Introduction
T-cell acute lymphoblastic leukemia (T-ALL) is a
type of malignant hematopoietic tumor caused by malignant
transformation of T lymphocytes. T-ALL has a poor prognosis and it
is difficult to achieve complete remission after recurrence through
chemotherapy alone (1). Therefore,
there is a requirement for novel therapeutic agents. Abnormal
proliferation, disordered differentiation and inhibition of
apoptosis are implicated in the pathogenesis and recurrence of
leukemia. Cellular Fas-associated death domain-like interleukin-1β
converting enzyme inhibitory protein long form (c-FLIPL)
is an important anti-apoptotic protein, which has been reported as
an important factor affecting apoptosis sensitivity and has been
demonstrated to exert a significant inhibitory effect on apoptosis
(2). A previous study revealed
that c-FLIPL is highly expressed in leukemia (3). It has been reported that
c-FLIPL is highly expressed in bone marrow mononuclear
cells obtained from patients with T-ALL and is associated with the
malignancy and prognosis of T-ALL.
Histone deacetylase (HDAC) inhibitors (HDACis)
comprise an important class of epigenetic drugs with high efficacy
and low toxicity, which are becoming valuable antitumor drugs. It
has been reported that HDACis successfully inhibit proliferation
and increase apoptosis in various malignant tumor cells by
decreasing c-FLIPL transcription and translation
(4). Chidamide is a novel HDACi
that has been independently researched and developed in China,
which has been approved by the US Food and Drug Administration.
Zheng et al (5) reported
that patients with peripheral T-cell lymphoma exhibited increased
c-FLIPL expression, and that HDACis inhibited the
expression of c-FLIPL and increased apoptosis in T-cell
lymphoma (5).
Necroptosis is a novel type of cell death that
exhibits characteristics of apoptosis and necrosis, and is
regulated by a series of signaling molecules independent of
caspases (6). Furthermore,
necroptosis is inhibited by the specific inhibitor necrostatin-1
(Nec-1) (7). Necroptosis has
attracted increasing attention due to its close association with
inflammation, ischemia-reperfusion injury, and tumor occurrence and
development (6,8). In general, activation of death
receptors induces apoptosis; however, cytokine-mediated apoptosis
can also initiate necroptosis if apoptosis is inhibited or blocked
(9). Certain leukemia cell lines
(such as Jurkat cells) have congenital caspase function deficiency;
hence, necroptosis is more likely to occur in these cell lines
(10). The present study
investigated the necroptosis-inducing effects of chidamide on
Jurkat and HUT-78 T-cell leukemia cells and explored the underlying
mechanism.
Materials and methods
Cell culture
Jurkat, HUT-78 and H9 cells (Shanghai Cell Bank of
Chinese Academy of Sciences) were cultured in RPMI-1640 medium
supplemented with 10% fetal bovine serum (both from Gibco; Thermo
Fisher Scientific, Inc.), 100 units penicillin and 100 mg/ml
streptomycin at 37°C in a humidified atmosphere containing 5%
CO2.
Reverse-transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from Jurkat, HUT-78 and H9
cells using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol. Total
RNA was reverse transcribed into cDNA using a Super MMLV RT kit
(Takara Bio, Inc.), according to the manufacturer's instructions.
The reaction was carried out at 37°C for 15 min, 85°C for 5 sec and
then terminated at 4°C. For qPCR, the reaction solution was
prepared on ice in five replicate wells per cell type. The 20-µl
reaction mixture consisted of 10 µl 2X SYBR Premix Ex Taq II
(Takara Bio, Inc.), 0.8 µl forward primer (10 µmol/l), 0.8 µl
reverse primer (10 µmol/l), 0.4 µl ROX Reference Dye or Dye II
(50X), 6 µl cDNA solution and 6 µl dH2O. The primer
sequences were as follows: c-FLIPL, forward,
5′-GCGAAGCCACCTGTTGATG-3′ and reverse, 5′-CTGCTCCGCAAAACCAGAA-3′;
and β-actin, forward, 5′-TGGCACCCAGCACAATGAA-3′ and reverse,
5′-CTAAGTCATAGTCCGCCTAGAAGCA-3′. The amplification conditions were
as follows: Pre-denaturation at 95°C for 30 sec, followed by 40
cycles at 95°C for 5 sec and 60°C for 34 sec. The mRNA expression
levels of c-FLIPL were quantified using the
2−ΔΔCq method and normalized to the internal reference
gene β-actin (11).
Western blotting
Cells were harvested and lysed in lysis buffer
(Beyotime Institute of Biotechnology). Lysates were centrifuged at
16,099.2 × g for 1 min at 4°C and the supernatants were collected.
Total cellular protein was extracted by using lysis buffer
(Wanleibio) and the protein concentration was determined by the
bicinchoninic acid assay. In total, 20 µl protein extracts were
separated by 10 or 12% polyacrylamide gel electrophoresis and
transferred onto polyvinylidene fluoride membranes (EMD Millipore).
The membranes were blocked in 5% skim milk solution and incubated
with primary antibodies against HDAC1 (ABclonal, Inc.; cat. no.
A0238; 1:500), HDAC3 (ABclonal, Inc.; cat. no. A2603; 1:500),
c-FLIPL (ProteinTech Group, Inc.; cat. no. 10394-1-AP;
1:800), mixed lineage kinase domain-like pseudokinase (MLKL) (Cell
Signaling Technology, Inc.; cat. no. 14993S; 1:1,000),
phosphorylated (p)-MLKL (Cell Signaling Technology, Inc.; cat. no.
91689S; 1:1,000), receptor-interacting protein kinase (RIP) 3 (BD
Bioscience; cat. no. 610458; 1:1,500), caspase-3 (ABclonal Inc.;
cat. no. A11021; 1:500), caspase-8 (ProteinTech Group, Inc.; cat.
no. 13423-1-AP; 1:1,500), RIP1 (ProteinTech Group, Inc.; cat. no.
17519-1-AP; 1:1,000), cyclin-dependent kinase inhibitor 1A (p21;
Wanleibio; cat. no. WL0362; 1:400), proliferating cell nuclear
antigen (PCNA; Wanleibio; cat. no. WL02208; 1:500), β-actin
(Wanleibio; cat. no. WL01845; 1:5,000) and GAPDH (ProteinTech
Group, Inc.; cat. no. 60004-1-Ig; 1:10,000) at 4°C overnight.
Following primary antibody incubation, the membrane was incubated
with a secondary antibody (Wanleibio; cat. no. WLA023; 1:5,000)
37°C for 45 min. Protein bands were visualized using enhanced
chemiluminescence (Wanleibio; cat. no. WLA003).
