Introduction
Aortic valve stenosis is a common disease of left
ventricular (LV) overload, resulting in heart failure (HF) and
mortality. Surgery is considered as the main treatment in the
unload process (1). The benefits
of surgery can differ greatly between patients. In a proportion of
patients who had a long course of disease, there was little
improvement in heart function (2).
Furthermore, these patients were subjected to higher perioperative
risk and surgical mortality (3).
Myocardial ultrastructural changes induced by LV pressure overload
was deemed to be a major molecular event hindering cardiac function
recovery. Therefore, it is essential to investigate the changes in
myocardial ultrastructure over different periods of LV overload, as
well as the underlying molecular mechanisms, in order to develop
clinical treatments.
Previous studies have demonstrated that following an
initial period of LV overload, adaptive left ventricular
hypertrophy (LVH) maintained the cardiac output against an
increasing resistance to ejection (4,5),
which was characterized by cardiomyocyte hypertrophy and
ventricular wall thickening. Gradually, the adaptive LVH changed
into cardiac insufficiency, and even heart failure (HF), if the
condition of pressure overload continued (6). A large number of studies have used
failing heart tissue to demonstrate pathological changes, including
severe interstitial fibrosis and excessive collagen I and III
deposition (7,8). The heart function was hardly improved
and was reversed at this stage, even if the pressure overload was
relieved (9). A study by Oka et
al (10) also demonstrated
that the density of capillaries was greatly decreased, and that the
microcirculation of myocardium was in a state of hypoxia.
Therefore, the present study speculated that various pathological
changes in the myocardial interstitium, rather than in the
cardiomyocytes, may be the cause of irreversible impairment of
cardiac function, however the molecular mechanism underlying the
progression of adaptive LVH to HF induced by LV overload remains
unclear.
Kruppel-like factor 15 (KLF15), a member of the
Kruppel-like factors (KLFs), which are a subclass of the
zinc-finger family of transcriptional regulators, is known to
participate in a diverse range of biological processes, such as
cell proliferation, differentiation, migration and apoptosis
(11,12). Over the past decade, research has
focused on its vital protective effect in the cardiovascular field.
It has previously been demonstrated that KLF15 is expressed in
myocardial tissue and notably decreased in HF tissue, both in
rodents and humans (13). A
previous study also revealed that the expression of KLF15 was
decreased in prolonged LV pressure overload in rats, and sustained
low expression after unloading, accompanied by severe collagen
deposition and interstitial fibrosis (9). Another study demonstrated that under
conditions of aortic constriction, KLF15 null(−/-) mice lapsed into
HF in a short time compared with wild-type (WT) mice (14). It was recently verified that KLF15
could stimulate the process of endothelial cell proliferation and
transformation into tube-like structures in vitro (15). Overall, the expression level of
KLF15 was negatively associated with the development of the HF
process, and the present study speculated that KLF15 could protect
heart function by means of inhibiting cardiac interstitial
remodeling and promoting angiogensis. However, the precise
underlying molecular mechanism remains unknown. The present study
constructed a mouse model of ascending aortic constriction (AAC)
(9) and included cardiac-specific
KLF15-overexpressed transgenic mice to clarify the pathological
progression and molecular mechanism underlying LV pressure
overload.
Materials and methods
Cardiac-specific KLF15-overexpressed
transgenic mice
The vector pRP.ExSi-αMHC-Klf15 was constructed and
microinjected into the oosperm of C57BL/6J mice. Following
surrogacy and PCR identification, two cardiac-specific
KLF15-overexpressed transgenic (TG) founders were obtained. All the
aforementioned processes were completed by Cyagen Biosciences, Inc.
The founders were mated with wild-type (WT) mice to obtain F1
generation mice. Tail DNA was extracted for PCR detection by Premix
Taq (Takara Bio). According to the protocol by Cyagen Biosciences,
Inc., the forward primer for the transgene was
5′-AGAAGCAGGCACTTTACATGG-3′, and the reverse primer was
5′-AGAAGCAGGCACTTTACATGG-3′. The transgene PCR product size was 333
bp. Total cardiac protein from the F1 generation of mice that was
identified as positive was extracted in order to assess the
expression of KLF15 via western blotting and for screening the
KLF15-overexpressed mice in the following study (16).
LV pressure overload animal model
establishment
All procedures abided by the Guide for the Care and
Use of Laboratory Animals (Department of Health and Human Services
publication no. NIH 78–23,1996) and were approved by the Committee
on Animal Research of Army Military Medical University, Chongqing,
China. Male C57BL/6J WT mice, 8–10-weeks of age, weighing 21.0–25.0
g, were provided by the Experimental Animal Center of Xinqiao
Hospital, and cardiac-specific KLF15-overexpressed transgenic (TG)
mice were provided by Cyagen Biosciences, Inc. The mice were used
to construct the LV overload models by means of an AAC surgery
without artificial ventilation. All mice were deprived of food and
allowed a small amount of water 8 h before surgery.
The present study used 1.2% tribromoethyl alcohol
(0.24 mg/g) as an anesthetic by intraperitoneal injection and fixed
the overturned mice. A transverse skin incision was then made in
the suprasternal fossa after shaving and sterilizing. The sternum
was cut lengthwise to the level of the second rib and the soft
tissue was blunt dissected from the superficial trachea. The upper
ascending aorta was exposed after the separation of the thymus, 4-0
sutures encircled the ascending aorta and a 27-gauge needle was
placed parallel to it. The needle was withdrawn rapidly after
binding. The aforementioned procedure was performed with care in
order to avoid damage to pleura and causing pneumothorax. The
diameter of the ascending aorta dropped to ~50% after banding and
the incision was closed when the respiration and heartbeat were
stable. Following surgery, the mice were allowed to eat and drink
freely. A proportion of mice underwent a sham operation as a
negative control.
