Introduction
Deficiency of adenosine deaminase 2 (DADA2) is an
autosomal recessive autoinflammatory disease, characterized by
early-onset vasculopathy appearing a wide range of clinical
manifestations, associated with mutations in adenosine deaminase 2
(ADA2) enzyme (1,2). This condition is mainly characterized
by an inflammatory vasculopathy resembling systemic polyarteritis
nodosa (PAN) (3). DADA2 has been
suggested to compromise endothelial integrity (2) or account for the development of
Sneddon's syndrome in some patients, which represents a poorly
understood disorder most common among middle-age women (4). Patients with DADA2 are characterized
by immunodeficiency of variable severity, which is most evident in
B cells 2 (2). Hematological
manifestations refer to pure red cell aplasia (PRCA),
thrombocytopenia, neutropenia and a severe type of anemia (2,5–7).
Other common manifestations include intermittent fever, arthralgia,
lymphadenopathy and early-onset stroke. In the majority of cases,
patients present with neurological manifestations in both the
peripheral and central nervous system (CNS) (8). CNS involvement is very frequent, as
presented by brain MRI studies that have revealed chronic ischemic
lesions in the basal ganglia, thalami and pons (3,9–11).
ADA2, a 59-kDa enzyme, consisting of 511 amino acid
residues, is encoded by the ADA2 locus, formerly known as
the cat eye syndrome chromosome region, candidate 1 (CECR1)
gene located on chromosome 22 (1).
It forms homodimers and is secreted (by antigen-presenting cells)
with high expression levels in plasma. ADA2 is considered to be
critical for the maintenance of vascular integrity, and to be
responsible for the extracellular degradation of adenosine. It has
also been implicated in the regulation of proliferation of
activated T cells and macrophages, as well as in the
differentiation of monocytes to macrophages (2,12).
Moreover, it has been found that ADA2 can bind to different cell
types through proteoglycans and with a higher specificity to T
cells via an unknown receptor (13). Adenosine is an important signaling
molecule and is normally found in low concentrations which may
increase significantly as a result of cell damage, inflammation,
oncogenesis and hypoxia (14).
ADA2 plays a role in a high spectrum of disorders, connecting
systemic inflammation, vascular pathology, and mild
immunodeficiency (2). It is a
highly polymorphic gene and >300 missense substitutions and
insertions/deletions (indels) have been identified thus far.
Importantly, a high number of copy number variants (CNVs) have been
detected across the ADA2 coding gene (http://exac.broadinstitute.org; http://gnomad.broadinstitute.org). However, as regards
DADA2, 61 disease-causing mutations have been described to date,
located over the entire coding region of ADA2, with the majority of
these being missense variants, although genomic deletions and
nonsense mutations have also been reported (15).
ADA2 belongs to the novel family of adenosine
deaminase growth factors (ADGFs), which play an important role in
tissue development. The three-dimensional (3D) structures of ADA2
reveal the structural basis of the catalytic/signaling activity of
ADGF/ADA2 proteins. The structure is composed of an 8-stranded,
parallel β-sheet that closes into a barrel and is surrounded by
classical α/β-TIM barrel motif helices and six additional α-helices
located at the N-terminus (NH1, NH2, NH3 and NH4) and at the C
terminus (H4 and H5) (Fig. 1).
Loops between β-strands and α-helices constitute most of the
essential features of the catalytic site. ADA2 consists of 4
domains: The signal sequence, the dimerization domain, the putative
receptor-binding (PRB) domain and the catalytic domain (3).
The ADA catalytic domain contains a deep
oblong-shaped active site cavity lined by the C-terminal segments
and connecting loops of the β-barrel strands, acting as a ‘floor’
and ‘walls’, and are capped from above with a ‘ceiling’ composed of
helices H3 and a7 and the hairpin loop between a7-1 and a7-2
helices (Figs. 1 and 2). The zinc ion sits in the deepest part
of the active site cavity in the center of the C-terminal end of
the barrel. It is coordinated to 4 invariant residues: His-86 and
His-88 in the b1 strand, His-330 in the b5 strand, and Asp-415 in
the loop between b1 strand and a8-1 helix (Fig. 3, left panel). The discovery of the
zinc ion confirms the earlier suggestion that ADGF/ADA2 as well as
ADA1 family proteins are zinc-dependent hydrolases (16,17).