Cell treatment
Jurkat and HUT-78 cells were divided into five
groups: i) Control group, untreated cells cultured at 37°C; ii)
chidamide-treated group [cells were treated with 2 µmol/l chidamide
(Sigma-Aldrich; Merck KGaA) at 37°C for 24 h]; iii)
Z-VAD-FMK-treated group [following pretreatment with 50 µmol/l
Z-VAD-FMK (Abcam) for 30 min, cells were treated with 2 µmol/l
chidamide at 37°C for 24 h]; iv) Nec-1-treated group [following
pretreatment with 20 µmol/l Nec-1 (Abcam) for 1 h, cells were
treated with 2 µmol/l chidamide at 37°C for 24 h]; and v) Z-VAD-FMK
and Nec-1-treated group (following pretreatment with 50 µmol/l
Z-VAD-FMK for 30 min and 20 µmol/l Nec-1 for 1 h sequentially,
cells were treated with 2 µmol/l chidamide at 37°C for 24 h).
Apoptosis analysis
Annexin V-FITC/propidium iodide (PI) staining kit
(BD Biosciences) was used to detect apoptosis in Jurkat and HUT-78
cells. Cells were collected by centrifugation at 71.552 × g for 5
min at 4°C. The supernatant was removed and the cells were
resuspended in 500 µl PBS. The cells were then incubated with 5 µl
Annexin V-FITC and PI staining reagent in the dark for 15 min at
room temperature. Living cells are negative to Annexin V-FITC and
PI (Annexin V−PI−), early apoptotic cells are
positive to Annexin V-FITC and negative to PI (Annexin
V+PI−), late apoptotic cells and necrotic
cells are double positive to Annexin V-FITC and PI (Annexin
V+PI+). Necroptosis and apoptosis were
subsequently analyzed using a flow cytometer (NovoCyte; ACEA
Biosciences, Inc.) and quantified by NovoExpress 1.2.5 (ACEA
Biosciences, Inc.).
Transmission electron microscopy
Jurkat and HUT-78 cells were fixed using 2.5%
glutaraldehyde (Servicebio) at 4°C for 2–4 h, dehydrated in a
graded series of ethanol, embedded in epoxy resin and sectioned
into ultra-thin sections (60–80 nm). The sections were subsequently
stained with 2% uranyl acetate and lead citrate at room temperature
for 15 min. Finally, features of necroptosis were visualized using
transmission electron microscopy (JEOL; cat. no. JEM-1400) and
analyzed by JEOL TEM Center Ver.1.7.15 (JEOL, Ltd.).
Mitochondrial function assay
JC-1 staining solution (Beyotime Institute of
Biotechnology) was used to determine the mitochondrial membrane
potential of treated Jurkat and HUT-78 cells, according to the
manufacturer's protocol. The change in mitochondrial membrane
potential was analyzed using a flow cytometer (NovoCyte) and
quantified by NovoExpress (Novoexpress 1.2.5). In order to assess
the production of reactive oxygen species (ROS), Jurkat and HUT-78
cells were treated with DCFH-DA solution at 37°C for 20 min
according to the manufacturer's instructions (KeyGEN BioTECH) and
were subsequently quantified using a flow cytometer (NovoCyte) and
quantified by NovoExpress (Novoexpress 1.2.5).
Patient samples
A total of 75 patients with T-ALL (46 men and 29
women; mean age, 20 years) were recruited from Shengjing Hospital
of China Medical University (Shenyang, China) between January 2014
and December 2015. A total of 30 healthy individuals (19 men and 11
women; mean age, 29 years) diagnosed with anemia or
lymphadenectasis served as the control group. Blood samples were
collected from the patients and controls. Blood sample collection
was conducted after the participants provided written informed
consent. Blood samples were analyzed using an automatic blood
analysis pipeline (XN-9000; SYSMEX Corporation). If the
participants were <18 years old, written informed consent was
obtained from their parents/guardians. The present study was in
accordance with the Declaration of Helsinki and was approved by the
Ethics Committee of Shengjing Hospital of China Medical
University.
Statistical analysis
Data are expressed as the mean ± SD. SPSS software
(version 17; SPSS, Inc.) was used to perform statistical tests. All
experiments were repeated three times (n=3). Two sample groups were
compared using an unpaired Student's t-test. Data from three or
more groups were analyzed by one-way analysis of variance followed
by Tukey's multiple comparison test. P<0.05 was considered to
indicate a statistically significant difference.
Results
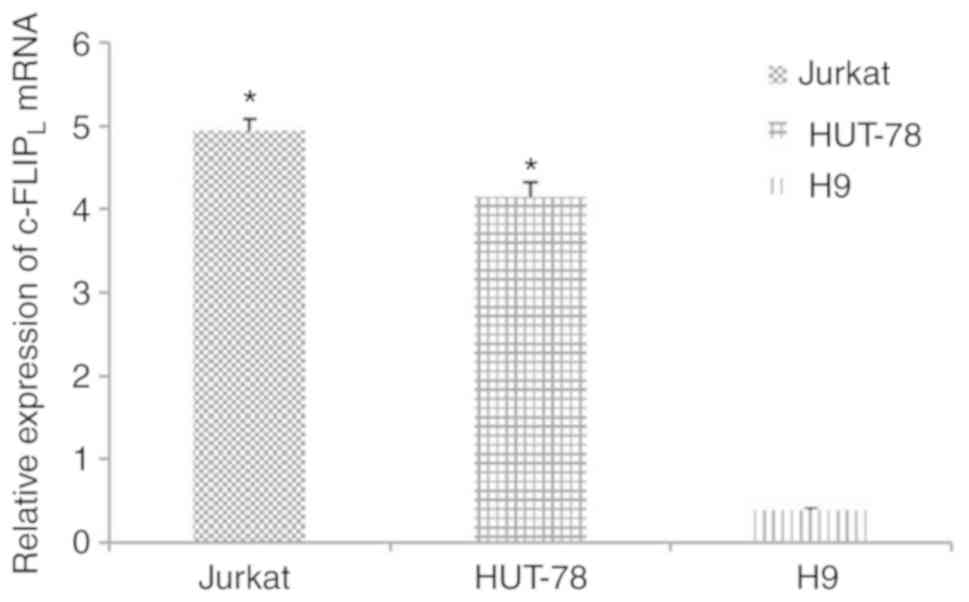
c-FLIPL is highly expressed
in Jurkat and HUT-78 cells
The mRNA expression levels of c-FLIPL
were significantly increased in Jurkat (4.94±0.14; n=5) and HUT-78
cells (4.15±0.17; n=5) compared with in human T lymphocyte H9 cells
(0.39±0.03; n=5; Fig. 1;
P<0.05).