The mice were divided into eight groups as follows:
i) WT mice 2-weeks sham group (WT sham2w); ii) WT mice 6-weeks sham
group (WT sham6w); iii) TG mice 2-weeks sham group (TG sham2w); iv)
TG mice 6-weeks sham group (TG sham6w); v) WT mice 2-weeks aortic
banding (WT AB2w); vi) WT mice 6-weeks aortic banding (WT AB6w);
vii) TG mice 2-weeks aortic banding (TG AB2w); viii) TG mice
6-weeks aortic banding (TG AB6w).
Echocardiographic measurements of
heart morphology and function
After being anesthetized by 1.2% tribromoethyl
alcohol intraperitoneal injection, each group of mice was assessed
using the M-mode echocardiography of VEVO 2100 Imaging system to
measure heart rate (HR), systolic left ventricular posterior wall
thickness (LVPWs), diastolic left ventricular posterior wall
thickness (LVPWd), systolic interventricular septal thickness
(IVSs), diastolic interventricular septal thickness (IVSd), left
ventricular end systolic inside diameter (LVIDs), left ventricular
end diastolic inside diameter (LVIDd), ejection fraction (EF) and
fractional shortening (FS) 2 and 6 weeks after surgery.
Heart-lung weight ratio
measurement
Each group of mice was anesthetized by 4% isoflurane
inhalation and decapitated. The hearts and lungs were removed at
the corresponding time. The attached adipose and vascular tissue
was excised and residual blood was washed away with
phosphate-buffered saline (PBS). The heart weight (HW) and lung
weight (LW) were measured using precision electronic scales
(Sartorius, Inc.) after drying them gently with sterile gauze and
the HW/LW ratio was calculated.
Histological analysis of heart
microstructural changes
Myocardial tissue was preserved with
polyformaldehyde and cryopreservation. Each specimen was sectioned
to a 5-µm thickness after embedding in paraffin. Hematoxylin and
eosin (H&E) (Beijing Solarbio Science & Technology Co.,
Ltd.) staining and Masson staining (Beijing Solarbio Science &
Technology Co., Ltd.) were used to assess the myocardial morphology
and level of fibrosis. For Masson staining, the paraffin sections
were deparaffinized, washed, and stained with hematoxylin staining
solution for 5–10 min at room temperature. Next, the sections were
stained with Masson Ponceau acid fuchsin solution for 5–10 min at
room temperature, followed by immersion cleaning in 2% glacial
acetic acid aqueous solution then differentiation with 1%
phosphomolybdic acid aqueous solution for 3–5 min, and staining
with aniline blue or light green solution for 5 min. Finally, the
sections were mounted in neutral gum. For H&E staining, the
paraffin sections were dewaxed twice in xylene (10 min each).
Sections were rehydrated sequentially in descending series of
alcohol for 5 min each in anhydrous, 90, 80 and 70% alcohol.
Sections were then treated with phosphate buffered saline for 2
min. Specimens were stained by immersing in hematoxylin for 10 min
at room temperature, treated with 1% acid alcohol for 3 sec, washed
with running water for 10 min, washed with distilled water for 2
min, stained with 0.5% eosin for 3 min and washed with distilled
water for 2 sec. Specimens were then dehydrated twice in 95%
ethanol for 2 min each and cleared by treating twice with xylene
for 5 min each. Sections were then mounted in neutral balsam. The
specific marker CD31 on vascular endothelial cells (VECs) was
exposed by immunohistochemical (IHC) staining to assess myocardial
vascular density and distribution (17). The changes were collected and
observed using a Leica DMIRB light microscope.
ELISA for assessment of collagen I and
III deposition
The myocardial tissue of each group was ground to
homogenate and the protein was extracted. Collagen type I and III
were analyzed using an ELISA kit (FANKEWEI Bioscience, Inc.)
according to the manufacturer's protocol, and statistical analyses
were performed by comparing the standard curve.
KLF15, CTGF and VEGF expression levels
and Smad3 and p38 phosphorylation levels detected via western
blotting
Western blotting was performed to identify the
differential expression levels of KLF15, CTGF, VEGF, p-p38, t-p38,
p-Smad3 and t-Smad3 in each group. Proteins were extracted from an
appropriate amount of ground frozen heart tissue by Cell Extraction
Buffer (Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. The protein concentrations of the
samples were measured using the BCA method. SDS-PAGE buffer was
added to the samples at a unified dilution, boiled for 8 min and
stored at 4°C. SDS-PAGE (10%) was performed on 20 µg of each sample
at 95 V for 120 min, followed by electrophoretic transfer to a
polyvinylidene difluoride membrane at 15 V for 50 min. The bands
were incubated at 4°C for 2 h. The samples were then incubated with
rabbit anti-mouse KLF15 (1:1,000, AV32587, Sigma-Aldrich; Merck
KGaA), CTGF (1:1,000, ab6992, Abcam), VEGF (1:2,000, ab46154,
Abcam), p-p38 (1:1,000, ab4822, Abcam), t-p38 (1:1,000, ab170099,
Abcam), p-Smad3 (1:2,000, ab52903, Abcam), t-Smad3 (1:1,000,
ab40854, Abcam) and GAPDH (1:10,000, ab181602, Abcam) antibodies at
4°C overnight. The following day, the samples were washed with PBST
three times, followed by the addition of horseradish
peroxidase-labeled goat anti-rabbit IgG (1:10,000, 31460
Invitrogen; Thermo Fisher Scientific, Inc.) and incubated at room
temperature for 1 h. The samples were rinsed again with PBST three
times, stained with ECL reagent (Thermo Fisher Scientific, Inc.),
and imaged with a Bio-Rad gel imaging system (Bio-Rad
Laboratories). Quantity One software was used to analyze the gray
value of the bands using GAPDH as the reference.