In addition to the catalytic domain, the 3D
structure has revealed two ADGF/ADA2-specific domains of novel
folds that mediate the protein dimerization and binding to the cell
surface receptors. The N-terminal extension forms by the N-terminal
α-helices HN1, HN2, HN3 and HN4, whereas the C-terminal extension
elongates the C-terminal helix H5, which together with the
N-terminal helices forms a unique helix-turn-helix arrangement
(Fig. 1). Two highly conserved
charged residues of helix HN1, Arg-8 and Glu-15, are engaged in
ionic interactions with the Asp-347 and His-365 of the neighboring
subunit, respectively. An extensive glycosylation and the presence
of a conserved disulfide bond and a signal peptide in ADA2 strongly
suggest that ADA2, in contrast to ADA1, is specifically designed to
act in the extracellular environment (12).
Recently, Gibson et al (18) performed the screening of an
international registry of children with systemic primary vasculitis
for variants in ADA2 and the subsequent genotyping of 9 children
identified with DADA2. By performing DNA sequencing of the coding
exons, they found rare variants of either known (p.Gly47Arg and
p.Gly47Ala) or novel (p.Arg8Trp, p.Leu351Gln and p.Ala357Thr)
associations with DADA2. Moreover, they assessed the functional
consequences of the identified variants by using specific ADA2
assays and immunoblotting. Prompted by these recent results, and
considering the suggestion that screening ADA2 among children with
vasculitis rash, unclassifiable vasculitis (UCV), PAN, or
unexplained early-onset CNS disease with systemic inflammation may
enable an earlier diagnosis of DADA2 (18), this study was performed in an
attempt to further elucidate the functional significance of these
mutations by using a structural biological approach.
Materials and methods
The three dimensional structure of human ADA2 in