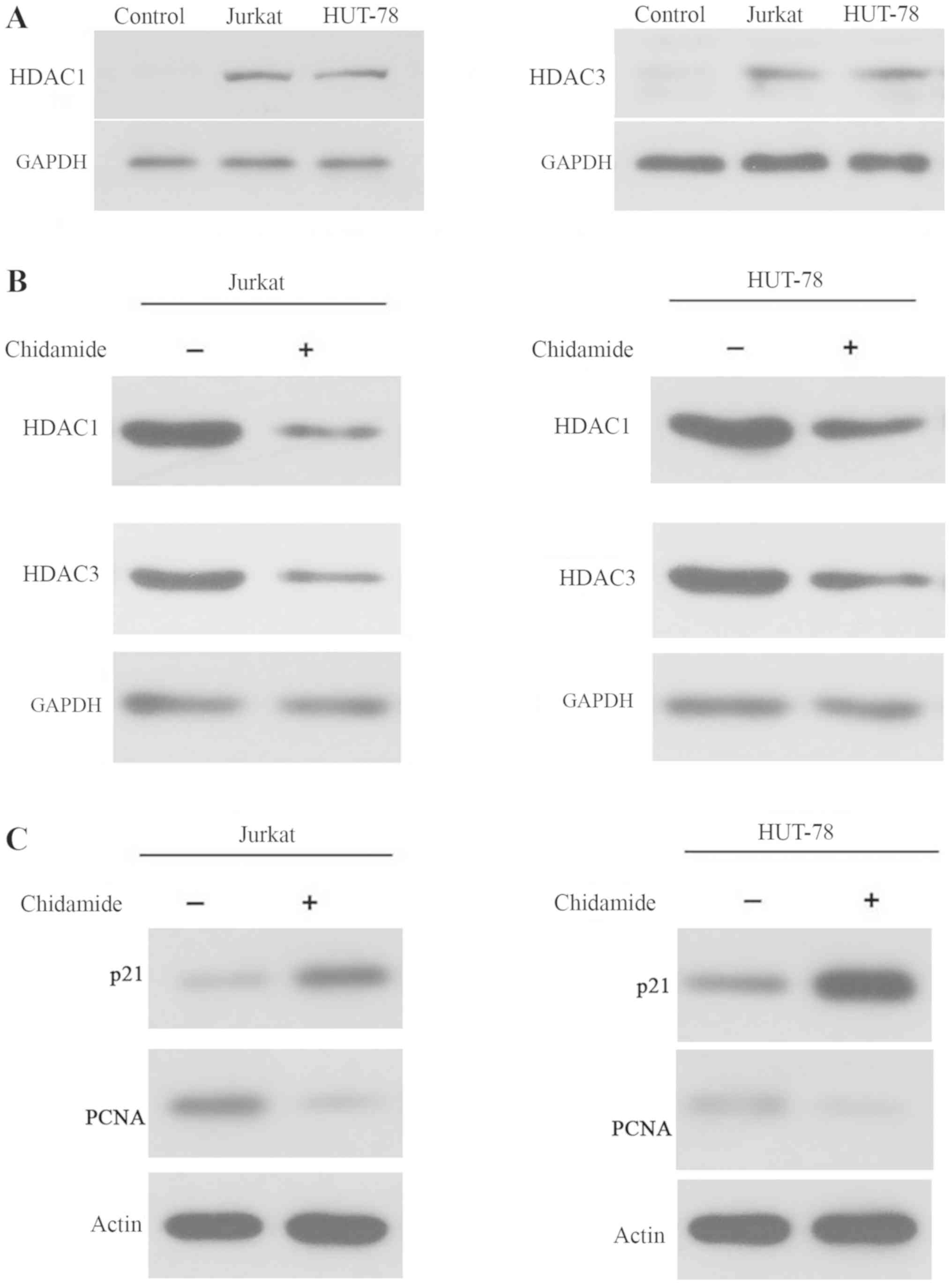
Chidamide inhibits histone
deacetylation in Jurkat and HUT-78 cell lines
As shown in Fig.
2A, the expression levels of HDAC1 and HDAC3 were increased in
Jurkat and HUT-78 cells, indicating that HDAC levels were elevated
in T-cell leukemia cell lines. Treatment with 2 µmol/l chidamide
markedly reduced the expression levels of HDAC1 and HDAC3 in Jurkat
and HUT-78 cells (Fig. 2B).
Additionally, p21 and PCNA levels were increased and decreased,
respectively, in 2 µmol/l chidamide-treated Jurkat and HUT-78 cells
(Fig. 2C). These data indicated
that chidamide inhibited histone deacetylation and regulated cell
cycle arrest, as p21 and PCNA are involved in regulation of the
cell cycle, in T-cell leukemia cell lines.
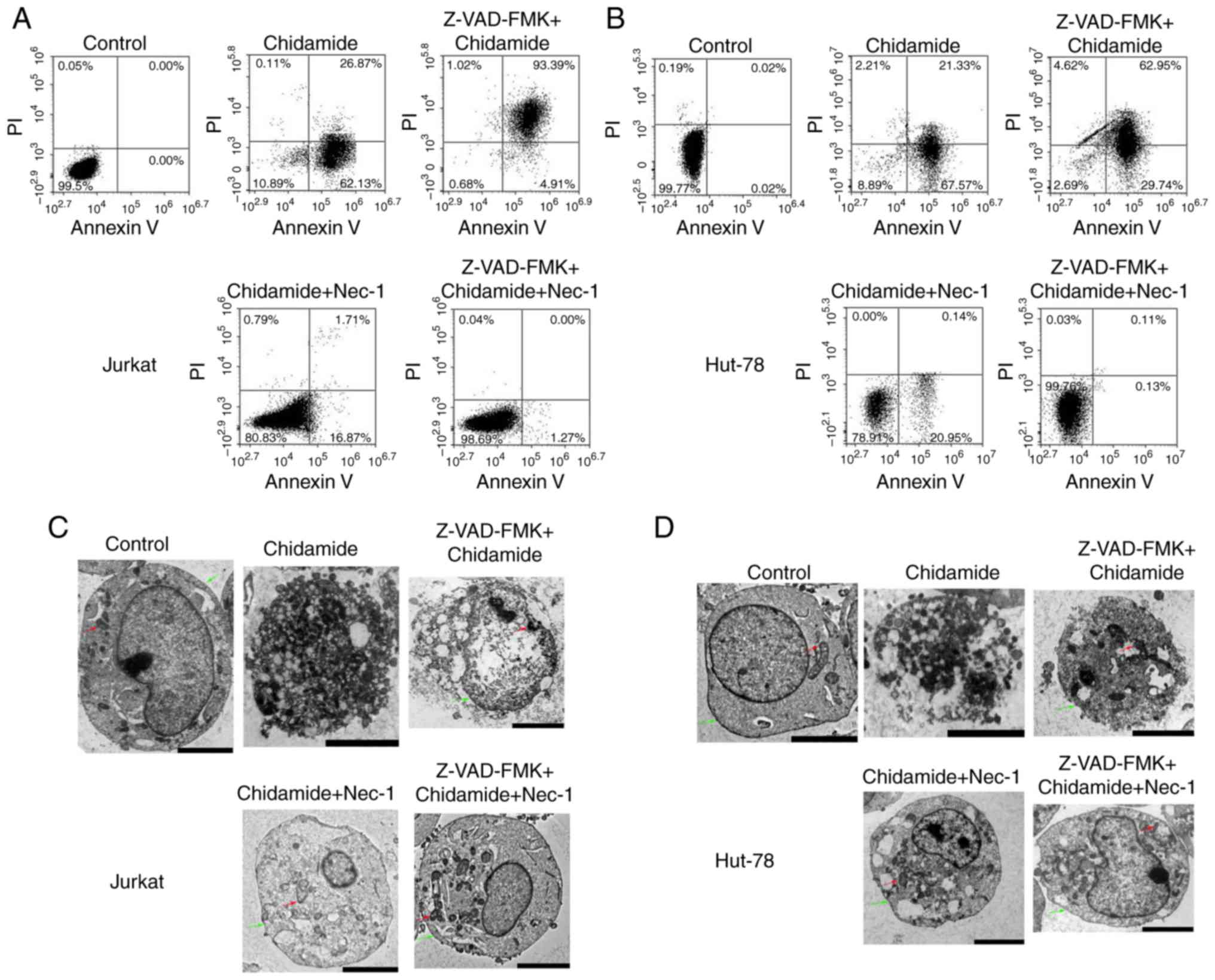
Chidamide induces necroptosis and
apoptosis in Jurkat and HUT-78 cell lines
According to the fluorescence intensities of Annexin
V and PI staining, Annexin V+/PI+ cells were
defined as late apoptotic and necroptotic cells, whereas Annexin
V+/PI− cells were categorized as early
apoptotic cells (12). As shown in
Fig. 3A, cell death was markedly
increased in Jurkat cells compared with in control cells following
treatment with 2 µmol/l chidamide for 24 h (early apoptotic cells,
62.13%; necroptotic and late apoptotic cells, 26.87%); the
proportion of early apoptotic cells was higher. Pretreatment of
cells with 50 µmol/l Z-VAD-FMK (pan-caspase inhibitor) for 30 min
followed by the addition of chidamide (2 µmol/l) significantly
increased necroptosis and late apoptosis (93.39%). The proportion
of necroptotic and late apoptotic cells induced by chidamide (2
µmol/l) was significantly reduced (1.71%) following Nec-1 treatment
(20 µmol/l) for 1 h. Compared with the control group,
Chidamide-induced cell death was not significantly different
following treatment with Z-VAD-FMK and Nec-1. HUT-78 cell death was
