Introduction
Human papillomavirus (HPV) infection is one of the
main causes of infection-related cancer in both men and women
(1). Among >200 types of HPVs,
high-risk HPV (HR-HPV-16, −18, −31, −33, −35, −39, −45, −51, −52,
−56, −58 and −59) infections are responsible for almost all
cervical cancer cases and there is growing evidence that they can
cause other anogenital cancers, including anal, vulval, vaginal and
penile, and head and neck cancers (1,2).
However, only a relatively small number of lesions associated with
HR-HPV infections evolve into high-grade lesions or cancer. In
addition to genetic and immunological factors, viral factors, such
as HR-HPV variants, viral load and viral integration, can increase
the risk of viral persistence and influence progression to cancer
(3,4). In particular, it is important to
monitor the genetic variability of HR-HPVs over time in order to
estimate the clinical course of HR-HPV infections, predict the
prognosis of benign and malignant lesions and define treatment
strategies, as genetic diversity can influence the long-term
efficacy of current HPV vaccines.
The α-HPV types have a circular, double-stranded DNA
genome of ~7,900 bp consisting of eight protein-coding genes (L1,
L2, E1, E2, E4, E5, E6 and E7), a non-coding region (NCR) and a
long control region (LCR) (5). The
LCR of HPVs contain the highest degree of genomic diversity and is
usually used to classify HPV variants into lineages and
sublineages, previously described as geographic origin lineages
(4,6–8). In
particular, HPV-16 LCR variants were grouped into four major
lineages and nine sublineages: i) Lineage A, including A1, A2, A3
(previously known as European) and A4 (Asian) sublineages; ii)
lineage B, including B1 (African-1a) and B2 (African-1b)
sublineages; iii) lineage C (African-2); and iv) lineage D,
including D1 (North American, NA1), D2 (Asian-American, AA2) and D3
(Asian-American, AA1) sublineages (4,7).
HPV-52 variants were grouped into four major lineages and 6
sublineages: i) Lineage A, including A1 and A2 (European)
sublineages; ii) lineage B, including B1 (African-1a) and B2
(African-1b) sublineages; iii) lineage C, including C1 (African-2a)
and C2 (African-2b) sublineages; and iv) lineage D (Asian-American)
(4). Moreover, the LCR contains
the early promoter and various transcriptional regulatory sites for
both viral and cellular proteins, such as E2, yin-yang 1 (YY1),
activator protein 1 (AP-1), octamer 1 (Oct-1), nuclear factor 1
(NF-1) and transcriptional enhancer factor 1 (TEF-1) (9). The L1 gene, encoding the major capsid
protein, is a conserved region that is used to classify HPVs into
species and types. Furthermore, the L1 protein can self-assemble
into virus-like particles (VLPs), which are used for producing
prophylactic vaccines (10). In
total, five hypervariable immune-dominant regions (BC, DE, EF, FG
and HI) show high levels of polymorphism in and among HPV types,
resulting in the generation of neutralizing antibodies of different
binding affinities (11). The
phylogenetic analysis of the LCR and L1 regions, and the study of
single nucleotide polymorphisms (SNPs) and amino acid mutations
allows the identification of the HR-HPV variants circulating in the
population. The description and understanding of HR-HPV genetic
variants is an important area for molecular pathogenesis, and for
the development of molecular diagnostics for HPV, vaccines and
other therapeutic approaches aimed at controlling and/or
eliminating virus-induced diseases.
Among HR-HPVs, HPV-16 and HPV-18 are responsible for
approximately 70% of cervical cancers worldwide (1). The HPV-16 and HPV-18 L1 regions are
the targets of the HPV vaccine and, therefore, the study of their
variants is a high priority. HPV-16 is the most common HPV type
worldwide, while other HR-HPVs, such as HPV-52 in HIV-positive
subjects (12), are particularly
prevalent among high-risk populations. These types can develop
persistent HPV infections and put the patients at risk of
progression to cancer, and so these patients should be closely
monitored.
The aim of the present study was to retrospectively
describe the genetic variability of the LCR region in the HPV-16
and HPV-52 types, and of the L1 region of the HPV-16 and HPV-18
types. In the present study, HPV sequences identified from subjects
enrolled in previous studies (13–17)
were analyzed.
Materials and methods
Study sequences
The LCR sequences of HPV-16 and HPV-52 (n=221 and
n=41, respectively), and L1 sequences of HPV-16 and HPV-18 (n=148
and n=48, respectively) were analyzed. The sequences were obtained
from 375 cervical and 83 anal swabs positive for HPV-16, HPV-18 and
HPV-52 in our previous studies (13–17).
No patients were enrolled in the present study.
In total, 95 of the 458 sequences analyzed were
identified from the general population [54 women; median age, 34
years; interquartile range (IQR)=29–41], whereas 363 were
identified in high-risk groups for the acquisition of HPV
infection, as adolescents/young people (36 girls; median age, 22
years; IQR=21–25), HIV positive subjects (114 women, median age 43
years, IQR=37–47; 69 men, median age 35 years, IQR=30–42) and
migrants (37 women, median age 29 years, IQR=25–40). Upon informed
consent of the participants, all the samples were collected at the
clinical centers that collaborated in the previous studies
(13–17). The approval of the ethics committee
was obtained for each of the previous studies (13–17).
Nucleic acid extraction and sequencing
amplification
Nucleic acids were extracted from the biological
samples using the NucliSENS® easyMAG™ automated platform
(BioMérieux Benelux B.V.), according to the off-board lysis
protocol (https://www.biomerieux-diagnostics.com/nuclisensr-easymagr).
HPV-16 LCR fragments were obtained using a previously described
in-house PCR (17) and HPV-52 LCR
was amplified using nested-PCR, as previously described (18). L1 genetic characterization was
performed by sequence analysis of a 1,488 bp L1 gene amplicon for
HPV-16 and a 1,489 bp L1 gene amplicon for HPV-18 obtained by two
partially overlapping fragments amplified using degenerate primers
(19). The primer sequences used
in the amplification protocols are presented in Table SI.
Each PCR run included both negative (water) and
positive controls (DNA extracted from HPV-16, HPV-18 and HPV-52
positive samples); each sample was tested three times to confirm
the mutations detected.