complex with coformycin, a transition state analog, (PDB code 3LGG)
was downloaded from the Protein Data Bank and used to analyze the
consequences to structure and function of the mutations p.Gly47Arg,
p.Gly47Ala, p.Arg8Trp, p.Leu351Gln and p.Ala357Thr. Mutants were
constructed using molecular modeling with the program Maestro
(Schrodinger, LLC) which was also used to analyze the
conformational changes caused by the mutation. Rotational
flexibility on mutated side chains was tested due to the restricted
space in the mutation vicinity and the conformation with the least
bad contacts was adopted. The electrostatic surface potential of
the models was calculated by the Adaptive Poisson-Boltzmann Solver
(APBS) using the PyMOL plug-in with the default parameter settings.
All figures depicting 3D models were created using the molecular
graphics program PyMOL V.2.2 (19).
Results
PAN-associated mutations in ADA2
structure
Taking into account the domain description detailed
in the ‘Introduction’ and the secondary structure elements involved
in their functionality, the 5 PAN-associated mutants examined seem
to be readily involved in functional changes.
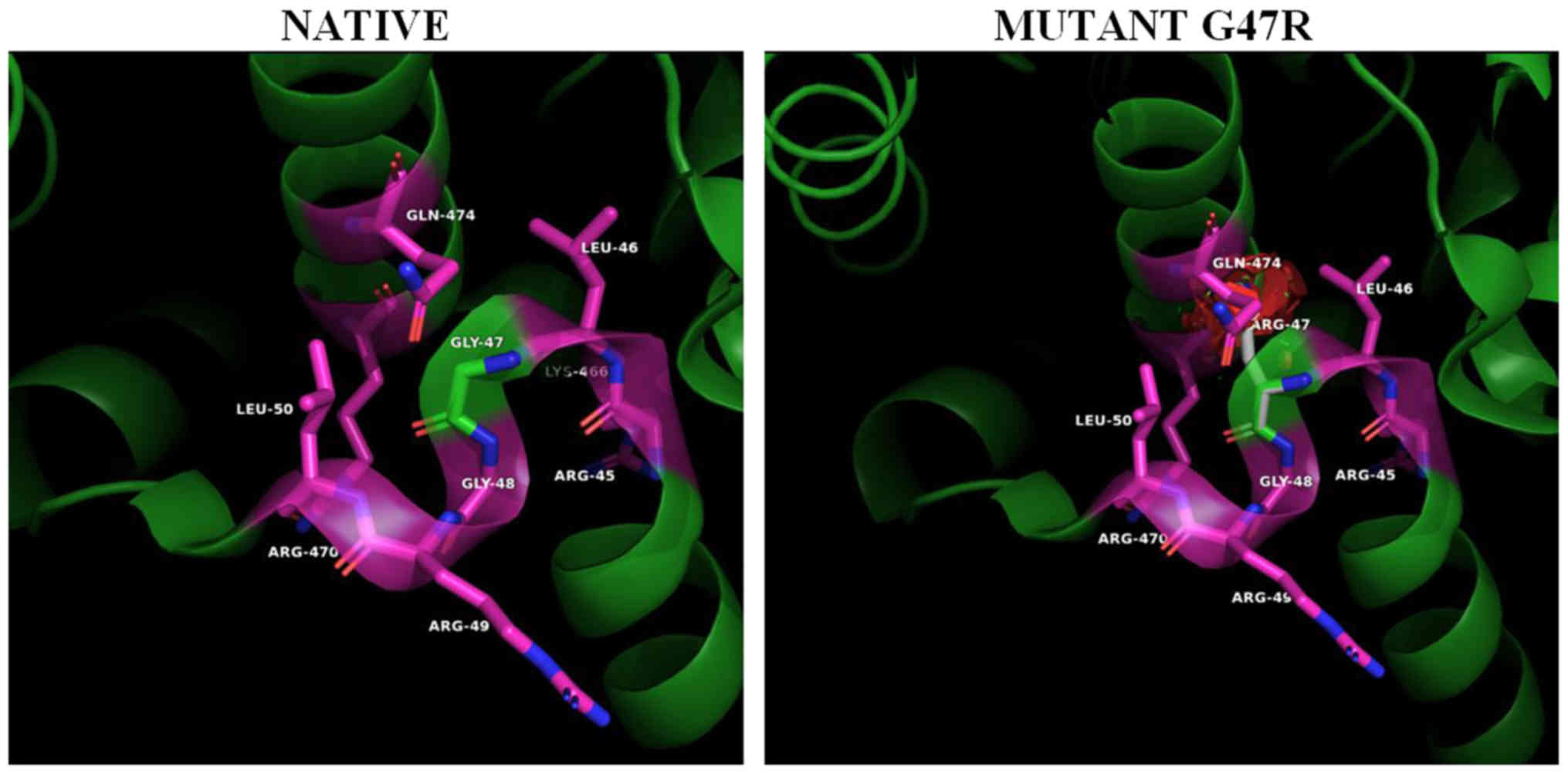
Helix HN1 projects as a finger from its own subunit
and almost entirely interacts with the ADA domain of the
neighboring subunit (Fig. 4, left
panel). This ‘helix anchor’ provides the major contact between
subunits in the dimer that contributes >40% of the hydrophobic
interactive area. Helix HN1 docks to the surface created by helices
a5 and a6. Two highly conserved charged residues of helix HN1,
Arg-34 and Glu-41, are engaged in ionic interactions with the
Asp-373 and Arg369 of the neighboring subunit, respectively
(Fig. 4, left panel). Hydrophobic
Ile-30, Leu-37, Leu- 38, as well as parts of aliphatic chains of
polar Thr-33 and Lys-14 form hydrophobic contacts with residues of