also significantly increased following treatment with 2 µmol/l
chidamide, and a high proportion of early apoptotic cells was
observed compared with the control group (early apoptotic cells,
67.57%; necroptotic and late apoptotic cells, 21.33%; Fig. 3B). Pretreatment with Z-VAD-FMK (50
µmol/l) significantly increased the proportion of necroptotic and
late apoptotic cells (62.95%). However, Nec-1 treatment (20 µmol/l)
significantly reduced the proportion of necroptotic and late
apoptotic cells (0.14%). Following pretreatment with Z-VAD-FMK (50
µmol/l) and Nec-1 (20 µmol/l), chidamide-induced necroptosis/late
apoptosis and early apoptosis was reduced to 0.13 and 0.11%,
respectively. As shown in Fig. 3C and
D, membranolysis, mitochondrial swelling and organelle
disappearance were observed in 2 µmol/l chidamide-treated cells. In
addition, representative apoptotic and necroptotic features could
be reversed by Z-VAD-FMK (50 µmol/l) and Nec-1 (20 µmol/l)
pretreatment. These results suggested that chidamide induced
necroptosis in Jurkat and HUT-78 cell lines.
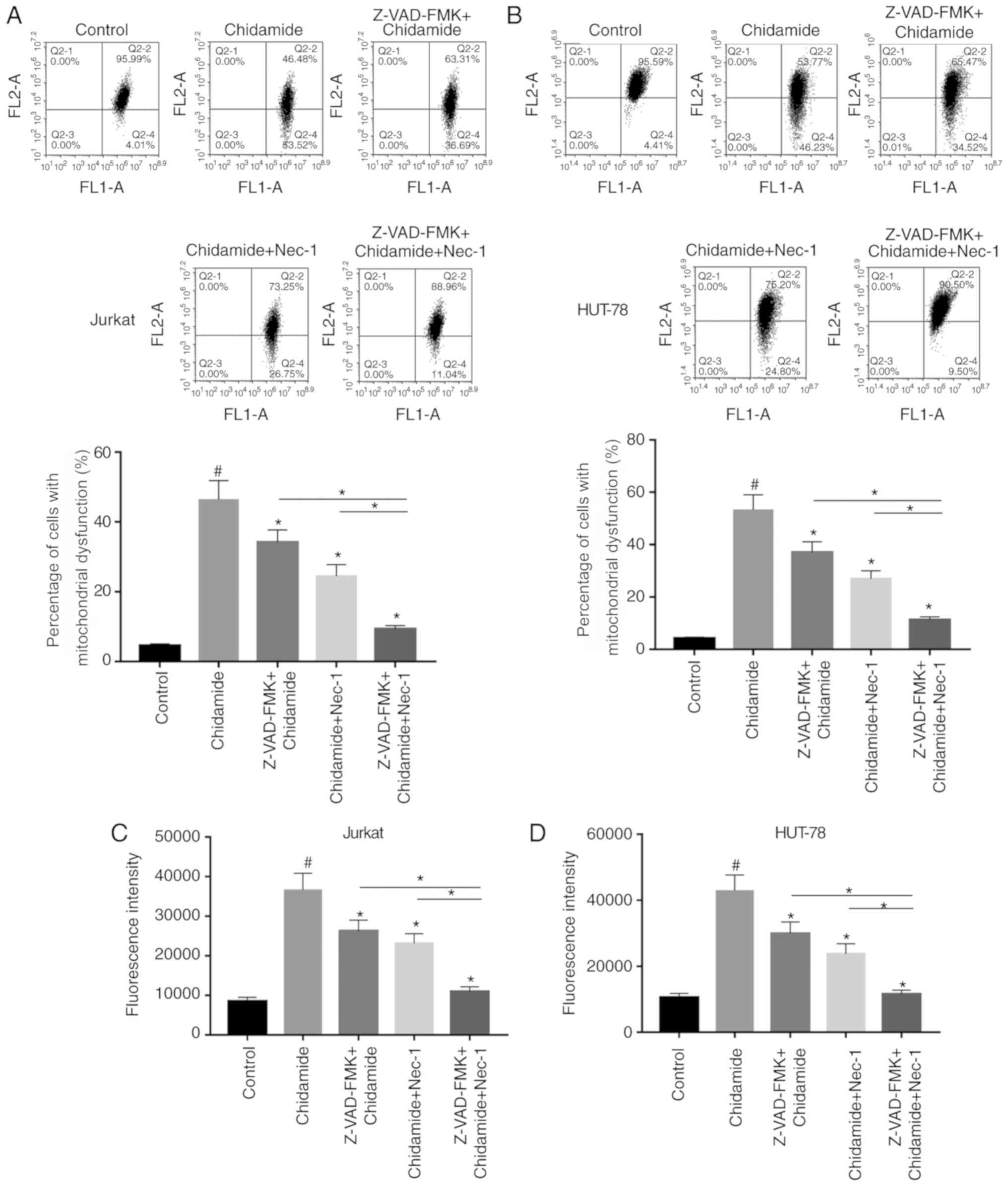
Chidamide aggravates mitochondrial
dysfunction in Jurkat and HUT-78 cells
The effects of chidamide on mitochondrial function
in Jurkat and HUT-78 cells were investigated. JC-1 fluorescent dye
was used to detect alterations in mitochondrial membrane potential
following chidamide treatment (2 µmol/l). Chidamide induced
mitochondrial dysfunction, which could be alleviated by single or
combined pretreatment with Z-VAD-FMK (50 µmol/l) and Nec-1 (20
µmol/l) (Fig. 4A and B;
P<0.05). ROS generation was also analyzed, and the results were
consistent with the results of mitochondrial function detection
mentioned above (Fig. 4C and D;
P<0.05). The present results suggested that chidamide induced
mitochondrial dysfunction in Jurkat and HUT-78 cells.
