Introduction
Asthma is a heterogeneous disease that involves an
imbalance of airway inflammation. The airway epithelium is not only
the first anatomical barrier to airborne pathogens and stimuli, but
it is also capable of mounting a number of immune responses
(1). When an inflammatory stimulus
activates epithelial cells, a series of inflammatory cascades,
including innate and adaptive immune responses, are initiated
(2). These important functions of
airway epithelial cells make them potential targets to treat
asthma.
The protein kinase B (AKT) family of kinases (AKT1,
−2 and −3) has a key role in the inflammatory response. However, it
is not clear which of the AKT family, if any, is a major player in
asthmatic airway inflammation. In our recent experiments, it was
observed that specific variants in the 3′untranslated region (UTR)
of AKT2 may influence susceptibility to asthma (unpublished
data); however, the details of the involvement of AKT2 activity are
unknown.
MicroRNAs (miRs) are short noncoding RNAs that pair
with the 3′UTR of target mRNAs to regulate gene expression through
mRNA degradation or translational repression (3). An increasing body of evidence has
suggested that dysregulation of the expression of miRs is
correlated with various physiological and pathological processes
(4), for example, in asthma
(5,6). Recent studies have demonstrated the
regulation of miR gene expression in bronchial epithelial cells
(BECs) (6–9). The expression levels of miR-18A,
−27A, −128, −155 and −181b-5p in asthmatic BECs decreased (6), while the level of miR-19a in the
epithelia of patients with severe asthma increased (7,8). A
functional study demonstrated that miR-19a inhibits the
proliferation of severe asthmatic BECs by targeting transforming
growth factor-β receptor 2 mRNA (9). miR-181b-5p affects eosinophilic
inflammation by targeting the gene encoding secreted phosphoprotein
1 (8). For other miRs, including
miR-18A, −27A, −128 and −155, no consensus miR binding sites were
observed in the 3′UTRs of the target genes (5).
miR-625 is a multifunctional miR that has been
implicated in carcinogenesis, including colorectal adenocarcinoma
(10), malignant pleural
mesothelioma (11) and
hepatocellular cancer (12). To
date, only one study has focused on the miR-625/AKT2 axis in
increasing the chemosensitivity of glioma (13). In terms of its association with
asthma, miR-625-5p is significantly downregulated in pediatric
asthma and targets the gene encoding estrogen receptor 1 (14). This suggested a role for this miR
in the pathogenesis of allergic diseases. Therefore, the present
study was designed to investigate the novel characteristics of
miR-625 in the potential pathogenic mechanism of asthma. It was
hypothesized that miR-625-5p may alter the inflammatory responses
in human BECs (HBECs) by targeting the AKT2 signaling pathway.
Materials and methods
Cell culture
The cell lines (16HBECs and A549 cells) were
purchased from the Type Culture Collection of the Chinese Academy
of Science (Shanghai, China) and grown in Dulbecco's modified
Eagle's medium (DMEM; Gibco®; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) with 10% fetal bovine serum (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) and 100 µg/ml streptomycin
(Beyotime Institute of Biotechnology, Shanghai, China) at 37°C in
5% CO2.
MTT assay
An MTT assay (Beyotime Institute of Biotechnology,
Shanghai, China) was used to measure cell viability. The cells were
seeded in 96-well plates with 1–3×104 cells in a volume
of 200 µl in each well and incubated for 24 h. The cells were then
incubated with different concentrations (0, 0.1, 1, 10 and 100
µg/ml) of lipopolysaccharide (LPS; Sigma-Aldrich; Merck KgaA) for
12 h at 37°C. The supernatant was removed from the cells and added
to medium with 5 mg/ml MTT for 4 h. The medium was discarded and
100 µl solubilization solution (included in MTT assay kit) was
added to dissolve the formazan crystals. The absorbance was
measured at a wavelength of 570 nm using a Multiskan Spectrum
instrument (Thermo Fisher Scientific, Inc.). The ratio of the
absorbance of the treatment group to that of the control group
represented the viability of the cells.
Transfection
Cells seeded in 6-well plates at a density of
1×106 cells/well were transfected with an miR-625-5p
mimic (5′-AGGGGGAAAGUUCUAUAGUCC-3′), miR-625-5p inhibitor
(5′-AGGGGGAAAGUUCUAUAGUCC-3′), a negative control miR
(5′-UUCUCCGAACGUGUCACGU-3′) and an inhibitor negative control
(5′-AAGAGGCUUGCACAGUGCA-3′), which were obtained from Guangzhou
Ribobio Co., Ltd., (Guangzhou, China); and with the AKT2
small interfering RNA (si-AKT2; sense,
5′-GCUCCUUCAUUGGGUACAATT-3′), scrambled negative control
(5′-UUCUCCGAACGUGUCACGUTT-3′), the AKT2 overexpression
plasmid (v-AKT2), and control vector, which were obtained from
Shanghai GenePharma Co., Ltd., (Shanghai, China). miR-625-5p mimic
(75 pmol/well) or miR-625-5p inhibitors (120 pmol/well) and their
controls (corresponding concentration) were mixed with 5 µl
Lipofectamine 2000™ (Thermo Fisher Scientific, Inc.), then
incubated with 1×106 cells in 6-well plates. Similarly,
si-AKT2 (75 pmol/well) or v-AKT2 (2.5 µg/well) were mixed with 5 µl
Lipofectamine 2000™ and transfected into cells. Following 6 h,
Opti-Minimum Essential Medium™ (Gibco; Thermo Fisher Scientific,
Inc.) was discarded and fresh DMEM was added. The cells were
incubated for another 24 h, then washed three times with cold PBS
and used for protein extraction immediately after cell
collection.