Statistical analysis
Experimental data were presented as the mean ±
standard error. The statistical difference was compared in each
group using a one-way ANOVA, followed by a post hoc Tukey's test.
All tests were performed using SPSS software (version 13.0; SPSS,
Inc.) and GraphPad Prism software (version 7.0; GraphPad Software,
Inc.). P<0.05 was considered to indicate a statistically
significant difference.
Results
Cardiac expression of KLF15 is
increased in TG mice compared with WT mice
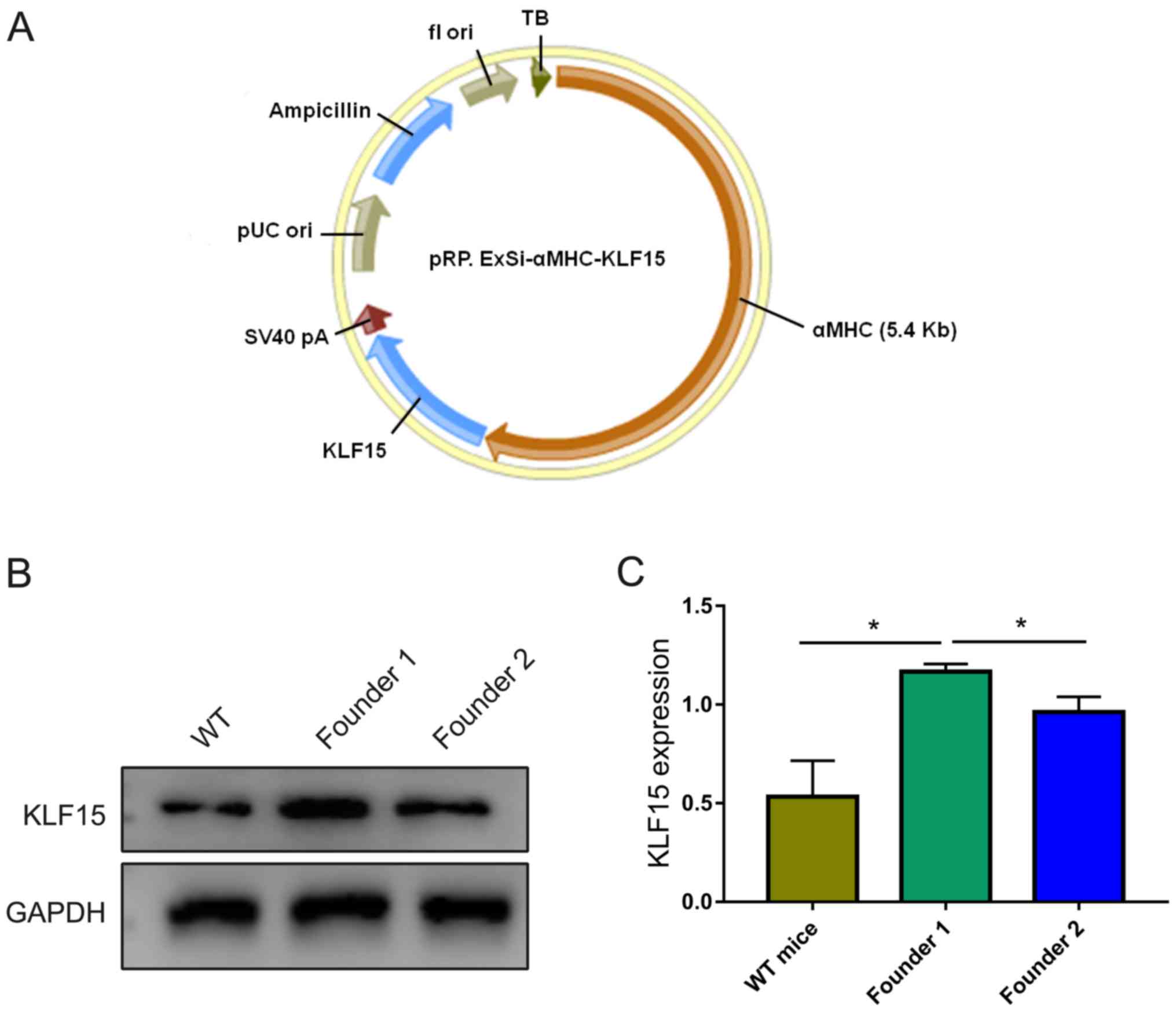
The construction of the vector pRP.ExSi-αMHC-Klf15
(Fig. 1A) and integration of
target genes was completed by Cyagen Biosciences, Inc. Ultimately,
the present study obtained two TG founders, named founder 1 and
founder 2. After mating with WT mice and identifying the F1
generation through tail DNA using PCR, 4 and 5 mice were identified
as positive from the founder 1 and founder 2 mice, respectively.