the neighboring subunit. A close examination of the interactions of
the ADA2 dimer interface (Fig.
4A), is illustrating the residue contacts between the two HN1
helix anchors, where Arg34 (blue-gray) is located. The Arg34Trp PAN
mutation (Fig. 4, right panel)
causes severe clashes between the bulky side chain of the
tryptophane 34 side chain and Leu372 of the homodimer's a5 helix as
well as loss of the homodimer stabilizing hydrogen bond interaction
(in yellow dashed lines) between Arg34 (blue-gray) and homomonomer
Asp373 (purple). It is worth noting that in cat's native sequence
where position 34 is occupied by a tryptophane residue the opposing
homodimer's a5 helix residue 373 has been replaced by a leucine
enhancing the hydrophobic interaction between HN1 and a5-helix.
Helix HN1 is followed by a sharp twist and 5-residue
loop, positioned at the right angle relative to the helix. The
twist and loop, as well as helices HN2, HN3 and HN4 pack on the
surface of the ADA domain of own subunit. The loop residues have
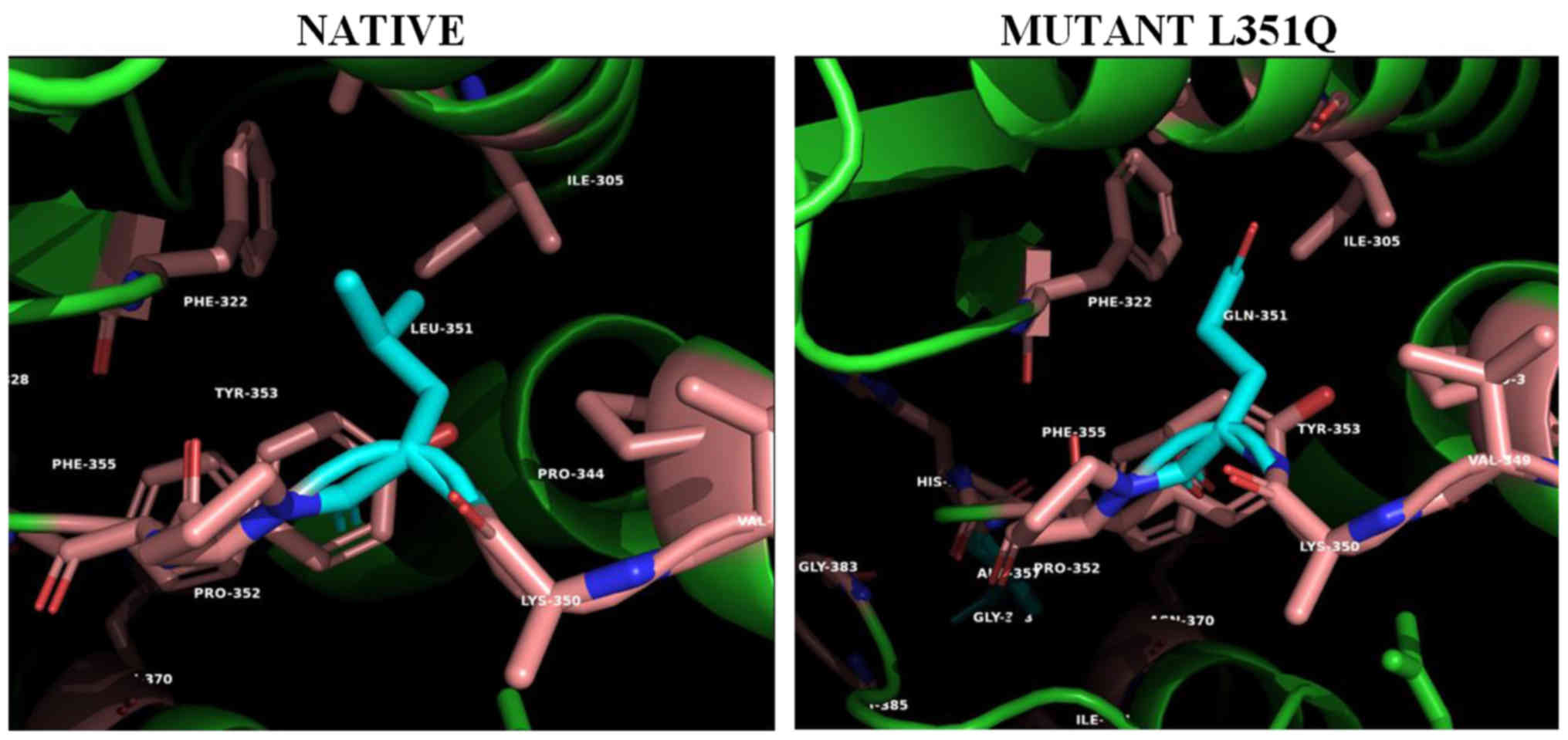
been observed with high conformational stability. Gly47 (Fig. 3, left panel) is located at the top
of the tight turn between the HN1 and HN2 α-helices that support
dimerization. Specifically, the Gly47 amino acid residue (Fig. 3, left panel) is located in the i+2
position of a tight double β turn regulating the position and angle
of the N-terminal HN1 dimer association helix (12). The substitution of the small
non-polar glycine with the much larger and positively charged
arginine (Fig. 3, right panel)
will strongly alter the relative position of HN1 helix with respect
to the a5 and a6 homo-monomer's interacting helices, reducing the
stability of the subunits, weakening the monomer to monomer
anchoring interactions and the formation of stable dimers (20).