ELISA
The cells seeded in 6-well plates at a density of
2×105 cells/well were cultured with the miR-625-5p mimic
or miR-625-5p inhibitor with LPS (1 µg/ml) for 24 h at 37°C. The
cells and the culture supernatants were collected separately. The
levels of interleukin (IL)-6 (cat. no. D6050) and tumor necrosis
factor-α (TNF-α) (cat. no. DTA00D) in the supernatant released from
16HBECs were evaluated using ELISA kits (R&D Systems, Inc.,
Minneapolis, MN, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA from cultured 16HBECs and isolated using
RNAiso Plus (Takara Biotechnology Co., Ltd., Dalian, China).
PrimeScript reagents (Takara Bio Inc., Otsu, Japan) were used to
synthesize cDNA at 37°C for 15 min followed by RT inactivation at
85°C for 5 sec. RT-qPCR was completed using a MxPRO 3000 real-time
PCR system and SYBR® Premix Ex Taq™ (TliRNaseH Plus;
cat. no. RR420) (Takara Bio, Inc.); 0.8 µl primers, 0.4 µl ROX
Reference Dye or Dye II 2 µl cDNA and 8 µl dH2O were
employed and the reaction mixture was made up to 20 µl. The
procedure was implemented according to the manufacturer's
protocols. The thermocycling conditions were as follows:
Denaturation at 95°C was for 5 sec, annealing at 60°C was for 34
sec min and extension at 60°C was for 1 min. The primers used are
presented in Table I. The
2−∆∆Cq relative quantification method was used to
calculate the mean fold expression difference between the groups
(15).
 | Table I.Quantitative polymerase chain reaction
primers. |
Table I.
Quantitative polymerase chain reaction
primers.
| Gene | Primer | Sequence
(5′-3′) |
|---|
| TNF-α | Forward |
CTCCTCACCCACACCATCA |
|
| Reverse |
GGAAGACCCCTCCCAGATAG |
| IL-6 | Forward |
TTCGGTCCAGTTGCCTTCT |
|
| Reverse |
GGTGAGTGGCTGTCTGTGTG |
| β-actin | Forward |
AGAGCTACGAGCTGCCTGAC |
|
| Reverse |
AGCACTGTGTTGGCGTACAG |
| U6 | Forward |
CTCGCTTCGGCAGCACA |
|
| Reverse |
AACGCTTCACGAATTTGCGT |
Western blotting
Proteins from 16HBECs were prepared routinely using
radioimmunoprecipitation lysis buffer (cat no. P0013C) and
phenylmethanesulfonylfluoride (cat no: ST506) kit (Beyotime
Institute of Biotechnology) according to the manufacturer's
protocol. A bicinchoninic assay kit (Beyotime Institute of
Biotechnology) was used to quantify the protein levels. Total
protein extracts (40 µg) were separated by 10% SDS-PAGE and
transferred onto a polyvinylidene fluoride membrane (EMD Millipore,
Billerica, MA, USA). Membranes were blocked in 5% bovine serum
albumin (Beyotime Institute of Biotechnology; cat no. P0007) for 1
h at room temperature, then incubated with primary antibodies
(1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA). The
primary antibodies were as follows: P-AKT2 (cat. no. 8599), AKT2
(cat. no. 2964), P-IκBα (cat. no. 9246), β-actin (cat. no. 4970)
overnight at 4°C, followed by incubation with horseradish
peroxidase-conjugated secondary antibodies (1:3,000; cat. nos. 7076
and 7074; Cell Signaling Technology, Inc.) for 2 h at room
temperature. The immunoreactive proteins on the blots were scanned
by FluorChem FC3 (Protein Simple, San Jose, CA, USA).
Plasmid construction
The AKT2 3′UTR was amplified using human DNA
as a template. The wild-type (WT) 3′UTR of AKT2 was inserted
into the luciferase gene in the pMIR-Report vector (Promega
Corporation, Madison, WI, USA; cat. no. E1330) and termed
pMIR-AKT2-WT. The primers used to amplify the wild-type sequence
were: 5′-CGAGCTCGGGAGGGGCCTGAAGAAGAACGA0-3′ forward, and
5′-CCAAGCTTCCTGGGCTTACTGGAGCTGCCA0-3′ reverse. pMIR-AKT2-MUT
contained a mutated sequence in the 3′UTR of AKT2 inserted
into the luciferase gene in the pMIR-Reporter vector. The primers
used to amplify the mutant sequences were:
5′-AAGTTATATATGCGAAACCACCCAGCGGTGATGGCAGCGAG-3′ (mutated site
underlined) forward;
5′-GTGGTTTCGCATATATAACTTTTTACTTAGCCTTTTTGGTT-3′ (mutated site
underlined) reverse. All constructs were verified by direct
sequencing by Sangon Biotech (Sangon Biotech Co. Ltd., Shanghai,
China).
Dual luciferase reporter assay
The biological software Microrna (http://www.mirbase.org/) and TargetScan release 7.2
(http://www.targetscan.org/) were used to
predicted as a target gene of miR-625-5p. Cells (2×104)
were co-transfected in 96-well plates in triplicate with a firefly
luciferase reporter plasmid (pMIR-AKT2-WT or pMIR-AKT2-MUT), a
Renilla luciferase vector (pRL-SV40; Promega Corporation,
Madison, WI, USA) and with miR-625-5p mimic or its control, using
Lipofectamine 2000™. pRL-SV40 was used as a normalization control.
Following 48 h, luciferase activity was determined using a Dual
Luciferase Reporter Assay System (Promega Corporation, cat. no.
E1910) according to the manufacturer's protocols and was expressed
as the ratio of the firefly luciferase activity to the
Renilla luciferase activity.