The present study then extracted total heart protein and examined
the expression levels of KLF15 factor in myocardial tissue using
western blot analysis, and quantified the average gray value with
WT mice as a control. The results revealed that a basic expression
level of KLF15 existed in WT mice, and the expression of KLF15 in
the F1 generation of founder 1 was higher than in the F1 generation
of founder 2 and WT mice (Fig.
1B). The difference was statistically significant (P<0.05;
Fig. 1C). Founder 1 and its F1
generation were identified as positive and considered a suitable
strain of KLF15 overexpression in the following study.
Mice survival
During the AAC surgery, the total mortality rate was
~16.7%. The main causes of death were pneumothorax (~4.2%), massive
hemorrhage (~8.3%) and acute heart failure (~4.2%). The mortality
rate of the sham operation was 0. All mice in each group survived
to the corresponding time-point after surgery, indicating that the
degree of constriction was appropriate.
TG mice have delayed LV diameter
increase and heart function decline
M-mode echocardiography is a dynamic curve image
formed by light spot groups of the heart movement via mono beam
scanning. IVS, LVID and LVPW are displayed from top to bottom.
Fig. 2A demonstrates that the wave
amplitude and ventricular wall thickness of WT AB2w and TG AB2w
groups were markedly increased compared with the sham groups. In
addition, AB2w induced a visible decrease in the LV chamber at the
systolic period. Conversely, AB6w groups displayed an eccentric
structural change featuring a weak amplitude and an enlargement of
the LV chamber that was more evident in WT mice than in TG
mice.
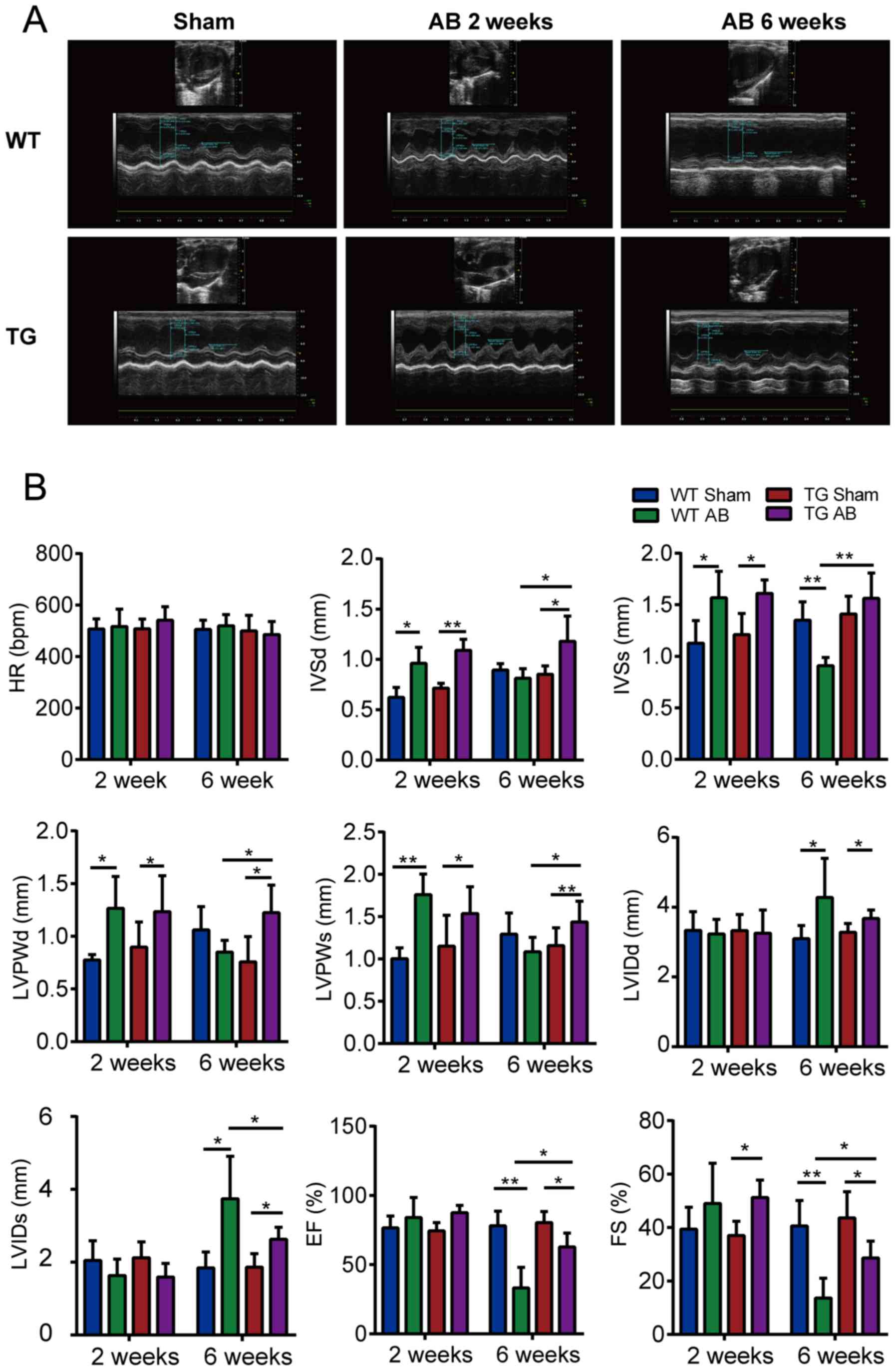 | Figure 2.The effects of KLF15 on cardiac
morphology and function measured by M-mode echocardiography. (A)
M-mode echocardiography images via long axial section of the left
ventricle. (B) Statistical results of the HR, IVSd, IVSs, LVPWd,
LVPWs, LVIDd, LVIDs, EF, and FS of indicated groups (n=5 mice per
group) by M-mode echocardiography of VEVO 2100 Imaging system. Data
are represented as the mean ± SEM. *P<0.05; **P<0.01.