Following PCR amplification, the amplicons were
purified using the NucleoSpin® Extract II purification
kit (Macherey-Nagel GmbH) and the nucleotide sequences were
obtained using automated DNA sequencing with an ABI PRISM 3100
genetic analyzer (Applied Biosystem; Thermo Fisher Scientific,
Inc.).
Sequence analysis
Multiple nucleotide sequences were aligned using
ClustalX version 2.0 (20). SNPs
and amino acid changes were determined by examining the sequence
chromatograms using the MEGA 6.0 software package (21). BioEdit version 7.2.5 (22) was used to assess the effects of LCR
and L1 variations on the binding sites for cellular transcription
factors and immune-dominant epitopes, respectively. Site-specific
entropy was estimated using BioEdit to evaluate genetic diversity
(22). A value of zero indicated
site-specific conservation and higher values indicated increasing
degrees of site-specific variation.
Phylogenetic analysis
Phylogenetic trees were constructed using the
Neighbor-Joining method (23), the
Kimura 2-Parameter model (24) and
the MEGA 6.0 software package (21). A bootstrap re-sampling analysis was
performed (1,000 replicates) to test tree robustness. The reference
viral strains used for constructing the phylogenetic trees,
selected according to the classification used by Burk et al
(4), were obtained from the NCBI
GenBank Database (https://www.ncbi.nlm.nih.gov/nucleotide/).
LCR sequences and cytology
In total, 117 (52.9%) of the 221 HPV-16 LCR
sequences were obtained from cervical/anal samples with cytological
abnormalities, 78 of which were low-grade squamous intraepithelial
lesions (LSIL) and 39 were high-grade squamous intraepithelial
lesions (HSIL). With regard to the 41 HPV-52 LCR sequences, 17
(41.5%) were obtained from samples with cytological abnormalities
(13 LSIL and 4 HSIL).
L1 sequences and selective
pressure
The partitioning approach to robust interference of
selection method, available on the DataMonkey 2.0 server
(www.datamonkey.org) (25), was used to detect whether a
proportion of sites in the alignment of HPV-16 and HPV-18 L1 gene
sequences evolved with a positive selective pressure, shown by a
dN/dS>1, which expresses the ratio of non-synonymous to
synonymous substitutions. The integrative analyses of four
different codon-based maximum likelihood methods, single likelihood
ancestor counting (SLAC), fixed effects likelihood (FEL), random
effects likelihood (REL) and mixed effect model of evolution
(MEME), all incorporating the HKY85 substitution model with
phylogenetic trees, inferred using the Neighbor-Joining method,
were carried out in order to estimate the dN/dS ratio for all codon
alignments.
Results
HPV-16 and HPV-52 LCR variants
HPV-16
Nucleotide polymorphisms analysis
A total of 221 HPV-16 partial LCRs were successfully
sequenced and analyzed. In total 80 SNPs were identified in 73
nucleotide sites of the 735 bases of HPV-16 LCR (10.9% variable
nucleotide positions), resulting in 62 unique variants identified
in one or more of the study samples (variant IDs 1 to 62; Table I). On comparing the sequences
analyzed with the reference sequence (accession number, K02718 A1),
it was observed that >40 SNPs had previously been identified and
reported, while 40 SNPs (40/80; 50%; Table SII) were, to the best of our
knowledge, identified for the first time in the present study.
 | Table I.Frequencies and GenBank accession
numbers of analyzed HPV-16, HPV-18 and HPV-52 variants. |
Table I.
Frequencies and GenBank accession
numbers of analyzed HPV-16, HPV-18 and HPV-52 variants.
| Target | Variant name | Frequency | GenBank accession
number |
|---|
| LCR HPV-16 | Variant 1 | 2 | EU650455 |
|
| Variant 2 | 62 | EU650473 |
|
| Variant 3 | 3 | EU650433 |
|
| Variant 4 | 1 | MH028439 |
|
| Variant 5 | 1 | EU650463 |
|
| Variant 6 | 2 | MH028440 |
|
| Variant 7 | 1 | MH028441 |
|
| Variant 8 | 1 | MH028442 |
|
| Variant 9 | 1 | MH028443 |
|
| Variant 10 | 1 | MH028444 |
|
| Variant 11 | 1 | MH028445 |
|
| Variant 12 | 4 | EU650446 |
|
| Variant 13 | 1 | MH028446 |
|
| Variant 14 | 2 | MH028447 |
|
| Variant 15 | 2 | MH028448 |