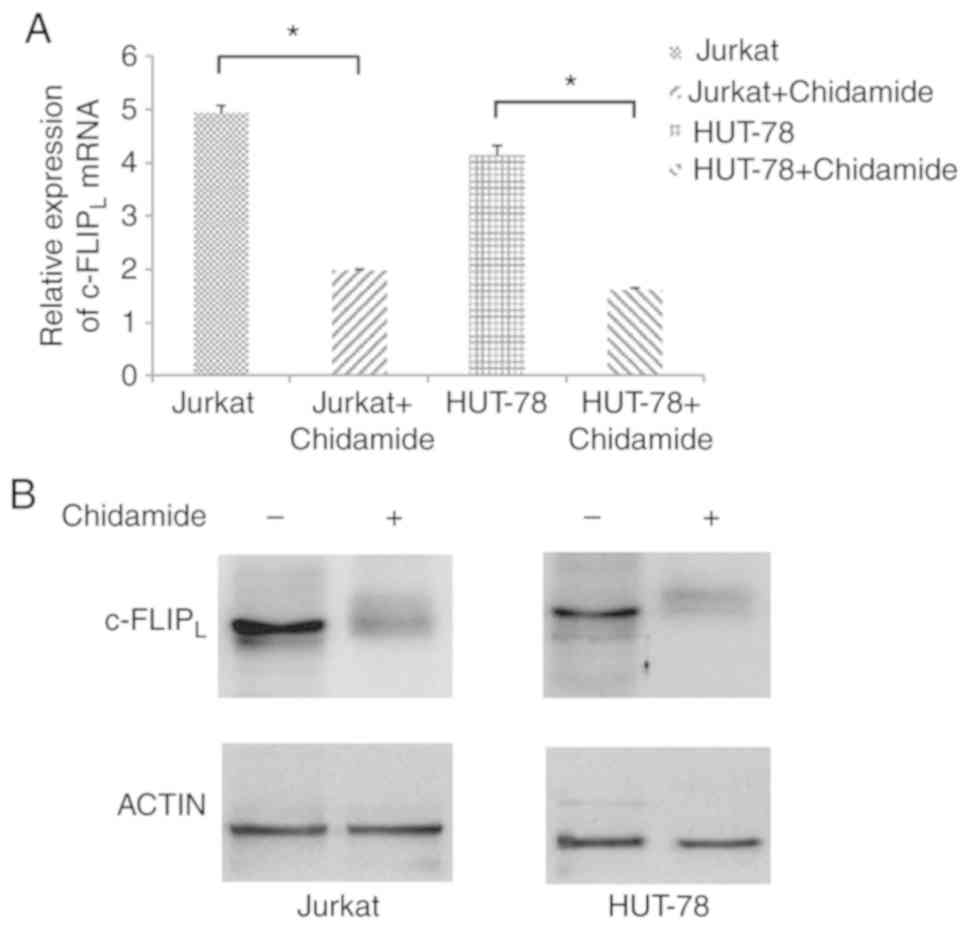
Chidamide downregulates
c-FLIPL expression in Jurkat and HUT-78 cells
The mRNA expression levels of c-FLIPL
were significantly decreased in Jurkat and HUT-78 cells following
chidamide treatment (2 µmol/l; Fig.
5A; P<0.05). In addition, the protein expression levels of
c-FLIPL were decreased in Jurkat and HUT-78 cell lines
following treatment with chidamide (2 µmol/l; Fig. 5B), indicating that chidamide
downregulated c-FLIPL mRNA and protein expression in
Jurkat and HUT-78 cells.
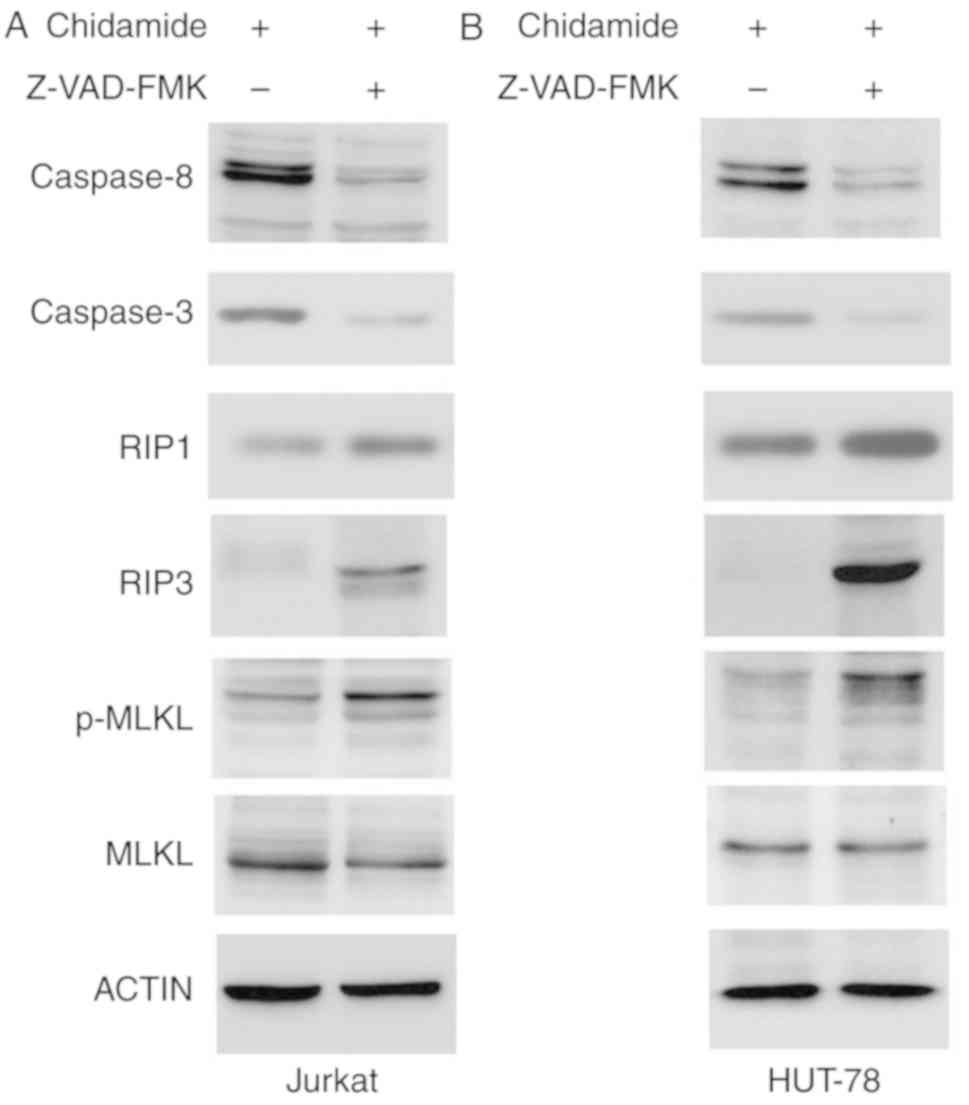
Effects of chidamide on caspase
activation and RIP3/MLKL signaling
Chidamide induced apoptosis of Jurkat and HUT-78
cells. The expression levels of caspase-3 and caspase-8 were
markedly reduced following Z-VAD-FMK pretreatment (50 µmol/l),
indicating that chidamide-induced apoptosis was inhibited by
caspase inhibition. Z-VAD-FMK pretreatment (50 µmol/l) also
increased the expression of the key necroptosis proteins RIP1 and
RIP3, and enhanced phosphorylation of MLKL (Fig. 6). These results indicated that
chidamide induced necroptosis via the RIP3/MLKL signaling pathway
when apoptosis was inhibited.