Kruppel-like factor 15; HR, heart rate; IVSd, diastolic
interventricular septal thickness; IVSs, systolic interventricular
septal thickness; LVPWd, diastolic left ventricular posterior wall
thickness; LVPWs, systolic left ventricular posterior wall
thickness; LVIDd, left ventricular end diastolic inside diameter;
LVIDs, left ventricular end systolic inside diameter; EF, ejection
fraction; FS, fractional shortening; WT, wild-type; TG; transgenic;
AB, aortic banding. |
In addition, the present study assessed the HR, IVS,
LVID, LVPW, EF and FS of each group for statistical analysis. As
presented in Fig. 2B, the
parameters did not differ between the WT sham and TG sham mice. At
2 weeks after aortic banding, the ventricular wall was
significantly thicker and heart function compensation had increased
in the AB groups compared with those in the corresponding sham
groups (P<0.05), however the chamber diameter exhibited no
significant difference. At 6 weeks after aortic banding, WT mice
rather than TG mice demonstrated a significant decrease of EF and
FS (P<0.01) and an increase of LVID compared with those in the
sham groups (P<0.05). HR did not differ among the groups
(P>0.05).
TG mice alleviate the HW/LW decline
induced by aortic banding for 6 weeks
Compared with the sham groups, the LW of WT mice and
TG mice did not significantly change under LV overload conditions
for 2 weeks, however, the HW and HW/LW increased. There was no
significant difference between the WT mice and TG mice in the sham
and AB groups, respectively (Table
I). However, the LW of WT mice and TG mice with LV overload for
6 weeks was significantly higher and the HW/LW was lower than that
of the sham mice, WT mice had a higher degree of change compared
with TG mice. There was no significant difference in HW among
groups (Table II).
 | Table I.Heart weight, lung weight and heart
weight/lung weight ratio at 2 weeks after aortic banding. |
Table I.
Heart weight, lung weight and heart
weight/lung weight ratio at 2 weeks after aortic banding.
|
| Sham | AB |
|---|
|
|
|
|
|---|
| Parameter | WT (n=5) | TG (n=5) | WT (n=5) | TG (n=5) |
|---|
| HW (mg) | 113.2±4.009 | 112.2±5.128 |
129.8±3.114a |
128.9±3.794a |
| LW (mg) | 136.5±2.368 | 134.1±2.183 | 133.9±3.270 | 135.1±1.497 |
| HW/LW (mg/mg) | 0.831±0.035 | 0.835±0.026 |
0.973±0.041a |
0.954±0.022a |
| Table II.The heart weight, lung weight and
heart weight/lung weight ratio at 6 weeks after aortic banding. |