|
| Variant 16 | 1 | MH028449 |
|
| Variant 17 | 1 | MH028450 |
|
| Variant 18 | 1 | MH028451 |
|
| Variant 19 | 1 | MH028452 |
|
| Variant 20 | 3 | MH028453 |
|
| Variant 21 | 1 | MH028454 |
|
| Variant 22 | 1 | MH028455 |
|
| Variant 23 | 1 | MH028456 |
|
| Variant 24 | 6 | EU650442 |
|
| Variant 25 | 4 | MH028457 |
|
| Variant 26 | 31 | EU650481 |
|
| Variant 27 | 1 | MH028458 |
|
| Variant 28 | 1 | MH028459 |
|
| Variant 29 | 2 | MH028460 |
|
| Variant 30 | 3 | EU650465 |
|
| Variant 31 | 1 | MH028461 |
|
| Variant 32 | 1 | MH028462 |
|
| Variant 33 | 1 | MH028463 |
|
| Variant 34 | 2 | MH028464 |
|
| Variant 35 | 1 | MH028465 |
|
| Variant 36 | 1 | MH028466 |
|
| Variant 37 | 1 | MH028467 |
|
| Variant 38 | 1 | MH028468 |
|
| Variant 39 | 38 | EU650484 |
|
| Variant 40 | 3 | MH028469 |
|
| Variant 41 | 1 | MH028470 |
|
| Variant 42 | 1 | MH028471 |
|
| Variant 43 | 2 | MH028472 |
|
| Variant 44 | 1 | MH028473 |
|
| Variant 45 | 1 | MH028474 |
|
| Variant 46 | 1 | MH028475 |
|
| Variant 47 | 1 | MH028476 |
|
| Variant 48 | 1 | MH028477 |
|
| Variant 49 | 1 | EU650448 |
|
| Variant 50 | 1 | MH028478 |
|
| Variant 51 | 1 | MH028479 |
|
| Variant 52 | 1 | MH028480 |
|
| Variant 53 | 1 | MH028481 |
|
| Variant 54 | 2 | EU650474 |
|
| Variant 55 | 2 | MH028482 |
|
| Variant 56 | 1 | MH028483 |
|
| Variant 57 | 1 | MH028484 |
|
| Variant 58 | 1 | MH028485 |
|
| Variant 59 | 1 | MH028486 |
|
| Variant 60 | 1 | MH028487 |
|
| Variant 61 | 1 | MH028488 |
|
| Variant 62 | 3 | MH028489 |
| L1 HPV-16 | Variant 1 | 1 | JF728181 |
|
| Variant 2 | 2 | JF728156 |
|
| Variant 3 | 1 | JF728169 |
|
| Variant 4 | 3 | JF728175 |
|
| Variant 5 | 2 | JF728173 |
|
| Variant 6 | 3 | JF728162 |
|
| Variant 7 | 5 | JF728179 |
|
| Variant 8 | 1 | JF728168 |
|
| Variant 9 | 1 | JF728165 |
|
| Variant 10 | 21 | EU650438 |
|
| Variant 11 | 2 | JF728176 |
|
| Variant 12 | 1 | JF728167 |
|
| Variant 13 | 2 | JF728163 |
|
| Variant 14 | 1 | JF728172 |
|
| Variant 15 | 76 | JF728182 |
|
| Variant 16 | 1 | JF728161 |
|
| Variant 17 | 1 | EU650439 |
|
| Variant 18 | 1 | JF728180 |
|
| Variant 19 | 1 | JF728159 |
|
| Variant 20 | 1 | JF728170 |
|
| Variant 21 | 1 | JF728158 |
|
| Variant 22 | 1 | JF728164 |
|
| Variant 23 | 8 | JF728171 |
|
| Variant 24 | 1 | JF728177 |
|
| Variant 25 | 1 | JF728155 |
|
| Variant 26 | 1 | JF728166 |
|
| Variant 27 | 1 | JF728160 |
|
| Variant 28 | 1 | EU650483 |
|
| Variant 29 | 1 | JF728174 |
|
| Variant 30 | 2 | JF728183 |
|
| Variant 31 | 1 | JF728157 |
| L1 HPV-18 | Variant 1 | 1 | MH028419 |
|
| Variant 2 | 1 | MH028420 |
|
| Variant 3 | 8 | MH028421 |
|
| Variant 4 | 1 | MH028422 |
|
| Variant 5 | 3 | MH028423 |
|
| Variant 6 | 1 | MH028424 |
|
| Variant 7 | 4 | MH028425 |
|
| Variant 8 | 1 | MH028426 |
|
| Variant 9 | 1 | JF728186 |
|
| Variant 10 | 2 | JF728184 |
|
| Variant 11 | 1 | MH028427 |
|
| Variant 12 | 1 | MH028428 |
|
| Variant 13 | 10 | MH028429 |
|
| Variant 14 | 1 | MH028430 |
|
| Variant 15 | 1 | JF728185 |
|
| Variant 16 | 1 | MH028431 |
|
| Variant 17 | 1 | MH028432 |
|
| Variant 18 | 2 | MH028433 |
|
| Variant 19 | 1 | MH028434 |
|
| Variant 20 | 2 | MH028435 |
|
| Variant 21 | 1 | MH028436 |
|
| Variant 22 | 1 | MH028430 |
|
| Variant 23 | 1 | MH028437 |
|
| Variant 24 | 1 | MH028438 |
| LCR HPV-52 | Variant 1 | 2 | MH028409 |
|
| Variant 2 | 20 | MH028410 |
|
| Variant 3 | 2 | MH028411 |
|
| Variant 4 | 1 | MH028412 |
|
| Variant 5 | 2 | MH028413 |
|
| Variant 6 | 5 | MH028414 |
|
| Variant 7 | 3 | MH028415 |
|
| Variant 8 | 2 | MH028416 |
|
| Variant 9 | 1 | MH028417 |
|
| Variant 10 | 3 | MH028418 |
The site-specific variations in the LCR fragment
varied from 0 (no variation) to 0.63. The most common LCR changes
with entropy scores>0.4 were as follows: G7521A (168/221; 76%),
C7764T (22/221; 9.9%), G7489A (21/221; 9.5%), C7786T (21/221;
9.5%), A7485C (20/221; 9%), C7669T (19/221; 8.6%), C7689A (19/221;
8.6%), G7834T (14/221; 6.3%), C14T/G (12/221; 5.4%), A7729C
(11/221; 5.0%), C32T (10/221; 4.5%), A7837C (9/221; 4.1%), A7839G
(9/221; 4.1%; Fig. S1). No
insertion or deletion mutation sites were identified.