Statistical analysis
All experiments were repeated three times. GraphPad
Prism version 5 (GraphPad Software, Inc., La Jolla, CA, USA) was
used to analyze the data. The data are presented as the mean ±
standard deviation for normally distributed data. A Student's
t-test was used to assess the difference between two groups, while
one-way analysis of variance was to analyze differences among three
or more groups with a post hoc Student-Newman-Keuls test. P<0.05
was considered to indicate a statistically significance
difference.
Results
Effect of LPS on the viability of
16HBECs
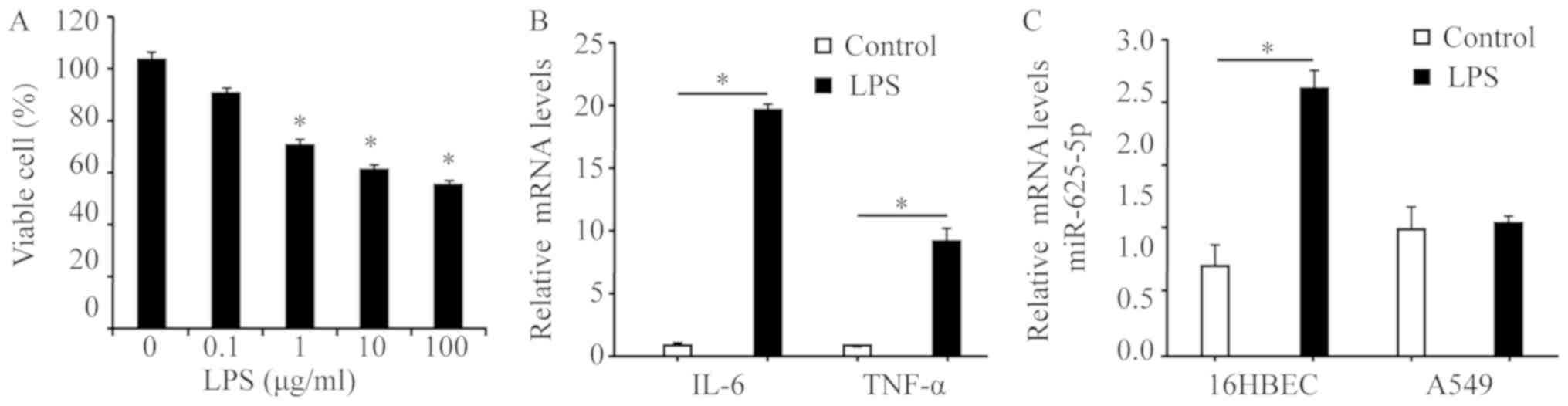
An MTT assay was performed to determine whether LPS
influenced the viability of 16HBECs following 24 h of treatment
with the various LPS concentrations (0, 0.1, 1, 10 and 100 µg/ml).
LPS demonstrated a dose-dependent cytotoxic effect. A total of 1
µg/ml LPS was chosen as the best stimulatory concentration for
further experiments (Fig. 1A). As
presented in Fig. 1B, 16HBECs
treated with LPS at 1 µg/ml significantly stimulated the cells'
ability to secrete inflammatory cytokines compared with the control
cells (IL-6 and TNF-α; P<0.05). 16HBECs expressed a
significantly increased level of miR-625-5p compared with the A549
cell line following LPS induction (P<0.05; Fig. 1C); therefore, 16HBECs were selected
for further analysis.
Effect of miR-625-5p on LPS-induced
cytokine expression
ELISA and RT-qPCR were used to confirm the
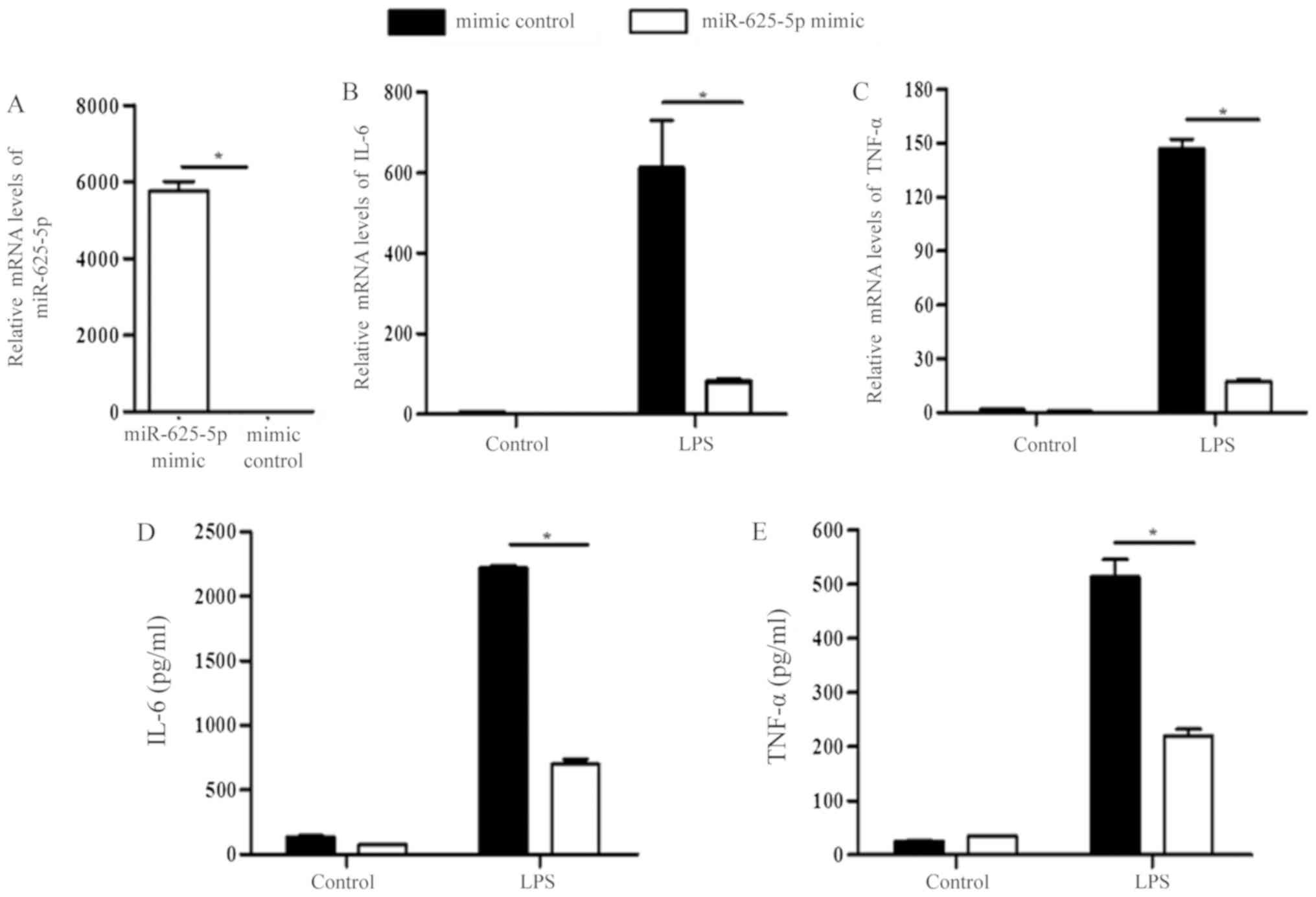
association between miR-625-5p and inflammatory cytokine secretion.