Table II.
The heart weight, lung weight and
heart weight/lung weight ratio at 6 weeks after aortic banding.
|
| Sham | AB |
|---|
|
|
|
|
|---|
| Parameter | WT (n=5) | TG (n=5) | WT (n=5) | TG (n=5) |
|---|
| HW (mg) | 121.2±4.028 | 120.9±4.574 |
131.9±1.375a | 128.0±2.975 |
| LW (mg) | 144.1±2.357 | 141.8±3.644 |
194.8±2.159b |
167.1±2.768b,c |
| HW/LW (mg/mg) | 0.843±0.041 | 0.852±0.020 |
0.678±0.010b |
0.768±0.025a,c |
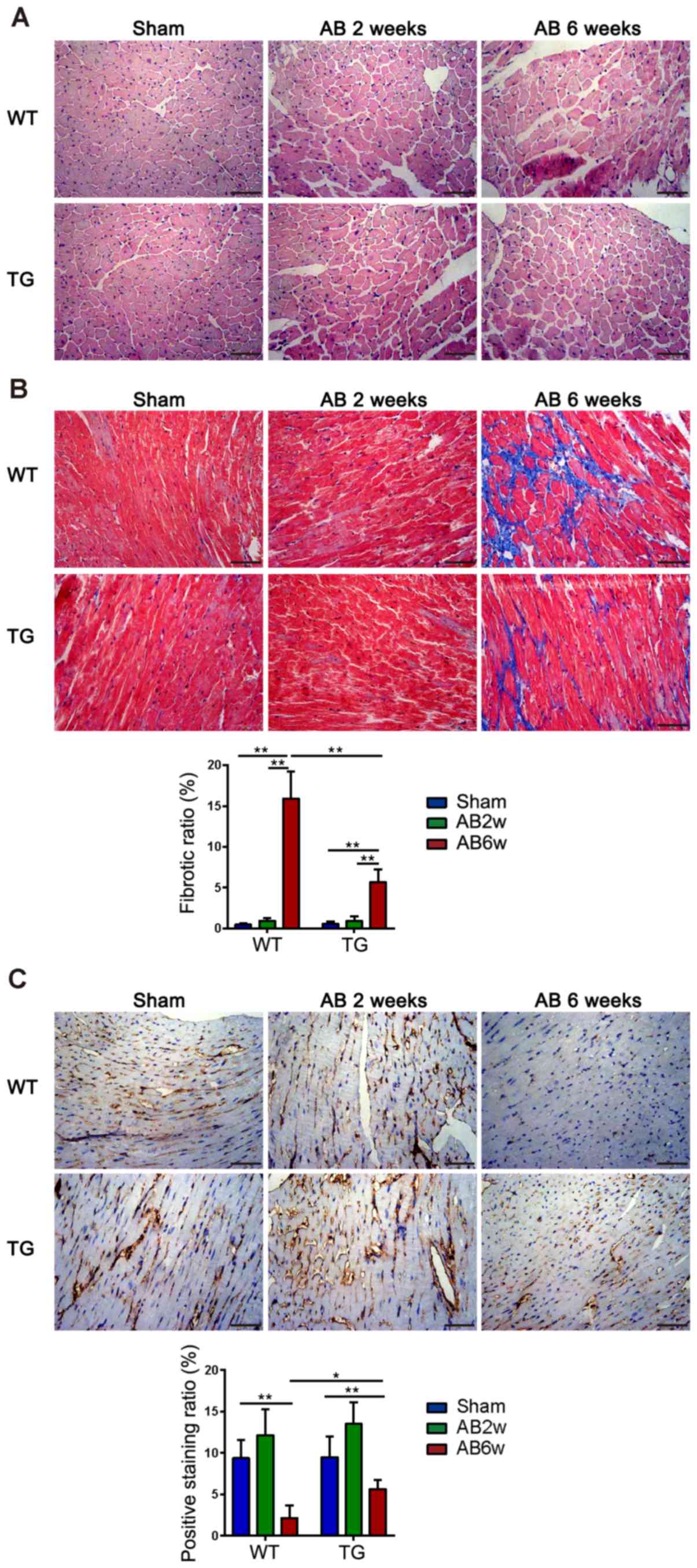
Milder pathological changes in the
myocardial interstitium of TG mice 6 weeks after aortic
banding
H&E staining of myocardial sections in WT AB2w
and TG AB2w groups revealed increased cardiomyocyte size and larger
nuclei compared with the sham groups, and the cells were arranged
regularly in both the sham and AB2w groups. Myocardial sections of
mice subjected to aortic banding for 6 weeks (WT AB6w and TG AB6w)
exhibited a shrinkage and irregular arrangement of cardiomyocytes
compared with AB2w groups, and this was more evident in WT mice
than in TG mice (Fig. 3A). Masson
staining revealed that aortic banding after 2 weeks did not induce
cardiac interstitial fibrosis in either WT or TG mice. WT mice
appeared to have severe fibrosis distributed in the myocardial
interstitium primarily around the blood vessels compared with TG
mice at 6 weeks after aortic banding, which was statistically
significant (P<0.01; Fig. 3B).
As a specific marker for VECs, CD31 was demonstrated in brown via
IHC staining. In WT and TG mice, the density was decreased in the
AB6w groups compared with the sham groups (P<0.01). In addition,
the TG AB6w group exhibited a higher density of CD31 than the WT
AB6w group (P<0.05). No significant difference was observed
between the sham groups and AB2w groups (P>0.05; Fig. 3C).
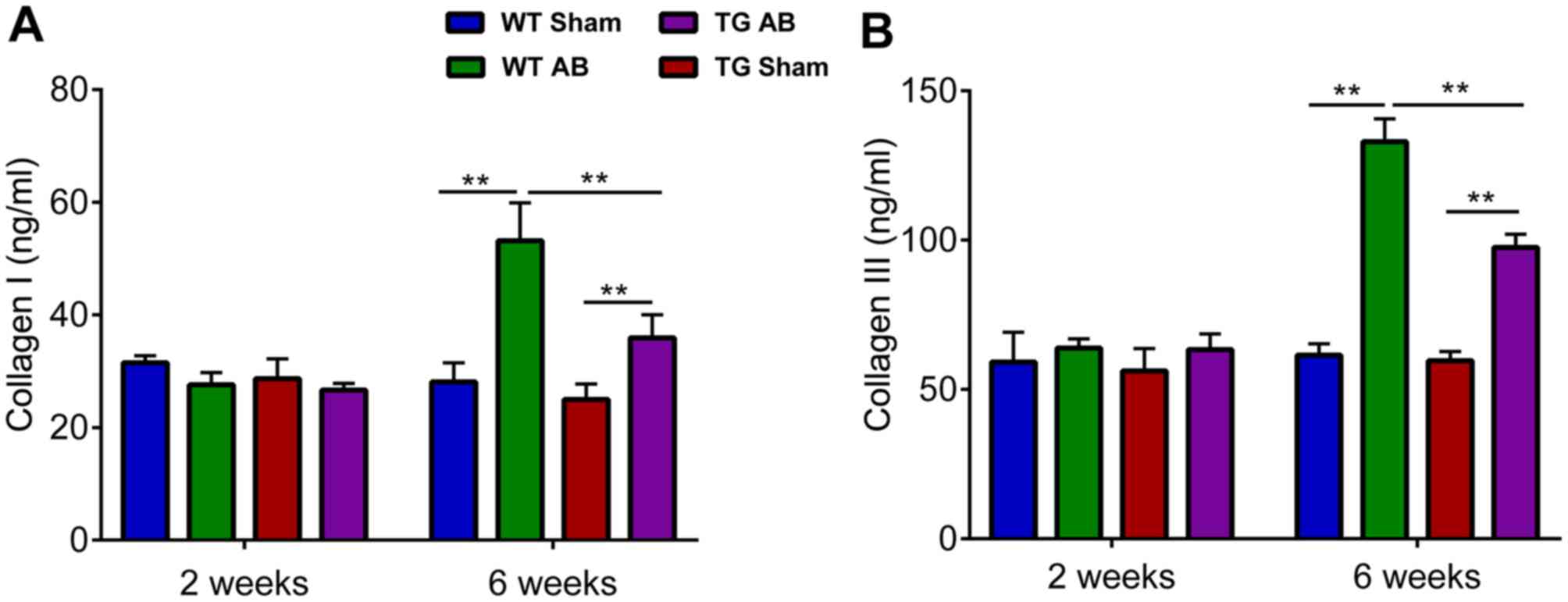
Decreased collagen deposition in TG
mice is induced by aortic banding for 6 weeks
When compared with the standard curve for
quantification, the content of collagen I and III in myocardial
tissue assessed by ELISA tended to coincide with the fibrosis
demonstrated by Masson staining. Collagen I and III levels of TG
and WT AB6w groups were increased compared to their corresponding
sham groups (P<0.01,) and there was also a statistically
significant difference observed between these two groups
(P<0.01) (Fig. 4). No
significant difference was observed among the sham groups.