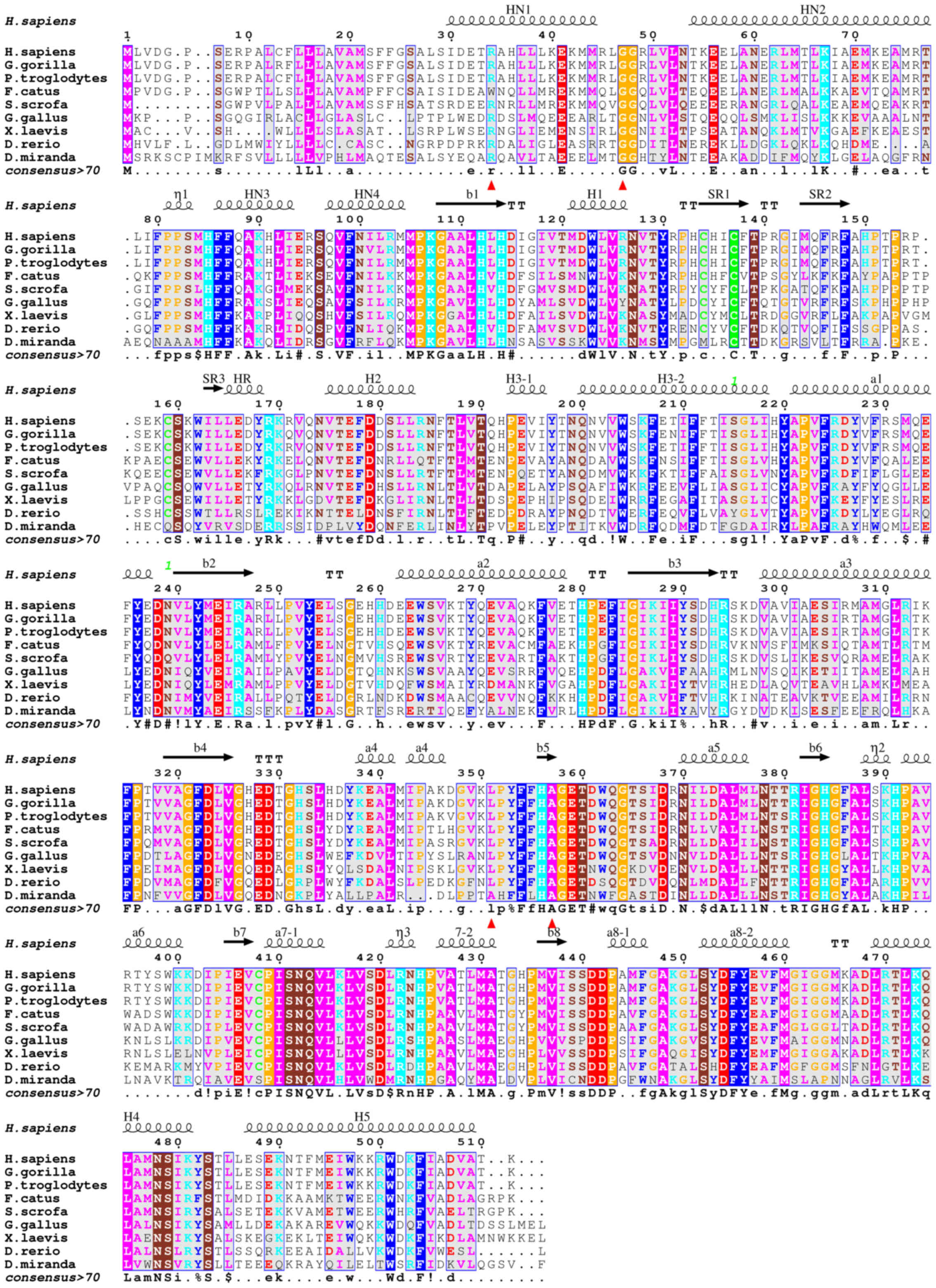
The hydrophobic cluster between helix a4 (residues
338–348) and strand b5 (residues 349–356) is being interrupted with
the introduction of the polar Gln351 (PAN mutant L351Q) in the
Ile305/Phe322-formed hydrophobic pocket (Fig. 2). This disruption could be
transmitted to the neighboring His356 coordinated to the metal ion
or affect the confirmed glycosylation at the next amino acid
residue Asn352 (12).
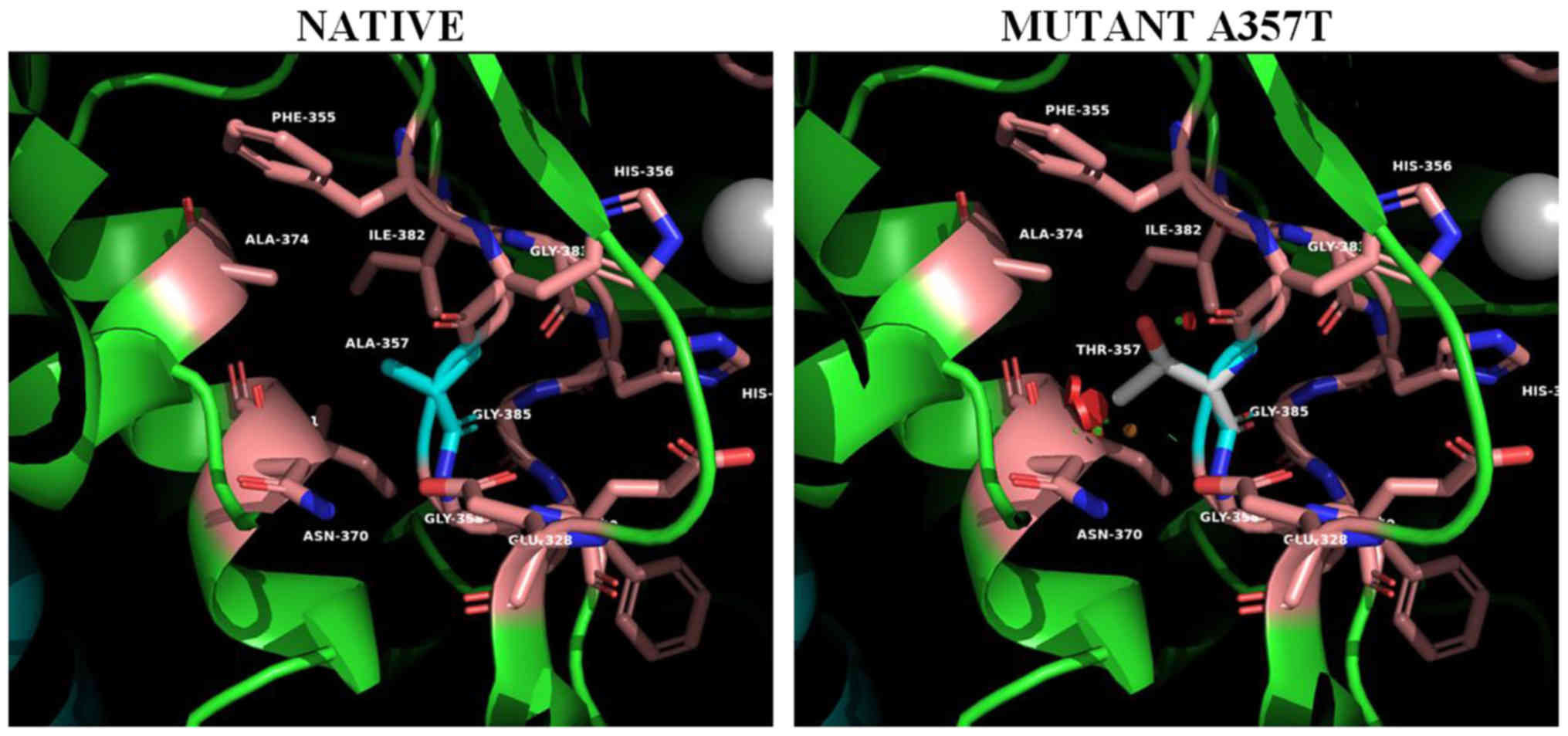
For the mutant on position 357 (Ala357Thr) (Fig. 5), the spatial distortion introduced
to the metal coordination site is even more apparent. The
introduction of the polar threonine side chain located on a β-bulge
(res.356-357) and in contact with the metal coordinated His 356
further distorts metal coordination at the active site influencing
the activity of the enzyme. In addition this mutation creates
unfavorable bad contacts with helix α5.
Discussion
In the present study, the authors sought to analyze
the functional role of rare variants of either known (p.Gly47Arg
and p.Gly47Ala) or novel (p.Arg34Trp, p.Leu351Gln and p.Ala357Thr)
of the ADA2 protein, by using 3-D modeling and assessing the
structural consequences of the respective amino acid substitutions.
The identified variants showing a novel association with DADA2 were
initially predicted to be damaging (18). ADA2 has been considered to be both
the major extracellular adenosine deaminase and an adenosine
deaminase-related growth factor (1). Analysis of the 3D protein structures
of ADA2 conducted by our group, regarding either already known or
novel mutations suggested that the novel mutations found may affect
the formation/stability of the homodimer or influence the active
site of the enzyme (15).
The variation observed in the clinical phenotype may
be attributed to the location of the mutations on the protein
domains. Thus, Navon Elkan et al (1) found that glycine at residue 47 of
ADA2 was highly conserved in a variety of sequenced species, thus
suggesting that its substitution with arginine is predicted to be
highly damaging. In particular, Gly47Arg and Gly47Val possibly
affect the stability of homodimers or their individual subunits and
seem to be the most prominent mutations causing PAN. However, the
disease severity was found to be highly varied from mild type of
the disease limited to the skin, without any constitutional
symptoms to severe, which was finally fatal in some cases (1). As regards the study conducted by
Gibson et al (18), the
authors suggested that Arg8Trp may have benign consequences and
both Leu351Gln and Ala357Thr probably damaging ones. It has been
suggested that the clinical significance of novel variants cannot
be assessed from their allele frequencies only (21). Therefore, their consequences on
protein function must be analyzed by proper biochemical assays
and/or in silico analysis using bioinformatics (22).