High levels of c-FLIPL
predict poor prognosis in patients with T-ALL
As shown in Table
I, the mRNA expression levels of c-FLIPL were not
associated with a specific age group or sex. However, according to
the National Comprehensive Cancer Network Guidelines for diagnosing
and treating pediatric acute lymphoblastic leukemia (13), high c-FLIPL mRNA
expression was closely associated with dangerous degree, complete
remission rate, levels of hydroxybutyrate and lactate
dehydrogenase, leukocyte counts, CD45 expression and silver (mouse
homolog) like-TAL bHLH transcription factor 1 erythroid
differentiation factor gene fusion.
 | Table I.Clinical features of patients with
T-ALL in the present study. |
Table I.
Clinical features of patients with
T-ALL in the present study.
| Clinical
feature | n | c-FLIPL
mRNA expression | P-value |
|---|
| Age (years) |
|
| 0.536 |
|
<1 | 1 | 3.16±0.00 |
|
|
1–14 | 25 | 2.94±0.25 |
|
|
≥14 | 20 | 3.80±0.34 |
|
| Sex |
|
| 0.066 |
|
Male | 28 | 3.65±0.22 |
|
|
Female | 18 | 2.80±0.03 |
|
| Dangerous
degree |
|
| <0.0001 |
|
High | 27 | 4.37±0.33 |
|
|
Low | 19 | 1.82±0.23 |
|
| Leukocyte count
(109/l) |
|
| <0.0001 |
|
≥100 | 11 | 4.90±0.37 |
|
|
<100 | 35 | 2.82±0.04 |
|
| HBDH (U/l) |
|
| <0.0001 |
|
≥1,000 | 17 | 4.37±0.41 |
|
|
<1,000 | 29 | 2.70±0.40 |
|
| LDH (U/l) |
|
| 0.002 |
|
≥1,000 | 14 | 4.37±0.45 |
|
|
<1,000 | 32 | 2.86±0.44 |
|
| CD45 |
|
| <0.0001 |
|
Positive | 33 | 3.99±0.29 |
|
|
Negative | 13 | 1.62±0.46 |
|
| Fusion gene |
|
| 0.003 |
|
Negative | 40 | 3.09±0.46 |
|
|
SIL-TAL1 | 5 | 5.22±0.47 |
|
| CR in initial
chemotherapy |
|
| 0.011 |
|
Yes | 41 | 3.15±0.54 |
|
| No | 5 | 4.65±0.64 |
|
Discussion
Aberrant apoptosis and proliferation are important
mechanisms underlying the pathogenesis and recurrence of T-ALL. The
majority of chemotherapeutic drugs used in leukemia induce
apoptosis of cancer cells, which are initially sensitive to
apoptosis inducers but may develop resistance following prolonged
treatment. Additionally, hyper-malignant T-ALL cells demonstrate
primary drug resistance. A previous study has revealed that
multidrug resistance in leukemia cells caused by P-glycoprotein,
multidrug resistance associated protein 1, ATP binding cassette
subfamily G member 2 (Junior blood group), BCL2 apoptosis regulator
and BCL2 like 1 may be overcome by inducing necroptosis (14).
c-FLIPL is an apoptosis-inhibiting
protein widely found in viruses, eukaryotes and mammals, which was
first identified in malignant melanoma by Irmler et al
(15) in 1997. Similar to the
function of pro-caspase-8, c-FLIP binds to Fas-associated protein
with death domain and competes with caspase-8 for a binding site on
the death-inducing signaling complex. This leads to incomplete
activation of caspase-8, thereby inhibiting apoptosis mediated by
death receptors, such as Fas cell surface death receptor, tumor
necrosis factor-related apoptosis-inducing ligand receptor and
tumor necrosis factor receptor 1. A recent study has demonstrated
that c-FLIPL expression is significantly increased in
certain solid tumors and is associated with advanced disease
progression (16).
c-FLIPL expression decreases during disease remission,
but remains high in drug-resistant tumor cells not undergoing
apoptosis. However, c-FLIPL expression is downregulated
by c-FLIPL inhibitors, which in turn induce apoptosis
(17). High c-FLIPL
expression is associated with a poor prognosis in patients with
leukemia (18), which is
consistent with a previous clinical investigation (4). Therefore, it is important to
investigate gene-targeting drugs that can specifically modulate the
expression levels of c-FLIPL. The results of the current
study revealed that Jurkat and HUT-78 cells expressed increased
levels of c-FLIPL compared with control cells.
Therefore, the effect of inducing necroptosis in Jurkat and HUT-78
cells by downregulating c-FLIPL expression was
investigated.