miR-625-5p expression was significantly increased in cells
transfected with the miR-625-5p mimic (P<0.05; Fig. 2A). miR-625-5p had no effect on the
production of IL-6 and TNF-α in control 16HBECs. However, the mRNA
and protein levels of IL-6 and TNF-α were significantly decreased
in miR-625-5p mimic transfected 16HBECs following LPS-induction
(P<0.05; Fig. 2B-E).
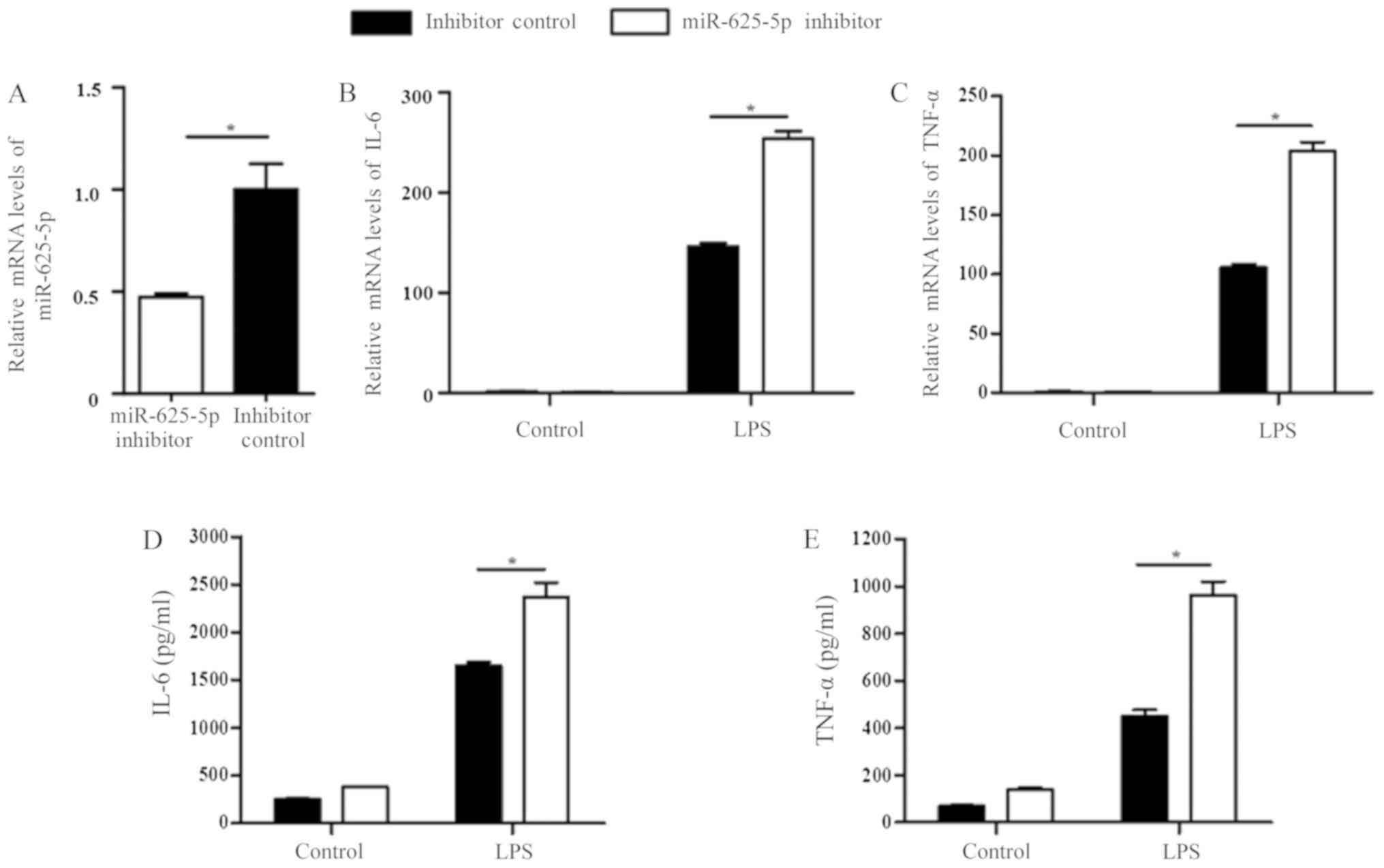
Transfection with the miR-625-5p inhibitor significantly decreased
miR-625-5p expression in 16HBECs when compared with the inhibitor
control following LPS induction (P<0.05; Fig. 3A). Transfection with the miR-625-5p
inhibitor resulted in the significantly increased expression of
inflammatory cytokine mRNAs (IL-6 and TNF-α) when compared with the
control in LPS treated cells (P<0.05; Fig. 3B and C). As presented in Fig. 3D and E, inhibition of miR-625-5p
expression significantly upregulated LPS-induced IL-6 and TNF-α
secretion.