Özen et al (23) attempted to perform a possible
genotype-phenotype correlation and reported that dimerization
domain mutations are associated with PAN-like phenotype, while
catalytic domain mutations are associated with hematological
manifestations. Furthermore, homozygous G47R mutation has been
detected in a Jewish patient diagnosed with HHV-8-negative
Castelman disease (24). In
another clinical study involving 10 DADA2 patients from The
Netherlands and Belgium, homozygosity for R169Q mutation was
associated with the presence of cytopenia (25). In two patients carrying a
homozygous deletion of the locus 22q11.1, harboring both copies of
the IL-17 receptor A (IL17RA) and the CECR1 gene,
muco-cutaneous infections and dermatitis were observed (26). By contrast, Gibson et al
(18) were not able to correlate
genotype to phenotype in nine children diagnosed with DADA2, thus
assuming that apart from the ADA2 mutations other factors such as
modifying genes, epigenetic modifications and possibly
environmental factors are also involved in the pathogenesis of
DADA2 and disease expressivity. Therefore, further studies are
warranted in order to better understand the genotype-phenotype
correlation in patients with DADA2.
The findings presented by Gibson et al
(18) are supportive of previous
reports that the disease under investigation appears an extensive
genotypic and phenotypic variability, which cannot be explained by
the importance of each causal mutation regarding the protein
function (1,6). The published case series of patients
with DADA2 revealed a large phenotypic variability that cannot be
fully explained by the impact of causal mutations on protein
function. To the best of our knowledge, this is the first study to
evaluate the structural significance of the ADA2 novel mutations
under discussion. It was concluded that Arg8Trp and Gly47Arg
mutations are affecting the position and interaction of the dimer
associated NH1 helical structure and therefore dimer formation and
stabilization, while Leu351Gln and Ala357Thr influence the metal
coordination in the active site. This information may aid in the
further interpretation of the recent findings of Gibson et
al (18) from the structural
biology point of view and justifies that screening ADA2 in children
with various types of pediatric vasculitis may enable an earlier
diagnosis of DADA2. Moreover, it is important to be aware of the
identity of these monogenic diseases with vasculitic features,
taking into account that any diagnostic delay may be considerable
and the alternative treatments for these diseases differs than
classical vasculitides.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
EE, CM, MM and GNG conceived and designed the study
and drafted the manuscript. EE, GNG, CM, DAS and MIZ searched the
literature. EE, MM, DAS and MIZ analyzed and interpreted the data.
MIZ and DAS critically revised the manuscript. All authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
DAS is the Editor-in-Chief for the journal, but had
no personal involvement in the reviewing process, or any influence
in terms of adjudicating on the final decision, for this article.
All the other authors declare that they have no competing
interests.
References
|
1
|
Navon Elkan P, Pierce SB, Segel R, Walsh
T, Barash J, Padeh S, Zlotogorski A, Berkun Y, Press JJ, Mukamel M,
et al: Mutant adenosine deaminase 2 in a polyarteritis nodosa
vasculopathy. N Engl J Med. 370:921–931. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhou Q, Yang D, Ombrello AK, Zavialov AV,
Toro C, Zavialov AV, Stone DL, Chae JJ, Rosenzweig SD, Bishop K, et
al: Early-onset stroke and vasculopathy associated with mutations
in ADA2. N Engl J Med. 370:911–920. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Caorsi R, Penco F, Schena F and Gattorno
M: Monogenic polyarteritis: The lesson of ADA2 deficiency. Pediatr
Rheumatol Online J. 14:512016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Francès C, Papo T, Wechsler B, Laporte JL,
Biousse V and Piette JC: Sneddon syndrome with or without
antiphospholipid antibodies. A comparative study in 46 patients.
Medicine (Baltimore). 78:209–219. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Van Eyck L Jr, Hershfield MS, Pombal D,
Kelly SJ, Ganson NJ, Moens L, Frans G, Schaballie H, De Hertogh G,
Dooley J, et al: Hematopoietic stem cell transplantation rescues
the immunologic phenotype and prevents vasculopathy in patients
with adenosine deaminase 2 deficiency. J Allergy Clin Immunol.
135:283–287.e5. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ben-Ami T, Revel-Vilk S, Brooks R, Shaag
A, Hershfield MS, Kelly SJ, Ganson NJ, Kfir-Erenfeld S, Weintraub
M, Elpeleg O, et al: Extending the Clinical Phenotype of Adenosine
Deaminase 2 Deficiency. J Pediatr. 177:316–320. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cipe FE, Aydogmus C, Serwas NK,
Keskindemirci G and Boztuğ K: Novel mutation in CECR1 leads to
deficiency of ADA2 with associated neutropenia. J Clin Immunol.
38:273–277. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Westendorp WF, Nederkoorn PJ,
Aksentijevich I, Hak AE, Lichtenbelt KD and Braun KP: Unexplained
early-onset lacunar stroke and inflammatory skin lesions: Consider
ADA2 deficiency. Neurology. 84:2092–2093. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Elbracht M, Mull M, Wagner N, Kuhl C,
Abicht A, Kurth I, Tenbrock K and Häusler M: Stroke as initial
manifestation of adenosinedeaminase 2 deficiency. Neuropediatrics.