Epigenetic alterations affect gene transcription
through the chemical modification of DNA without altering the DNA
sequence (19). Histone
modifications, including phosphorylation, methylation, acetylation
and ribosylation, represent important epigenetic mechanisms that
serve an important role in the regulation of gene expression
(20). Acetylation by histone
acetyltransferase promotes gene expression and HDAC gene expression
can lead to silencing of tumor suppressor genes (21). As a key enzyme regulating gene
expression, HDAC has an important role in the occurrence and
development of hematological malignancies via the promotion of cell
proliferation and inhibition of cell apoptosis (22). There are 18 types of HDACs in
mammals, which are divided into four groups: Class I, IIa, IIb and
IV, according to their structure and function. Class I HDACs,
including HDAC1, HDAC2, HDAC3 and HDAC8, are mainly distributed in
the nucleus and regulate histone acetylation levels. Recent studies
have demonstrated that class I HDACs serve an important role in
regulating differentiation, proliferation and apoptosis (23–25).
HDACis target and inhibit HDACs to increase histone acetylation
levels and trigger chromatin remodeling, thereby regulating the
expression levels of key proto-oncogenes or tumor suppressor genes.
Furthermore, HDACis may potentially inhibit proliferation, and
induce differentiation and apoptosis. HDACis have exhibited
efficacy in the treatment of hematopoietic malignancies and four
HDACis, including vorinostat, romidepsin, panobinostat and
belinostat, have been approved by the US Food and Drug
Administration for clinical use (26).
Chidamide is a benzamide HDACi developed in China,
which selectively inhibits HDAC class I subtypes 1, 2 and 3, and
class IIb subtype 10. Numerous in vitro experiments have
demonstrated the effects of chidamide on various tumors (27–29).
The results of the present study revealed that Jurkat and HUT-78
cells expressed high levels of HDAC1 and HDAC3, which were
significantly decreased following treatment with chidamide.
Additionally, chidamide regulated cell cycle arrest, as p21 and
PCNA are involved in regulation of the cell cycle, and induced
mitochondrial dysfunction and necroptosis by downregulating
c-FLIPL transcription and translation in Jurkat and
HUT-78 cells.
Degterev et al (30) proposed the term necroptosis in 2005
and revealed that the small molecule Nec-1 acted specifically on
RIP1 without affecting apoptosis. Necroptosis is defined as cell
death mediated by death receptors, which is specifically inhibited
by Nec-1, but not the apoptosis inhibitor Z-VAD-FMK. The current
study revealed that the proportions of Jurkat and HUT-78 cells
undergoing necroptosis and late apoptosis were significantly
increased following pretreatment with the apoptosis inhibitor
Z-VAD-FMK. Furthermore, treatment with the necroptosis-specific
inhibitor Nec-1 significantly reduced the proportion of cells
undergoing necroptosis induced by chidamide. Chidamide-induced cell
death was also significantly inhibited in Jurkat and HUT-78 cells
following pretreatment with both Z-VAD-FMK and Nec-1. Similarly,
membranolysis, mitochondrial swelling and organelle disappearance
were observed following chidamide treatment. However,
representative apoptotic and necroptotic features, and
mitochondrial dysfunction were alleviated by Z-VAD-FMK and Nec-1
pretreatment.
The expression levels of caspase-3 and caspase-8
were markedly decreased after Z-VAD-FMK treatment in
chidamide-treated cells. Chidamide increased the expression of the
key necroptosis proteins RIP1 and RIP3 and phosphorylation of MLKL
in Jurkat and HUT-78 cells, when apoptosis was inhibited. The RIP3
gene is located on human chromosome 2q33, which is subject to
multiple gene mutations and chromosomal damage, particularly in
nasopharyngeal carcinoma and leukemia (31). Additionally, RIP3 is a key molecule
that regulates the transformation between apoptosis and necrosis,
and RIP3-deficient cells are only able to undergo apoptosis.
Downregulation of RIP3 prevents necroptosis without affecting
apoptosis (32). Tumor necrosis
factor-α, which is considered to be a specific factor controlling
necroptosis, does not induce necroptosis in mouse embryonic
fibroblasts lacking the RIP3 gene but with wild type RIP1
expression (33). In addition,
RIP3 expression is positively correlated with necroptosis and its
expression level is a key factor determining the ability of cells
to undergo necroptosis (34,35).
MLKL is an important downstream effector of RIP3, and serves as an
important switch for RIP3-mediated necroptosis (36,37).
In the present study, Z-VAD-FMK pretreatment followed by chidamide
treatment increased RIP3 expression and MLKL phosphorylation in
Jurkat and HUT-78 cells, indicating that chidamide induced
necroptosis when apoptosis was inhibited in T-ALL cells.
In conclusion, the present study revealed that
Jurkat and HUT-78 cells expressed high levels of
c-FLIPL. The HDACi chidamide induced necroptosis in
T-ALL cells by suppressing the transcription and translation of
c-FLIPL, which may serve as a novel target for the
treatment of T-ALL.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WY designed and directed the study. ZC was the major
contributor in writing the manuscript and analyzed the data. HG,
HL, BZ and MG participated in performing the experiments. BW
participated in writing and was involved in the design of the
experiments. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was in accordance with the
Declaration of Helsinki and was approved by The Ethics Committee of
Shengjing Hospital of China Medical University. All patients and
controls, or their parents/guardians, provided written informed
consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Beesley AH, Firth MJ, Ford J, Weller RE,
Freitas JR, Perera KU and Kees UR: Glucocorticoid resistance in
T-lineage acute lymphoblastic leukaemia is associated with a
proliferative metabolism. Br J Cancer. 100:1926–1936. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
He MX and He YW: CFLAR/c-FLIPL: A star in
the autophagy, apoptosis and necroptosis alliance. Autophagy.
9:791–793. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sachanas S, Levidou G, Angelopoulou MK,
Moschogiannis M, Yiakoumis X, Kalpadakis C, Vassilakopoulos TP,
Kontopidou F, Tsirkinidis P, Dimitrakopoulou A, et al: Apoptotic
and proliferative characteristics of proliferation centers in lymph
node sections of patients with chronic lymphocytic leukemia. Leuk
Lymphoma. 55:571–582. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Venza I, Visalli M, Oteri R, Teti D and
Venza M: Class I-specific histone deacetylase inhibitor MS-275
overrides TRAIL-resistance in melanoma cells by downregulating
c-FLIP. Int Immunopharmacol. 21:439–446. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zheng Z, Cheng S, Wu W, Wang L, Zhao Y,
Shen Y, Janin A and Zhao WL: C-FLIP is involved in tumor
progression of Peripheral T-cell lymphoma and targeted by histone
deacetylase inhibitors. J Hematol Oncol. 7:882014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Su Z, Yang Z, Xu Y, Chen Y and Yu Q:
Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol
Cancer. 14:482015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Deng XX, Li SS and Sun FY: Necrostatin-1
prevents necroptosis in brains after ischemic stroke via inhibition
of RIPK1-mediated RIPK3/MLKL signaling. Aging Dis. 10:807–817.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pasparakis M and Vandenabeele P:
Necroptosis and its role in inflammation. Nature. 517:311–320.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang B, Cao K, Liu Z, Shan W, Wen Q and
Wang R: Receptor interacting protein kinase 3 promotes
cisplatin-induced necroptosis in apoptosis-resistant HepG2/DDP
cells. Neoplasma. 2019:180710N4662019.PubMed/NCBI
|
|
10
|
Fu D, Jordan JJ and Samson LD: Human
ALKBH7 is required for alkylation and oxidation-induced programmed
necrosis. Genes Dev. 27:1089–1100. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pietkiewicz S, Schmidt JH and Lavrik IN:
Quantification of apoptosis and necroptosis at the single cell
level by a combination of imaging flow cytometry with classical
Annexin V/propidium iodide staining. J Immunol Methods. 423:99–103.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
NCCN Clinical Practice Guidelines in
Oncology, . Pediatric Acute Lymphoblastic Leukemia. Version1. 2018
March 12;2018.