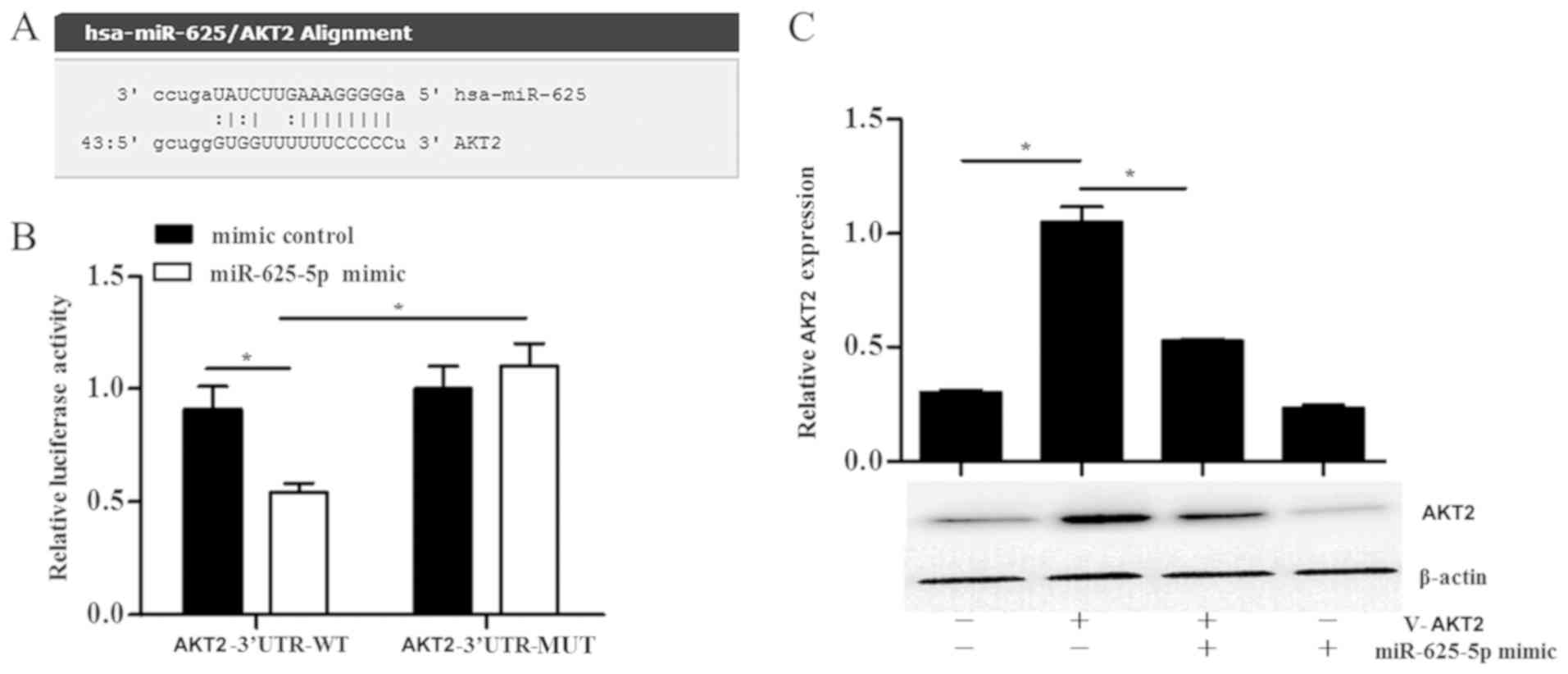
miR-625-5p targets AKT2
The AKT2 gene was predicted as a target gene
of miR-625-5p using the biological software Microrna and
TargetScan. A miR-625-5p binding site was identified in the 3′UTR
of AKT2 (Fig. 4A).
Overexpression of miR-625-5p significantly reduced the luciferase
activity in cells transfected with the pMIR-AKT2-WT vector when
compared with cells transfected with the pMIR-AKT2-MUT vector
(P<0.05; Fig. 4B). Western
blotting was then conducted to assess the effect of miR-625-5p on
the AKT2 protein level. In the miR-625-5p mimic transfected cells,
the protein level of AKT2 was significantly decreased when compared
with the vector only cells (P<0.05; Fig. 4C).