Phylogenetic analysis
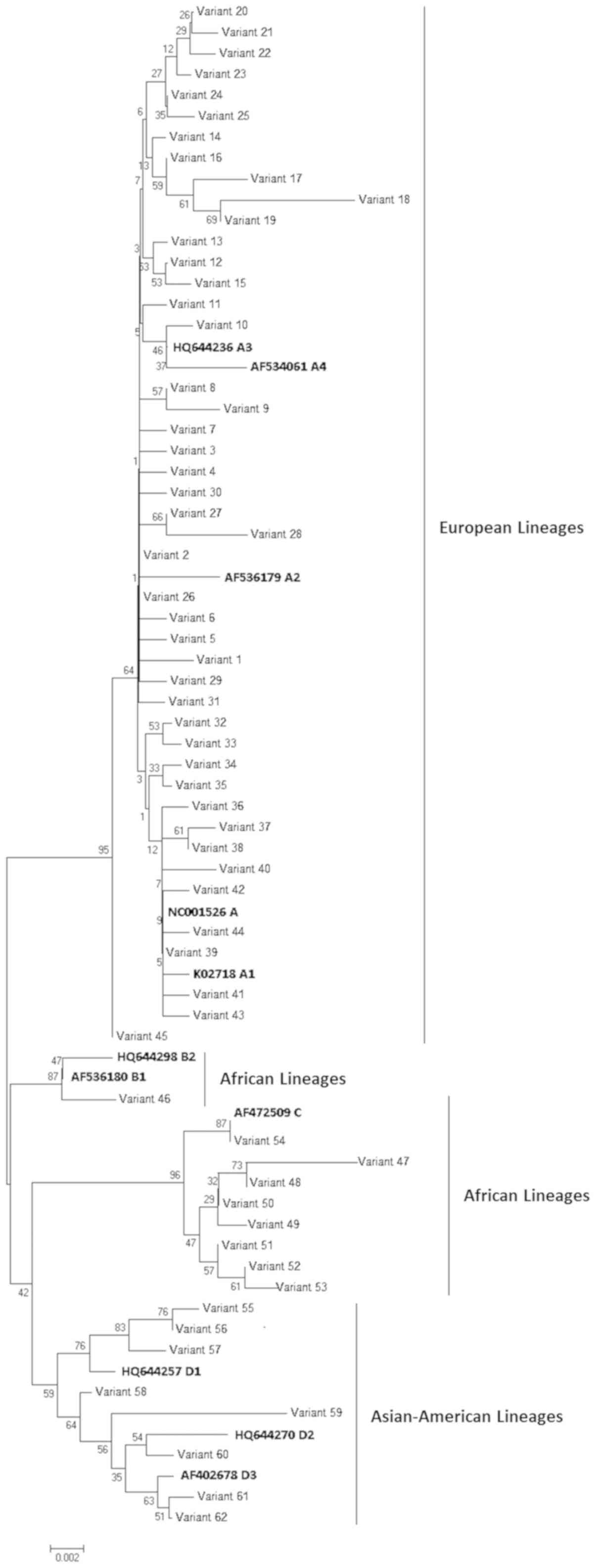
Phylogenetic analysis showed that the 62 variants
clustered into four main groups, corresponding to lineages A, B, C
and D (Fig. 1). In particular, 45
of the variants identified in 200 of the 221 HPV-16 partial LCRs
analyzed (200/221; 90.5%) clustered within lineage A (European
lineage), 1 variant (1/221; 0.4%) within lineage B (African 1
lineage), 8 variants (9/221; 4.1%) within lineage C (African 2
lineage) and 8 variants (11/221; 4.9%) within lineage D
(Asian-American lineage). With regards to the samples in lineage D,
36.3% (4/11) belonged to the sub-lineage D1 (North-American
sub-lineages), 27.3% (3/11) to D2 (Asian-American 2 sub-lineages)
and 36.3% (4/11) to D3 (Asian-American 1 sub-lineages). All of the
17 variants belonging to lineages B, C and D (Non-European) were
detected in the groups of people at higher risk for acquiring HPV
infection, such as migrants, HIV+ subjects and adolescent/young
people.
SNPs in the transcription factor
binding sites
Analysis of the SNPs revealed that 19 fell into the
HPV-16 LCR binding sites of transcription factors. In total, three
SNPs were located in the TEF-1 binding site, four SNPs were located
in the NF-1 binding site, six were located in the YY1 binding site,
one SNP fell into the AP-1 binding site, one SNP fell into the
Oct-1 binding site and four SNPs were located in the E2 binding
site (Table II).
| Table II.Single nucleotide polymorphisms in
the human papillomavirus-16 long control region binding sites of
transcription factors. |
Table II.
Single nucleotide polymorphisms in
the human papillomavirus-16 long control region binding sites of
transcription factors.
| Binding sites | Variant ID | Lineage |
|---|
| TEF-1 T7469A | 41 | A |
| TEF-1 C7689A | 46, 47–53,
55–62 | B, C, D |
| TEF-1 G7826A | 32, 33, 47–54 | A, C |
| NF-1 T7475G | 59 | D |
| NF-1 G7478A | 17 | A |
| NF-1 G7677A | 18, 19 | A |
| NF-1 C7748A | 59 | D |
| YY1 A7485C | 47-54, 55–62 | C, D |
| YY1 G7521A | 1-32, 35, 45, 46,
47–54, 55–62 | A, B, C, D |
| YY1 C7786T | 46, 47–54,
55–62 | B, C, D |
| YY1 G7826A | 32-33, 47–54 | A, C |
| YY1 A7837C | 47–54 | C |
| YY1 A7839G | 47–54 | C |
| AP-1 T7637A | 59 | D |
| Oct-1 A7839G | 47–54 | C |
| E2 G7869A | 25, 46, 51–53 | A, B, C |
| E2 G7869C | 50 | C |
| E2 A35C | 6 | A |
| E2 C37T | 47, 48, 53 | C |
LCR variants and cytology
Overall, the 117 LCR sequences detected from
subjects with cytological abnormalities clustered into the four
main lineage groups A, B, C and D.
In total, 36.3% (4/11) of the LCR sequences
belonging to lineage D (Asian-American lineage) were identified in
subjects with cytological abnormalities, all classified as LSIL.
With respect to lineage A (European lineage), 51.5% (104/200) of
the LCR sequences were from subjects with cytological lesions, 74
of which were LSIL and 30 were HSIL.
Almost all (9/10; 90%) of the African variants
(lineages B and C) were detected in subjects with HSIL. The eight
sequences belonging to lineage C were characterized by nine
mutations in the transcription factor binding sites (one in the
TEF-1 site, six in YY1 sites, one in Oct-1 and one in E2), whereas
the single sequence clustering within lineage B showed four
mutations in the transcription factor binding sites (one in the
TEF-1 site, two in YY1 sites and one in E2).
HPV-52
Nucleotide polymorphisms analysis
In total, 41 HPV-52 partial LCR regions were
successfully sequenced. On comparing the analyzed sequences with
the reference sequence (accession number, X74481 A1), 21 SNPs were
identified in the 19 nucleotide sites among the 726 bases of the
HPV-52 LCR (2.9% variable nucleotide positions), resulting in 10
unique variants identified in one or more of the study samples
(variant IDs 1 to 10; Table I). In
total, 13 SNPs had already been identified and reported, while 6
SNPs (6/19; 31.6%, Table SIII)
were identified, to the best of our knowledge, for the first time
in the present study.
Site-specific variation in the LCR fragment ranged
from 0 (no variation) to 1.0 (Fig.
