for Prognosis in Patients With
Severely Decreased GFR. Kidney Int Rep. 3:625–637. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Savira F, Magaye R, Hua Y, Liew D, Kaye D,
Marwick T and Wang BH: Molecular mechanisms of protein-bound uremic
toxin-mediated cardiac, renal and vascular effects: Underpinning
intracellular targets for cardiorenal syndrome therapy. Toxicol
Lett. 308:34–49. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Guo J, Lu L, Hua Y, Huang K, Wang I, Huang
L, Fu Q, Chen A, Chan P, Fan H, et al: Vasculopathy in the setting
of cardiorenal syndrome: Roles of protein-bound uremic toxins. Am J
Physiol Heart Circ Physiol. 313:H1–H13. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Niwa T: Uremic toxicity of indoxyl
sulfate. Nagoya J Med Sci. 72:1–11. 2010.PubMed/NCBI
|
|
42
|
Barreto FC, Barreto DV, Liabeuf S, Meert
N, Glorieux G, Temmar M, Choukroun G, Vanholder R and Massy ZA:
European Uremic Toxin Work Group (EUTox): Serum indoxyl sulfate is
associated with vascular disease and mortality in chronic kidney
disease patients. Clin J Am Soc Nephrol. 4:1551–1558. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lekawanvijit S, Adrahtas A, Kelly DJ,
Kompa AR, Wang BH and Krum H: Does indoxyl sulfate, a uraemic
toxin, have direct effects on cardiac fibroblasts and myocytes? Eur
Heart J. 31:1771–1779. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fujii H, Nishijima F, Goto S, Sugano M,
Yamato H, Kitazawa R, Kitazawa S and Fukagawa M: Oral charcoal
adsorbent (AST-120) prevents progression of cardiac damage in
chronic kidney disease through suppression of oxidative stress.
Nephrol Dial Transplant. 24:2089–2095. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Brito JS, Borges NA, Esgalhado M, Magliano
DC, Soulage CO and Mafra D: Aryl hydrocarbon receptor activation in
chronic kidney disease: Role of Uremic Toxins. Nephron. 137:1–7.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Asai H, Hirata J and Watanabe-Akanuma M:
Indoxyl glucuronide, a protein-bound uremic toxin, inhibits
hypoxia-inducible factor-dependent erythropoietin expression
through activation of aryl hydrocarbon receptor. Biochem Biophys
Res Commun. 504:538–544. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ito S, Osaka M, Edamatsu T, Itoh Y and
Yoshida M: Crucial role of the aryl hydrocarbon receptor (AhR) in
indoxyl sulfate-induced vascular inflammation. J Atheroscler
Thromb. 23:960–975. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Barisione C, Ghigliotti G, Canepa M, Balbi
M, Brunelli C and Ameri P: Indoxyl sulfate: A candidate target for
the prevention and treatment of cardiovascular disease in chronic
kidney disease. Curr Drug Targets. 16:366–372. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Esser C and Rannug A: The aryl hydrocarbon
receptor in barrier organ physiology, immunology, and toxicology.
Pharmacol Rev. 67:259–279. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang N and Walker MK: Crosstalk between
the aryl hydrocarbon receptor and hypoxia on the constitutive
expression of cytochrome P4501A1 mRNA. Cardiovasc Toxicol.
7:282–290. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ramezan A, Massy ZA, Meijers B, Evenepoel
P, Vanholder R and Raj D: Role of the gut microbiome in uremia: A
potential therapeutic target. Am J Kidney Dis. 67:483–498. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Dou L, Sallée M, Cerini C, Poitevin S,
Gondouin B, Jourde-Chiche N, Fallague K, Brunet P, Calaf R, Dussol
B, et al: The cardiovascular effect of the uremic solute indole-3
acetic acid. J Am Soc Nephrol. 26:876–887. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lund AK, Goens MB, Nuñez BA and Walker MK:
Characterizing the role of endothelin-1 in the progression of
cardiac hypertrophy in aryl hydrocarbon receptor (AhR) null mice.
Toxicol Appl Pharmacol. 212:127–135. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Fernandez-Salguero PM, Ward JM, Sundberg
JP and Gonzalez FJ: Lesions of aryl-hydrocarbon receptor-deficient
mice. Vet Pathol. 34:605–614. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wang Q, Chen J, Ko CI, Fan Y, Carreira V,
Chen Y, Xia Y, Medvedovic M and Puga A: Disruption of aryl
hydrocarbon receptor homeostatic levels during embryonic stem cell
differentiation alters expression of homeobox transcription factors
that control cardiomyogenesis. Environ Health Perspect.
121:1334–1343. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zordoky BN and El-Kadi AO: Effect of
cytochrome P450 polymorphism on arachidonic acid metabolism and
their impact on cardiovascular diseases. Pharmacol Ther.
125:446–463. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Amara IE, Elshenawy OH, Abdelrady M and
El-Kadi AO: Acute mercury toxicity modulates cytochrome P450,
soluble epoxide hydrolase and their associated arachidonic acid
metabolites in C57Bl/6 mouse heart. Toxicol Lett. 226:53–62. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kaur-Knudsen D, Bojesen SE and
Nordestgaard BG: Cytochrome P450 1B1 and 2C9 genotypes and risk of
ischemic vascular disease, cancer, and chronic obstructive
pulmonary disease. Curr Vasc Pharmacol. 10:512–520. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
El-Sherbeni AA and El-Kadi AO: Alterations
in cytochrome P450-derived arachidonic acid metabolism during
pressure overload-induced cardiac hypertrophy. Biochem Pharmacol.
87:456–466. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Malik KU, Jennings BL, Yaghini FA,
Sahan-Firat S, Song CY, Estes AM and Fang XR: Contribution of
cytochrome P450 1B1 to hypertension and associated pathophysiology:
A novel target for antihypertensive agents. Prostaglandins Other
Lipid Mediat. 98:69–74. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ravarotto V, Simioni F, Pagnin E, Davis PA
and Calò LA: Oxidative stress-chronic kidney disease-cardiovascular
disease: A vicious circle. Life Sci. 210:125–131. 2018. View Article : Google Scholar : PubMed/NCBI
|