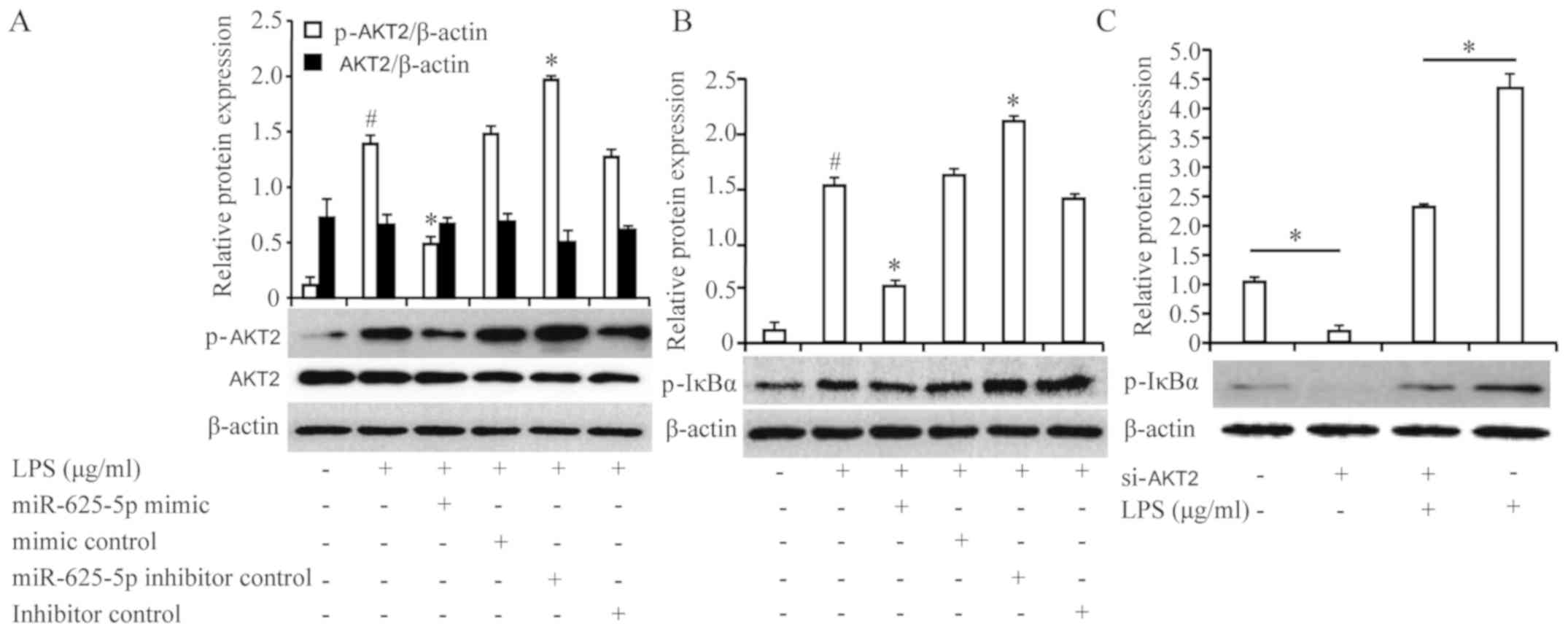
Effect of miR-625-5p on signaling
pathways
AKT2 is involved in triggering the nuclear factor-κB
(NF-κB) signaling pathway and induces inflammatory cytokines
(16). To investigate whether
miR-625-5p influenced the regulation of the phophoinositol-3-kinase
(PI3K)/AKT pathway, the levels of phosphorylated AKT2 were
quantified. The level of phosphorylated AKT2 pretreatment with the
miR-625-5p mimic was significantly decreased compared with LPS
treatment alone (P<0.05; Fig.
5A). Conversely, inhibition of miR-625-5p expression
significantly increased the LPS-induced phosphorylation of AKT2
(P<0.05). The phosphorylation/degradation of inhibitor of κB
(IκBα) is an essential step in the NF-κB signaling pathway
(17). Phosphorylation of IκBα was
assessed to determine whether the NF-κB signaling pathway is
involved in the protective effect of miR-625-5p. The LPS-induced
phosphorylation of IκBα was significantly suppressed by miR-625-5p
overexpression and significantly increased by miR-625-5p inhibition
compared with LPS treatment alone (P<0.05; Fig. 5B). To further confirm whether
activation of the NF-κB pathway was dependent on the
phosphorylation of AKT2, NF-κB activation (i.e., the
phosphorylation of IκBα) was detected in LPS-stimulated 16HBECs
pretreated for 24 h with si-AKT2 to knockdown AKT2
expression. Notably, treatment with si-AKT2 significantly inhibited
the LPS-stimulated phosphorylation of IκBα (P<0.05), which
demonstrated that the PI3K/AKT axis may be involved in NF-κB
regulation in 16HBECs (Fig.
5C).
Discussion
In the present study, it was demonstrated that
miR-625-5p inhibits the secretion of inflammatory mediators in
HBECs. Furthermore, AKT2 was revealed to be a direct target
gene of miR-625-5p. The results indicated that miR-625-5p
suppresses the airway inflammatory response by downregulating AKT2,
which inhibits the NF-κB signaling pathway. Therefore, miR-625-5p
may function as an inhibitor of asthma airway inflammation by
suppressing the inflammatory response in HBECs by directly
targeting AKT2.
Bioinformatics analysis identified that the 3′UTR of
human AKT2 paired miR-625-5p binding sites. The luciferase
reporter assay verified the association between miR-625-5p and
AKT2. Furthermore, the interactions between AKT2 and
miR-625-5p in 16HBECs using a miR-625-5p mimic and inhibitor were
demonstrated.
AKT2 is involved in insulin-mediated regulation of
glucose homeostasis. However, a number of studies on AKT2 have
focused on its role in tumors and demonstrated that AKT2 is
essential for tumor growth, colony formation and cancer cell
proliferation, including ovarian cancer (18), breast cancer (19), and hepatocellular carcinoma
(20). According to these studies,
an AKT isoform has a vital role in tumor growth, colony formation
and cancer cell proliferation in other lung diseases, in addition
to lung tumors; however, there are contradictory results concerning
its functions. Previous studies have demonstrated that AKT2 is
involved in tumor growth and colony formation, and inhibiting AKT2
decreased cellular motility and migration (21–24).