S2). The most common LCR changes, with entropy scores >0.4,
were C7188A (4/41; 9.8%), G7354T (4/41; 9.8%), G7605A (9/41; 22%),
T7607C (16/41; 39%), A7640C (4/41; 9.8%), T7642C (4/41; 9.8%),
G7695C (4/41; 9.8%), C7726T (10/41; 24.4%), T7727C (5/41; 12.2%),
G7844A (4/41; 9.8%), A7848G (4/41; 9.8%). On comparing the analyzed
sequences with the reference sequence (accession number, X74481
A1), four sequences with deletions were found. All of the variants
showed deletion sites in position 7370–7374; variant ID 9 presented
a deletion site in position 7173–7180, variant IDs 7 and 8 were
characterized by a deletion in the nucleotide 7626, and variants
IDs 7–9 were characterized by deletion sites at position 7681–7683.
No insertion mutation sites were identified.
Phylogenetic analysis
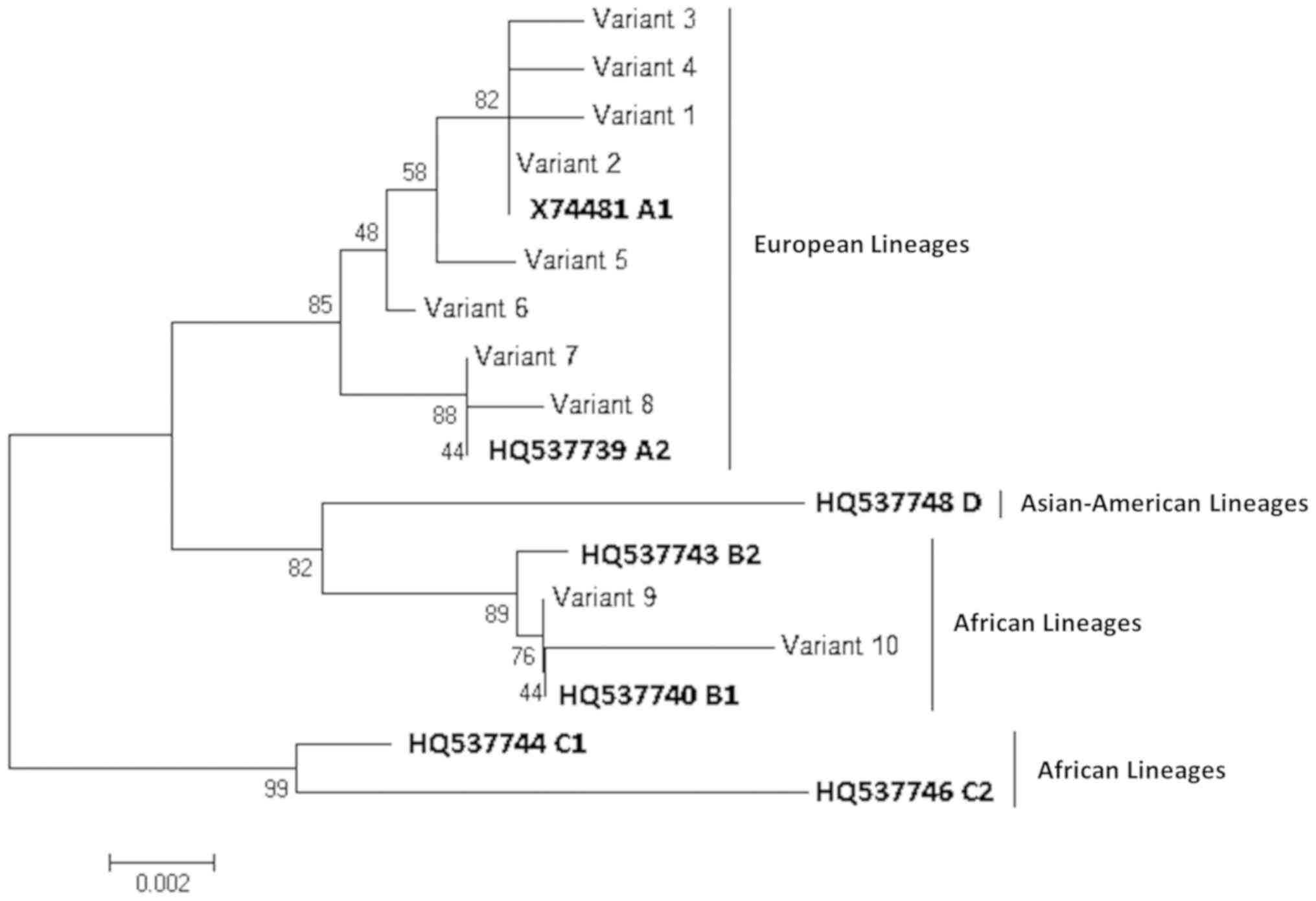
Phylogenetic analysis showed that the 10 variants
clustered into two main groups, corresponding to lineages A and B
(Fig. 2). In total, eight variants
identified in 37 of the 41 HPV-52 partial LCRs analyzed (37/41;
90.2%) clustered within lineage A (formerly European lineage) and 2
variants (4/41; 9.6%) clustered within lineage B (Asian-American
lineage).
SNPs in the transcription factor
binding sites
On analyzing the SNPs, it was observed that none of
them fell within the HPV-52 LCR binding sites of transcription
factors.
LCR variants and cytology
The 17 LCR sequences isolated from subjects with
cytological abnormalities, 13 LSIL and 4 HSIL, clustered into
lineage A.
HPV-16 and HPV-18 L1
HPV-18 L1 variants
In total, 48 HPV-18 partial L1 regions were
successfully sequenced. The phylogenetic analysis showed that 24
different variants were identified in one or more of the study
samples (variant IDs 1 to 24; Table
I).