|
|
14
|
Hu X and Xuan Y: Bypassing cancer drug
resistance by activating multiple death pathways-A proposal from
the study of circumventing cancer drug resistance by induction of
necroptosis. Cancer Lett. 259:127–137. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Irmler M, Thome M, Hahne M, Schneider P,
Hofmann K, Steiner V, Bodmer JL, Schröter M, Burns K, Mattmann C,
et al: Inhibition of death receptor signals by cellular FLIP.
Nature. 388:190–195. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hsu TS, Mo ST, Hsu PN and Lai MZ: c-FLIP
is a target of the E3 ligase deltex1 in gastric cancer. Cell Death
Dis. 9:1352018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Park SJ, Kim MJ, Kim HB, Sohn HY, Bae JH,
Kang CD and Kim SH: Trichostatin A sensitizes human ovarian cancer
cells to TRAIL-induced apoptosis by down-regulation of c-FLIPL via
inhibition of EGFR pathway. Biochem Pharmacol. 77:1328–1336. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mclornan D, Hay J, Mclaughlin K, Holohan
C, Burnett AK, Hills RK, Johnston PG, Mills KI, McMullin MF,
Longley DB and Gilkes A: Prognostic and therapeutic relevance of
c-FLIP in acute myeloid leukaemia. Br J Haematol. 160:188–198.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Florean C, Schnekenburger M, Grandjenette
C, Dicato M and Diederich M: Epigenomics of leukemia: From
mechanisms to therapeutic applications. Epigenomics. 3:581–609.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xiao PF, Tao YF, Hu SY, Cao L, Lu J, Wang
J, Feng X, Pan J and Chai YH: mRNA expression profiling of histone
modifying enzymes in pediatric acute monoblastic leukemia.
Pharmazie. 72:177–186. 2017.PubMed/NCBI
|
|
21
|
Haery L, Mussakhan S, Waxman DJ and
Gilmore TD: Evidence for an oncogenic modifier role for mutant
histone acetyltransferases in diffuse large B-cell lymphoma. Leuk
Lymphoma. 57:2661–2671. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Valdez BC, Li Y, Murray D, Liu Y, Nieto Y,
Champlin RE and Andersson BS: Combination of a hypomethylating
agent and inhibitors of PARP and HDAC traps PARP1 and DNMT1 to
chromatin, acetylates DNA repair proteins, down-regulates NuRD and
induces apoptosis in human leukemia and lymphoma cells. Oncotarget.
9:3908–3921. 2017.PubMed/NCBI
|
|
23
|
Zhou H, Cai Y, Liu D, Li M, Sha Y, Zhang
W, Wang K, Gong J, Tang N, Huang A and Xia J: Pharmacological or
transcriptional inhibition of both HDAC1 and 2 leads to cell cycle
blockage and apoptosis via p21 Waf1/Cip1 and p19 INK4d upregulation
in hepatocellular carcinoma. Cell Prolif. 51:e124472018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li S, Wang F, Qu Y, Chen X, Gao M, Yang J,
Zhang D, Zhang N, Li W and Liu H: HDAC2 regulates cell
proliferation, cell cycle progression and cell apoptosis in
esophageal squamous cell carcinoma EC9706 cells. Oncol Lett.
13:403–409. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ahn MY and Yoon JH: Histone deacetylase 8
as a novel therapeutic target in oral squamous cell carcinoma.
Oncol Rep. 37:540–546. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yoon S and Eom GH: HDAC and HDAC
Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med J.
52:1–11. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chan TS, Tse E and Kwong YL: Chidamide in
the treatment of peripheral T-cell lymphoma. Onco Targets Ther.
10:347–352. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lu CT, Leong PY, Hou TY, Huang SJ, Hsiao
YP and Ko JL: Ganoderma immunomodulatory protein and chidamide
down-regulate integrin-related signaling pathway result in
migration inhibition and apoptosis induction. Phytomedicine.
51:39–47. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang S, Nan P, Li C, Lin F, Li H, Wang T,
Zhou C, Zhang X, Meng X, Qian H, et al: Inhibitory effect of
chidamide on the growth of human adenoid cystic carcinoma cells.
Biomed Pharmacother. 99:608–614. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Degterev A, Huang Z, Boyce M, Li Y, Jagtap
P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA and Yuan J:
Chemical inhibitor of nonapoptotic cell death with therapeutic
potential for ischemic brain injury. Nat Chem Biol. 1:112–119.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Papaemmanuil E, Hosking FJ, Vijayakrishnan
J, Price A, Olver B, Sheridan E, Kinsey SE, Lightfoot T, Roman E,
Irving JA, et al: Loci on 7p12.2, 10q21.2 and 14q11.2 are
associated with risk of childhood acute lymphoblastic leukemia. Nat
Genet. 41:1006–1010. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ,
Lin SC, Dong MQ and Han J: RIP3, an energy metabolism regulator
that switches TNF-induced cell death from apoptosis to necrosis.
Science. 325:332–336. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Qiu X, Zhang Y and Han J: RIP3 is an
upregulator of aerobic metabolism and the enhanced respiration by
necrosomal RIP3 feeds back on necrosome to promote necroptosis.
Cell Death Differ. 25:821–824. 2018.PubMed/NCBI
|
|
34
|
Cook WD, Moujalled DM, Ralph TJ, Lock P,
Young SN, Murphy JM and Vaux DL: RIPK1-and RIPK3-induced cell death
mode is determined by target availability. Cell Death Differ.
21:1600–1612. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xu B, Xu M, Tian Y, Yu Q, Zhao Y, Chen X,
Mi P, Cao H, Zhang B, Song G, et al: Matrine induces RIP3-dependent
necroptosis in cholangiocarcinoma cells. Cell Death Discov.
3:160962017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kim SK, Kim WJ, Yoon JH, Ji JH, Morgan MJ,
Cho H, Kim YC and Kim YS: Upregulated RIP3 Expression Potentiates
MLKL phosphorylation-mediated programmed necrosis in toxic
epidermal Necrolysis. J Invest Dermatol. 135:2021–2030. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Moujalled DM, Cook WD, Murphy JM and Vaux
DL: Necroptosis induced by RIPK3 requires MLKL but not Drp1. Cell
Death Dis. 5:e10862014. View Article : Google Scholar : PubMed/NCBI
|