By contrast, another study revealed that AKT2 expression may be
increased in lung tumors in a tobacco-associated model; however,
loss of AKT2 expression did not lead to mutant
K-ras-mediated lung tumors in a genetic model (25). In acute lung injury, Vergadi et
al (26) reported that
depletion of AKT2 kinase activity resulted in light acid-induced
lung injury and protected mice from acid-induced lung injury
(27).
The results of the present study demonstrated that
miR-625-5p inhibits the secretion of TNF-α and IL-6 in 16HBECs.
This was in agreement with other studies (6,28,29)
in which the downregulation of multiple miRs, including
microRNAs-18a, 21, −27a, −128 and −155 and miR-218 also resulted in
the upregulation of IL-6 levels. However, a study by Jardim et
al (30) confirmed that the
expression of TNF-α was increased in asthmatic BECs, while the
level of IL-6 was downregulated compared with the samples from
healthy donors. This discrepancy may have been caused by the
different research methods used in the two studies. Asthma is a
complex disease with a variety of phenotypes (31) and there may be two possible
explanations for the discrepancy between these studies. The
majority of the patients selected by Jardim et al (30) had mild asthma, whereas the present
study was only an in vitro simulation that did not involve
samples of asthmatic tissue. In addition, in Jardim's study, the
majority of the patients with asthma were receiving medication and
this may have altered the expression of certain miRs. Further study
is required to determine the underlying functions of miRs in
asthmatic airway inflammation, which could lead to improved
diagnostic technologies and subcategorization of asthma phenotypes,
ultimately leading to targeted therapies for asthma.
The results of the present study demonstrated that
miR-625-5p suppresses the inflammatory response of 16HBECs by
silencing AKT2, which may be involved in regulating asthma.
Similarly, another study revealed that the expression of miR-625-5p
is markedly reduced in dust mite-induced pediatric asthma.
Therefore, it has been speculated that miR-625-5p may exert
protective effects in asthma (14). Indeed, lung tissue undergoes active
differentiation of cells and miR-625 may serve a role in this
process. Given the dysregulation of active inflammation in the
asthmatic bronchi, it would not be surprising if the inaccurate
differentiation of HBECs resulted in certain pathologies of asthma.
In the present study, one miR targeting a single gene was focused
on. However, a gene can be targeted by a number of miRs and,
conversely, an miR can regulate a number of target genes.
Therefore, it would be useful to characterize other miRs that may
be associated with asthma to provide further options for targeted
therapy. Knowledge of the function of miR-625 in inflammation is
based on cell cultures. The authors' future work will demonstrate
the association between miR-625 and AKT2 in murine models, which
will increase our understanding of the role of miRs in asthma.
In conclusion, miR-625-5p may protect LPS-induced
16HBECs from inflammation by targeting AKT2 and inhibiting
the NF-κB signaling pathway.
Acknowledgements
Not applicable.
Funding
The project was supported by the National Natural
Science Foundation of China (grant no. 81370119), and the Zhenjiang
Science & Technology Program (grant no. SH2015044).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
F-HQ and XD made substantial contributions to the
design of the present study. XD, Q-XZ, BW and D-DZ performed the
experiments. XD analyzed the data. F-HQ and XD wrote and revise the
manuscript. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare they have no competing
interests.
References
|
1
|
Schleimer RP, Kato A, Kern R, Kuperman D
and Avila PC: Epithelium: At the interface of innate and adaptive
immune responses. J Allergy Clin Immunol. 120:1279–1284. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fang R, Cui Q, Sun J, Duan X, Ma X, Wang
W, Cheng B, Liu Y, Hou Y and Bai G: PDK1/Akt/PDE4D axis identified
as a target for asthma remedy synergistic with β2 AR agonists by a
natural agent arctigenin. Allergy. 70:1622–1632. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Meister G and Tuschl T: Mechanisms of gene
silencing by double-stranded RNA. Nature. 431:343–349. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lu J, Getz G, Miska EA, Alvarez-Saavedra
E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA,
et al: MicroRNA expression profiles classify human cancers. Nature.
435:834–838. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Solberg OD, Ostrin EJ, Love MI, Peng JC,
Bhakta NR, Hou L, Nguyen C, Solon M, Nguyen C, Barczak AJ, et al:
Airway epithelial miRNA expression is altered in asthma. Am J
RespirCrit Care Med. 186:965–974. 2012. View Article : Google Scholar
|
|
6
|
Martinez-Nunez RT, Bondanese VP, Louafi F,
Francisco-Garcia AS, Rupani H, Bedke N, Holgate S, Howarth PH,
Davies DE and Sanchez-Elsner T: A microRNA network dysregulated in
asthma controls IL-6 production in bronchial epithelial cells. PLoS
One. 9:e1116592014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Matsukura S, Osakabe Y, Sekiguchi A, Inoue
D, Kakiuchi Y, Funaki T, Yamazaki Y, Takayasu H, Tateno H, Kato E,
et al: Overexpression of microRNA-155 suppresses chemokine
expression induced by Interleukin-13 in BEAS-2B human bronchial
epithelial cells. Allergol Int. 65 (Suppl):S17–S23. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huo X, Zhang K, Yi L, Mo Y, Liang Y, Zhao
J, Zhang Z, Xu Y and Zhen G: Decreased epithelial and plasma
miR-181b-5p expression associates with airway eosinophilic
inflammation in asthma. Clin Exp Allergy. 46:1281–1290. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Haj-Salem I, Fakhfakh R, Bérubé JC,
Jacques E, Plante S, Simard MJ, Bossé Y and Chakir J: MicroRNA-19a
enhances proliferation of bronchial epithelial cells by targeting
TGFβR2 gene in severe asthma. Allergy. 70:212–219. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rasmussen MH, Lyskjær I,
Jersie-Christensen RR, Tarpgaard LS, Primdal-Bengtson B, Nielsen
MM, Pedersen JS, Hansen TP, Hansen F, Olsen JV, et al: miR-625-3p
regulates oxaliplatin resistance by targeting MAP2K6-p38 signalling
in human colorectal adenocarcinoma cells. Nat Commun. 7:124362016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kirschner MB, Cheng YY, Badrian B, Kao SC,
Creaney J, Edelman JJ, Armstrong NJ, Vallely MP, Musk AW, Robinson
BW, et al: Increased circulating miR-625-3p: A potential biomarker
for patients with malignant pleural mesothelioma. J Thorac Oncol.