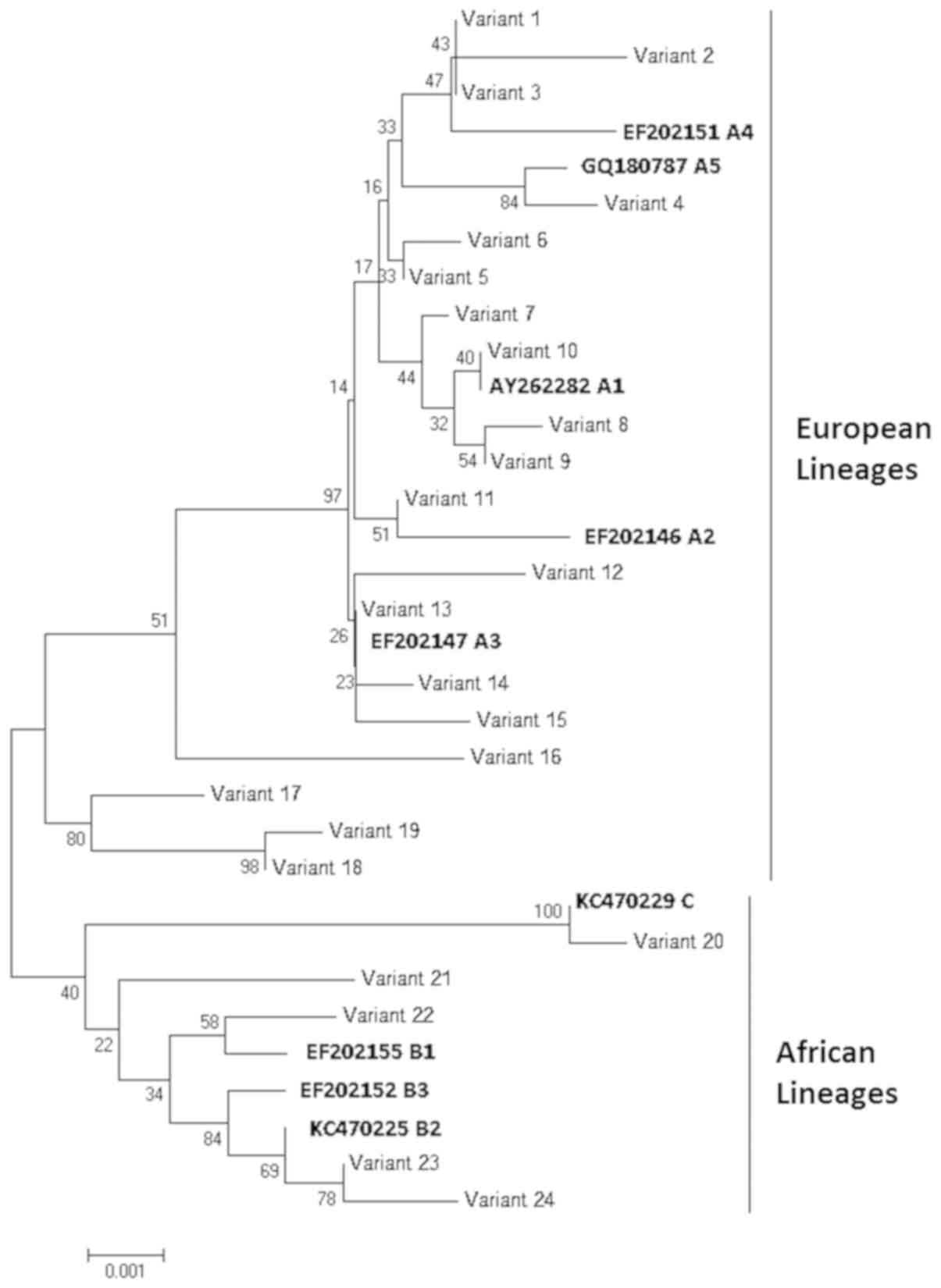
These variants clustered into three main groups,
corresponding to lineages A, B, and C (Fig. 3). More specifically, 19 variants,
representing 43 samples (43/48; 89.6%), clustered into lineage A
(European lineage), while four variants (4/48; 8.3%) clustered into
lineage B and one variant (1/48; 2.1%) clustered into lineage C
(both African lineage). All except one of these non-European HPV-18
variants were detected in HIV+ subjects; the exception was detected
in a subject in the adolescent/young people age group.
Amino acid changes in the
immune-dominant epitopes
A total of 148 HPV-16 partial L1 open reading frames
were successfully sequenced. On comparing the sequences analyzed
with the reference sequence (accession number, K02718 A1), 33 amino
acidic changes were identified among the 330 amino acids of the
HPV-16 L1 protein (Table SIV),
resulting in 31 unique variants identified in one or more of the
study samples (variant IDs 1 to 31; Table I). In total, 10 of these amino acid
changes (10/33; 30.3%) occurred in sequences encoding
immune-dominant loops. More specifically, the H76Y mutation
occurred in the BC loop, and the amino acid mutations A139E, T176N
and N177T occurred in the DE loop, H202D and Q214H fell into the EF
loop, while mutations A287T, P293S, T294S and Q314R were observed
in the FG loop.
In total, 96.7% (30/31) of the identified variants
(140 sequences) showed >1 amino acid substitution in an
immune-dominant loop. In particular, 7 (variant IDs 2, 8, 11, 16,
19 and 29) and 5 (variant IDs 4–7 and 9) were characterized by two
and three amino acid substitutions in the immune-dominant loop,
respectively.
On comparing the analyzed sequences with the
reference sequence (accession number, AY262282 A1), a total of 15
amino acidic substitutions were identified among the 441 amino
acids of the HPV-18 L1 protein (Table
SV). No insertion or deletion mutation sites were found. In
total, four (4/15; 26.7%) of these amino acid substitutions
occurred in sequences encoding immune-dominant loops. More
specifically, the R112K mutation occurred in the BC loop, while the
amino acid mutations Q334P, I338L and R344P occurred in the FG
loop.
In total, 29.2% (7/24) of the variants identified
(14 sequences) showed at>1 amino acid substitution in an
immune-dominant loop.
Selective pressure analysis
MEME found evidence of episodic positive selection
at one site of HPV-16 L1 (P<0.1), while there was no evidence
for positive selection in the analyzed sequence alignment of
HPV-18. The integrative selection analysis (SLAC, P=0.1; FEL, P=0.1
and REL Bayes factor=50) identified eight negatively selected
codons in HPV-16 L1 sequences (55, 93, 109, 130, 145, 216, 308 and
351), none of which fell into the immune-dominant loops, and 23
negatively selected codons in HPV-18 L1 sequences (85, 100, 126,
135, 144, 165, 168, 171, 191, 196, 201, 234, 239, 252, 296, 324,
369, 399, 405, 416, 430, 500 and 519), nine of which fell into four
different immune-dominant loops (loop BC, 126; loop DE, 171, 191,
196, 201; loop EF, 234 and 239; loop HI, 416) but none caused amino
acid changes.
Discussion
The present study identified and analyzed a large
number of HPV-16, HPV-18 and HPV-52 sequences (n=458) obtained from
subjects belonging to both general and high-risk populations in
Italy. Furthermore, to the best of our knowledge, this is the first
Italian study reporting phylogenetic data on HPV-52 variants.