48:111–114. 2017.PubMed/NCBI
|
|
10
|
Sahin S, Adrovic A, Barut K, Ugurlu S,
Turanli ET, Ozdogan H and Kasapcopur O: Clinical, imaging and
genotypical features of three deceased and five surviving cases
with ADA2 deficiency. Rheumatol Int. 38:129–136. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bulut E, Erden A, Karadag O, Oguz KK and
Ozen S: Deficiency of adenosine deaminase 2; special focus on
central nervous system imaging. J Neuroradiol. 46:193–198. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zavialov AV, Gracia E, Glaichenhaus N,
Franco R, Zavialov AV and Lauvau G: Human adenosine deaminase 2
induces differentiation of monocytes into macrophages and
stimulates proliferation of T helper cells and macrophages. J
Leukoc Biol. 88:279–290. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zavialov AV and Engström A: Human ADA2
belongs to a new family of growth factors with adenosine deaminase
activity. Biochem J. 391:51–57. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Haskó G, Linden J, Cronstein B and Pacher
P: Adenosine receptors: Therapeutic aspects for inflammatory and
immune diseases. Nat Rev Drug Discov. 7:759–770. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Meyts I and Aksentijevich I: Deficiency of
Adenosine Deaminase 2 (DADA2): Updates on the Phenotype, Genetics,
Pathogenesis, and Treatment. J Clin Immunol. 38:569–578. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wilson DK, Rudolph FB and Quiocho FA:
Atomic structure of adenosine deaminase complexed with a
transition-state analog: Understanding catalysis and
immunodeficiency mutations. Science. 252:1278–1284. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Charlab R, Valenzuela JG, Andersen J and
Ribeiro JM: The invertebrate growth factor/CECR1 subfamily of
adenosine deaminase proteins. Gene. 267:13–22. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gibson KM, Morishita KA, Dancey P,
Moorehead P, Drögemöller B, Han X, Graham J, Hancock REW, Foell D,
Benseler S, et al PedVas Investigators Network, : Identification of
novel adenosine deaminase 2 gene variants and varied clinical
phenotype in pediatric vasculitis. Arthritis Rheumatol.
71:1747–1755. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schrödinger LLC: The PyMOL Molecular
Graphics System 2016 version 2.2. simplepymol.org/2/support.htmlMarch 5–2019
|
|
20
|
Kinoshita T, Nakanishi I, Terasaka T, Kuno
M, Seki N, Warizaya M, Matsumura H, Inoue T, Takano K, Adachi H, et
al: Structural basis of compound recognition by adenosine
deaminase. Biochemistry. 44:10562–10569. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Meyts I, Bosch B, Bolze A, Boisson B, Itan
Y, Belkadi A, Pedergnana V, Moens L, Picard C, Cobat A, et al:
Exome and genome sequencing for inborn errors of immunity. J
Allergy Clin Immunol. 138:957–969. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Caorsi R, Penco F, Grossi A, Insalaco A,
Omenetti A, Alessio M, Conti G, Marchetti F, Picco P, Tommasini A,
et al: ADA2 deficiency (DADA2) as an unrecognised cause of early
onset polyarteritis nodosa and stroke: A multicentre national
study. Ann Rheum Dis. 76:1648–1656. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Özen S, Batu ED, Taskiran EZ, Özkara HA,
Ünal S, Güleray N, Erden A, Karadağ Ö, Gümrük F, et al: A monogenic
disease with a variety of phenotypes: deficiency of adenosine
deaminase 2. J Rheum. May 1–2019.(Epub ahead of print). View Article : Google Scholar
|
|
24
|
Van Eyck L, Liston A and Wouters C: Mutant
ADA2 in vasculopathies. N Engl J Med. 371:4802014.PubMed/NCBI
|
|
25
|
Van Montfrans JM, Hartman EA, Braun KP,
Hennekam EA, Hak EA, Nederkoorn PJ, Westendorp WF, Bredius RG,
Kollen WJ, Schölvinck EH, et al: Phenotypic variability in patients
with ADA2 deficiency due to identical homozygous R169Q mutations.
Rheumatology (Oxford). 55:902–910. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fellmann F, Angelini F, Wassenberg J,
Perreau M, Arenas Ramirez N, Simon G, Boyman O, Demaria O,
Christen-Zaech S, Hohl D, et al: IL-17 receptor A and adenosine
deaminase 2 deficiency in siblings with recurrent infections and
chronic inflammation. J Allergy Clin Immunol. 137:1189–1196.e2.
2016. View Article : Google Scholar : PubMed/NCBI
|