7:1184–1191. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhou X, Zhang CZ, Lu SX, Chen GG, Li LZ,
Liu LL, Yi C, Fu J, Hu W, Wen JM and Yun JP: miR-625 suppresses
tumour migration and invasion by targeting IGF2BP1 in
hepatocellular carcinoma. Oncogene. 34:965–977. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang J, Zhang J, Zhang J, Qiu W, Xu S, Yu
Q, Liu C, Wang Y, Lu A, Zhang J and Lu X: MicroRNA-625 inhibits the
proliferation and increases the chemosensitivity of glioma by
directly targeting AKT2. Am J Cancer Res. 7:1835–1849.
2017.PubMed/NCBI
|
|
14
|
Dong X, Xu M, Ren Z, Gu J, Lu M, Lu Q and
Zhong N: Regulation of CBL and ESR1 expression by microRNA-22-3p,
513a-5p and 625-5p may impact the pathogenesis of dust mite-induced
pediatric asthma. Int J Mol Med. 38:446–456. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ozes ON, Mayo LD, Gustin JA, Pfeffer SR,
Pfeffer LM and Donner DB: NF-kappaB activation by tumour necrosis
factor requires the Akt serine-threonine kinase. Nature. 401:82–85.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sizemore N, Lerner N, Dombrowski N,
Sakurai H and Stark GR: Distinct roles of the Ikappa B kinase alpha
and beta subunits in liberating nuclear factor kappa B (NF-kappa B)
from Ikappa B and in phosphorylating the p65 subunit of NF-kappa B.
J Biol Chem. 277:3863–3869. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Linnerth-Petrik NM, Santry LA, Moorehead
R, Jücker M, Wootton SK and Petrik J: Akt isoform specific effects
in ovarian cancer progression. Oncotarget. 7:74820–74833. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Riggio M, Perrone MC, Polo ML, Rodriguez
MJ, May M, Abba M, Lanari C and Novaro V: AKT1 and AKT2 isoforms
play distinct roles during breast cancer progression through the
regulation of specific downstream proteins. Sci Rep. 7:442442017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang Q, Yu WN, Chen X, Peng XD, Jeon SM,
Birnbaum MJ, Guzman G and Hay N: Spontaneous hepatocellular
carcinoma after the combined deletion of Akt isoforms. Cancer Cell.
29:523–535. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Attoub S, Arafat K, Hammadi NK, Mester J
and Gaben AM: Akt2 knock-down reveals its contribution to human
lung cancer cell proliferation, growth, motility, invasion and
endothelial cell tube formation. Sci Rep. 5:127592015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim CK, Nguyen TL, Lee SB, Park SB, Lee
KH, Cho SW and Ahn JY: Akt2 and nucleophosmin/B23 function as an
oncogenic unit in human lung cancer cells. Exp Cell Res.
317:966–975. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee MW, Kim DS, Lee JH, Lee BS, Lee SH,
Jung HL, Sung KW, Kim HT, Yoo KH and Koo HH: Roles of AKT1 and AKT2
in non-small cell lung cancer cell survival, growth, and migration.
Cancer Sci. 102:1822–1828. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Linnerth-Petrik NM, Santry LA, Petrik JJ
and Wootton SK: Opposing functions of akt isoforms in lung tumor
initiation and progression. PLoS One. 9:e945952014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hollander MC, Maier CR, Hobbs EA, Ashmore
AR, Linnoila RI and Dennis PA: Akt1 deletion prevents lung
tumorigenesis by mutant K-ras. Oncogene. 30:1812–1821. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vergadi E, Vaporidi K, Theodorakis EE,
Doxaki C, Lagoudaki E, Ieronymaki E, Alexaki VI, Helms M, Kondili
E, Soennichsen B, et al: Akt2 deficiency protects from acute lung
injury via alternative macrophage activation and miR-146a induction
in mice. J Immunol. 192:394–406. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gauna AE and Cha S: Akt2 deficiency as a
therapeutic strategy protects against acute lung injury.
Immunotherapy. 6:377–380. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xu H, Sun Q, Lu L, Luo F, Zhou L, Liu J,
Cao L, Wang Q, Xue J, Yang Q, et al: MicroRNA-218 acts by
repressing TNFR1-mediated activation of NF-κB, which is involved in
MUC5AC hyper-production and inflammation in smoking-induced
bronchiolitis of COPD. Toxicol Lett. 280:171–180. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Luo F, Xu Y, Ling M, Zhao Y, Xu W, Liang
X, Jiang R, Wang B, Bian Q and Liu Q: Arsenite evokes IL-6
secretion, autocrine regulation of STAT3 signaling, and miR-21
expression, processes involved in the EMT and malignant
transformation of human bronchial epithelial cells. Toxicol Appl
Pharmacol. 273:27–34. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jardim MJ, Dailey L, Silbajoris R and
Diaz-Sanchez D: Distinct microRNA expression in human airway cells
of asthmatic donors identifies a novel asthma-associated gene. Am J
Respir Cell Mol Biol. 47:536–542. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gauthier M, Ray A and Wenzel SE: Evolving
concepts of asthma. Am J Respir Crit Care Med. 192:660–668. 2015.
View Article : Google Scholar : PubMed/NCBI
|