The phylogenetic analysis showed that ~90% of HPV-16
(90.5%), HPV-18 (89.6%) and HPV-52 (90.2%) variants clustered into
lineage A, previously defined as the European lineage, as expected
for the geographical area under study.
Non-European variants (belonging to lineages B, C
and D) were only detected in populations at higher risk of HPV
infection, such as migrants, HIV+ subjects and adolescent/young
people. In particular, 53% of the non-European HPV-16 sequences
were detected in migrant women, 29% in HIV+ subjects and 18% in
adolescent/young people. With regards to the five non-European
HPV-18 sequences, four were detected in HIV+ subjects, while one
was found in an adolescent. Non-European HPV-52 sequences were all
identified in HIV+ subjects. The identification of non-European
variants in populations engaging in risky sexual behavior is
associated with a high risk of contracting several HPV infections
supported by different types/variants (13–15,17,18,26).
The LCR region of HPV-16 and HPV-52 types showed
high levels of genetic diversity (10.9 and 2.9%, respectively) and
a large number of new non-lineage-specific SNPs (50 and 31.6%,
respectively) were identified in the sequences analyzed.
Furthermore, HPV-52 LCR sequences were characterized by various
sequence deletion sites. However, at present, these SNPs and
deletions have not shown evidence of determining phylogenetic
groups, therefore, functional studies are required to clarify
whether these changes can affect any HPV variant phenotype.
With regards to the HPV-16 LCR sequences, 23.7%
(n=19) of the identified SNPs fell within the binding sites for
cellular and viral transcriptional factors, such as E2, YY1, AP-1,
Oct-1, NF-1, and TEF-1. Sequence changes in these sites can alter
carcinogenic potential by modulating virus replication and
transcription (9,27). In particular, SNPs at E2
transcription factor sites may affect the repression of E6/E7
oncoproteins, known as landmarks in cancer progression, and
multiple disruptions to YY1 binding sites, such as the six SNPs
identified in the sequences analyzed in the present study, are
required to significantly upregulate E6 promoter activity by three-
to six-fold (27). The results of
the present study showed that several mutations in transcription
factor sites characterized variants belonging to the African
lineages B and C isolated from subjects with HSIL. However, it is
important to carry out a thorough transcriptional analysis of the
HPV-16 LCR in order to investigate the association between LCR SNPs
and the carcinogenicity of HPV-16 variants.
A sequence analysis of the HPV-16 and HPV-18 L1
proteins determined that ~30% of the amino acid mutations fell
within the immune-dominant epitope loops (30.3 and 26.7%,
respectively). Although these mutations were neutral and not under
positive selection, HR-HPVs use this strategy to evade recognition
by neutralizing antibodies. In fact, with respect to HPV-16, six
out of 10 identified amino acid mutations (A139E, N181S, A266T, G28
1W, S282P and T353P) were involved in the loss of reactivity of a
specific monoclonal antibody (MAb), due to the loss of direct
interactions with the MAb and for the conformational changes that
indirectly disrupt interactions with the Mab (28). Therefore, the real-time monitoring
of mutations is essential in this post-vaccination era and it is
important to highlight their ability to fix in the viral
population. In fact, L1 proteins self-assemble into VLPs, which are
components of prophylactic vaccines, and amino acid mutations in
the L1 protein may affect viral antigenicity and limit vaccine
efficacy (3).
A limitation of the present study was that the
HPV-16, HPV-18 and HPV-52 genomes were only partially analyzed.
Complete sequencing of the genome would provide better insights
into the different HPV types, thus enhancing the phylogenetic
classification of HPV in relation to oncogenic risk (29). However, the present study provided,
to the best of our knowledge, the first data on the circulation and
characterization of HPV-52 variants in Italy. Due to the ongoing
implementation of vaccination programs with the 9vHPV vaccine,
which also include the HPV-52 type, it is important to monitor all
HR-HPV variants, especially in vaccinated populations. In fact,
awareness of the viruses currently circulating in the population
will facilitate the medium- to long-term monitoring of genetic
viral evolution, thus enabling predictions about the potential of
evading the vaccine-induced immune response.
As the study of HR-HPV variants is important for
understanding the pathogenic role of the virus in malignant
lesions, well-designed and extensive epidemiological and clinical
studies are required in order to better determine the strength of
the correlation between the cytology and the identified SNPs, and
between the amino acid mutations and the oncogenic risks.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The sequence datasets analyzed in the present study
are available at the NCBI GenBank database (https://www.ncbi.nlm.nih.gov/nucleotide/).
Authors' contributions
ERF, AA and ET conceived and designed the
experiments and wrote the manuscript. SB and DC performed the
experiments. SB, ERF, FP and GZ analyzed the data. GZ supervised
the findings. ET supervised the study. SB and FP critically
evaluated the literature and revised the manuscript. All of the
authors reviewed and approved the final manuscript.
Ethics approval and consent to
participate
The approval from the ethical committee was obtained
for each of the previous studies. No subjects were enrolled in the
present study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Serrano B, Brotons M, Bosch FX and Bruni
L: Epidemiology and burden of HPV-related disease. Best Pract Res
Clin Obstet Gynaecol. 47:14–26. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
De Martel C, Plummer M, Vignat J and
Franceschi S: Worldwide burden of cancer attributable to HPV by
site, country and HPV type. Int J Cancer. 141:664–670. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Westrich JA, Warren CJ and Pyeon D:
Evasion of the host immune defences by human papillomavirus. Virus
Res. 231:21–33. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Burk RD, Harari A and Chen Z: Human
papillomavirus genome variants. Virology. 445:232–243. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Smith B, Chen Z, Reimers L, van Doorslaer
K, Schiffman M, Desalle R, Herrero R, Yu K, Wacholder S, Wang T and
Burk RD: Sequence imputation of HPV16 genomes for genetic
association studies. PLoS One. 6:e213752011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mammas IN, Spandidos DA and Sourvinos G:
Genomic diversity of human papillomaviruses (HPV) and clinical
implications: An overview in adulthood and childhood. Infect Genet
Evol. 21:220–226. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cornet I, Gheit T, Franceschi S, Vignat J,
Burk RD, Sylla BS, Tommasino M and Clifford GM; IARC HPV Variant
Study Group, : Human papillomavirus type 16 genetic variants:
Phylogeny and classification based on E6 and LCR. J Virol.