TG mice suppress CTGF upregulation and
VEGF downregulation mediated by p-Smad3 and p-p38
The present study assessed the expression levels of
KLF15, CTGF, VEGF, Smad3 and p38 phosphorylation in each group
using western blot analysis, and quantified the expression using
the average gray value to investigate the role of each protein in
the pathological progression of LV overload. The results revealed
that under 2 weeks overload, the expression levels of CTGF and
p-p38 were low and there was no significant difference between each
group. The expression of VEGF and p-Smad3 were increased in the WT
AB2w and TG AB2w groups compared with the corresponding sham
groups, but there was no statistically significant difference
between the two groups. Compared with the WT sham2w group, the
expression of KLF15 was upregulated in the TG sham2w, WT AB2w and
TG AB2w groups, but there was no statistically significant
difference among the three groups.
Under 6 weeks overload, the expression of p-p38 and
p-Smad3 were upregulated in the WT AB6w and TG AB6w groups compared
with the corresponding sham groups, but there was no statistically
significant difference between the two groups. Furthermore, AB
induced increased expression of CTGF and decreased expression of
VEGF compared with the corresponding sham groups, which was even
more evident in WT mice than in TG mice. The expression of KLF15
was downregulated in the WT AB6w group compared with the sham and
TG AB6w groups (Fig. 5B).
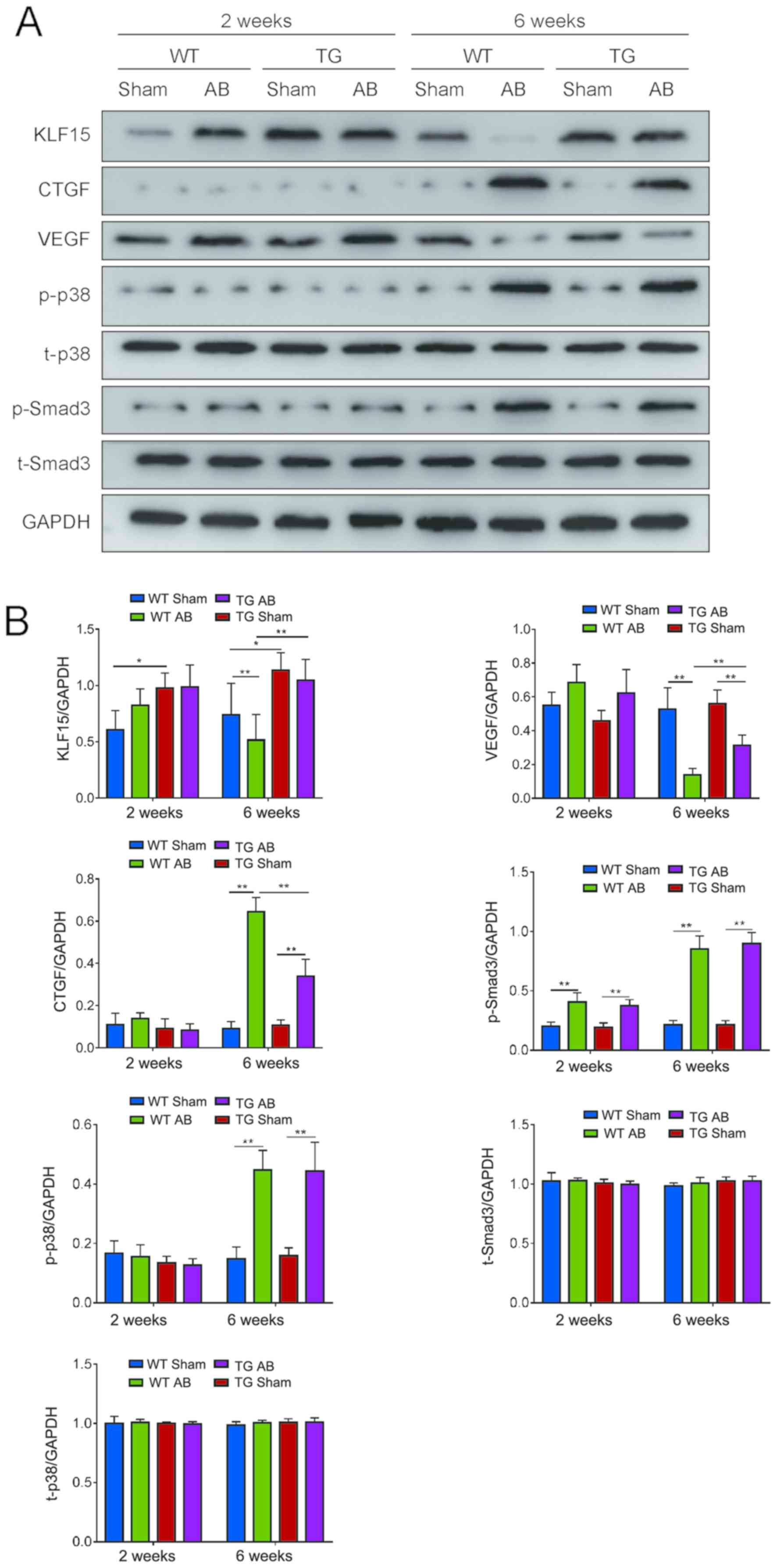 | Figure 5.KLF15, CTGF, VEGF expression levels
and Smad3, P38 phosphorylation levels in myocardial tissue detected
by western blot analysis. (A) KLF15, CTGF, VEGF, p-Smad3, t-Smad3,
p-p38, t-p38 expression in each group. (B) Quantitative measurement
of KLF15, CTGF, VEGF, p-Smad3, t-Smad3, p-p38, t-p38 expression
levels in each group. *P<0.05; **P<0.01. Kruppel-like factor
15; VECs, vascular endothelial cells; VEGF, vascular endothelial
growth factor; p-p38, phosphorylated p-38; p-Smad3, phosphorylated
Smad3; WT, wild-type; TG; transgenic; AB, aortic banding. |
Discussion
The present study demonstrated the histological and
molecular expression changes in the pathological development
process of LV overload in mice. An AAC operation was used instead
of traverse aortic constriction (TAC) operation, which is regarded
as a classic way to construct the ventricular hypertrophy model
(18) to simulate the pathological
process of LV pressure overload. The results of the present study
demonstrated that in WT mice subjected to aortic banding for 2
weeks (WT AB2w), ventricular thickness, heart weight and capillary
density increased, and LV systolic diameter decreased for enhancing
contractility, EF, FS and KLF15 expression increased without other
pathological changes. Gradually, the adaptive hypertrophy was
turning into decompensation. At 6 weeks of aortic banding, WT mice
exhibited a notable increase in LV diameter and lung weight, and a
decrease in EF and FS, with an irregular arrangement of
cardiomyocytes, severe interstitial fibrosis, collagen deposition
and capillary density decline, accompanied by downregulated
expression of KLF15 and VEGF, and upregulated expression of CTGF,
p-p38 and p-Smad3. Notably, TG mice exhibited an improved level of
resistance than WT mice to prolonged LV pressure overload, and