86:6855–6861. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen AA, Gheit T, Franceschi S, Tommasino
M and Clifford GM: Human papillomavirus 18 genetic variation and
cervical cancer risk worldwide. J Virol. 89:10680–10687. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
O'Connor M, Chan SY and Bernard HU:
Transcription factor binding sites in the long control region of
genital HPVs. Human papillomaviruses. A compilation and analysis of
nucleic acid and amino acid sequences Los Alamos, New Mexico: Los
Alamos National Laboratory; pp. III21–III41. 1995
|
|
10
|
Kirnbauer R, Booy F, Cheng N, Lowy DR and
Schiller JT: Papillomavirus L1 major capsid protein self-assembles
into virus-like particles that are highly immunogenic. Proc Natl
Acad Sci USA. 89:12180–12184. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Christensen ND, Dillner J, Eklund C,
Carter JJ, Wipf GC, Reed CA, Cladel NM and Galloway DA: Surface
conformational and linear epitopes on HPV-16 and HPV-18 L1
virus-like particles as defined by monoclonal antibodies. Virology.
223:174–184. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Martins AE, Lucena-Silva N, Garcia RG,
Welkovic S, Barboza A, Menezes ML, Maruza M, Tenorio T and Ximenes
RA: Prevalence of human papillomavirus infection, distribution of
viral types and risk factors in cervical samples from human
immunodeficiency virus-positive women attending three human
immunodeficiency virus-acquired immune deficiency syndrome
reference centres in northeastern Brazil. Mem Inst Oswaldo Cruz.
109:738–747. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bianchi S, Boveri S, Igidbashian S,
Amendola A, Urbinati AM, Frati ER, Bottari F, Colzani D, Landoni F,
Tanzi E, et al: Chlamydia trachomatis infection and HPV/Chlamydia
trachomatis co-infection among HPV-vaccinated young women at the
beginning of their sexual activity. Arch Gynecol Obstet.
294:1227–1233. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Panatto D, Amicizia D, Bianchi S, Frati
ER, Zotti CM, Lai PL, Domnich A, Colzani D, Gasparini R and Tanzi
E: Chlamydia trachomatis prevalence and chlamydial/HPV co-infection
among HPV-unvaccinated young Italian females with normal cytology.
Hum Vaccin Immunother. 11:270–276. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Orlando G, Fasolo M, Mazza F, Ricci E,
Esposito S, Frati E, Zuccotti GV, Cetin I, Gramegna M, Rizzardini
G, et al: Risk of cervical HPV infection and prevalence of
vaccine-type and other high-risk HPV types among sexually active
teens and young women (13–26 years) enrolled in the VALHIDATE
study. Hum Vaccin Immunother. 10:986–994. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Orlando G, Tanzi E, Rizzardini G,
Chatenoud L, Zanchetta N, Esposito S, Tisi G, Fasolo M, Bosari S,
Boero V, et al: Modifiable and Non-modifiable factors related to
HPV infection and cervical abnormalities in women at high risk: A
cross-sectional analysis from the VALHIDATE Study. Ann Virol Res.
2:10132016.
|
|
17
|
Tanzi E, Amendola A, Bianchi S, Fasolo MM,
Beretta R, Pariani E, Zappa A, Frati E and Orlando G: Human
papillomavirus genotypes and phylogenetic analysis of HPV-16
variants in HIV-1 infected subjects in Italy. Vaccine. 27 (Suppl
1):A17–A23. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang C, Park JS, Grce M, Hibbitts S,
Palefsky JM, Konno R, Smith-McCune KK, Giovannelli L, Chu TY,
Picconi MA, et al: Geographical distribution and risk association
of human papillomavirus genotype 52-variant lineages. J Infect Dis.
210:1600–1604. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Frati ER, Bianchi S, Colzani D, Zappa A,
Orlando G and Tanzi E: Genetic variability in the major capsid L1
protein of human papillomavirus type 16 (HPV-16) and 18 (HPV-18).
Infect Genet Evol. 11:2119–2124. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Larkin MA, Blackshields G, Brown NP,
Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm
A, Lopez R, et al: Clustal W and Clustal X version 2.0.
Bioinformatics. 23:2947–2948. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tamura K, Stecher G, Peterson D, Filipski
A and Kumar S: MEGA6: Molecular evolutionary genetics analysis
version 6.0. Mol Biol Evol. 30:2725–2729. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hall TA: BioEdit: A user-friendly
biological sequence alignment editor and analysis program for
Windows 95/98/NT. Nucl Acids Symp. 41:95–98. 1999.
|
|
23
|
Saitou N and Nei M: The neighbor-joining
method: A new method for reconstructing phylogenetic trees. Mol
Biol Evol. 4:406–425. 1987.PubMed/NCBI
|
|
24
|
Kimura M: A simple method for estimating
evolutionary rate of base substitutions through comparative studies
of nucleotide sequences. J Mol Evol. 16:111–120. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Weaver S, Shank SD, Spielman SJ, Li M,
Muse SV and Kosakovsky Pond SL: Datamonkey 2.0: A modern web
application for characterizing selective and other evolutionary
processes. Mol Biol Evol. Jan 2–2018.doi: 10.1093/molbev/msx335
(Epub ahead of print). View Article : Google Scholar
|
|
26
|
Frati ER, Fasoli E, Martinelli M, Colzani
D, Bianchi S, Carnelli L, Amendola A, Olivani P and Tanzi E:
Sexually transmitted infections: A novel screening strategy for
improving women's health in vulnerable populations. Int J Mol Sci.
18:E13112017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Marongiu L, Godi A, Parry JV and Beddows
S: Human papillomavirus type 16 long control region and E6 variants
stratified by cervical disease stage. Infect Genet Evol. 26:8–13.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ning T, Wolfe A, Nie J, Huang W, Chen XS
and Wang Y: Naturally occurring single amino acid substitution in
the L1 major capsid protein of human papillomavirus type 16:
Alteration of susceptibility to antibody-mediated neutralization. J
Infect Dis. 216:867–876. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Van der Weele P, Meijer CJLM and King AJ:
Whole-genome sequencing and variant analysis of human
papillomavirus 16 infections. J Virol. 91(pii): e00844–17.
2017.PubMed/NCBI
|