alleviated LV chamber enlargement, interstitial fibrosis, collagen
deposition and capillary density decrease, and improved heart
function compared with the WT mice. This model demonstrated
different changes in compensatory and decompensatory stages at two
time-points, including cardiac morphology, ultrastructure and
molecular expression.
Compared with the classical TAC model, the AAC model
could highlight the effect of mechanical pressure on the
development of ventricular hypertrophy and decrease the
interference of neurohumoral factors induced by increasing blood
pressure in the brain. In addition, the whole procedure avoided
endotracheal intubation and artificial ventilation, and increased
the efficiency of model construction. This model effectively
revealed myocardial interstitial remodeling rather than enlargement
of individual cardiomyocytes associated with heart function
decline, and non-cardiomyocytes in the heart played a significant
role in the development process from the compensatory period to the
decompensatory period. In fact, non-cardiomyocytes account for ~70%
of the total cell number in cardiac tissue, with the majority being
fibroblasts and endotheliocytes (19). Previous studies have confirmed that
under prolonged mechanical stimulation, cardiac fibroblasts
transform into myofibroblasts participating in collagen secretion,
elastin synthesis and interstitial fibrosis (20). Excessive collagen deposition and
formation of collagen fibers resulting in extracellular matrix
(ECM) remodeling promote LV inflexibility and the compressive
deformation of cardiomyocytes contribute to cardiac insufficiency
(21). Meanwhile, anatomic
remodeling leads to electrophysiological changes and aggravated
heart function (22). Furthermore,
increased myocardial oxygen consumption, diffusion distance and
capillary constriction caused by pressure overload place the
myocardium in a state of microcirculation hypoxia and contractile
dysfunction (23). Angiogenesis,
as a vital physiological and pathological process in the
inflammatory response and oncology (24), may be a therapeutic strategy
towards HF recovery. In the present study, the density of vascular
endotheliocyte specific marker CD31 in each group was positively
associated with the corresponding EF and FS values. The
proliferation and migration of endotheliocytes involved in
angiogenesis display an effective compensation to a certain
degree.
KLF15 has been demonstrated to be a key factor in
cardiac remodeling (25,26). A previous study demonstrated that
under LV pressure overload, activated Smad3 promoted the expression
of downstream CTGF, which was considered to be a classic pathway in
the initiation of interstitial remodeling (27). Other studies have demonstrated that
prolonged aortic constriction in rats could activate p38
mitogen-activated protein kinase (p38-MAPK) in cardiomyocytes, and
would remain in sustainable activation even after LV pressure
unload (25). The present study
also revealed several new findings associated with KLF15. For one,
the expression of KLF15 was revealed to be negatively associated
with myocardial interstitial remodeling, rather than adaptive LVH.
Under 2 weeks of overload, a compensatory increase of KLF15
expression inhibited p-Smad3/CTGF pathway and promoted angiogenesis
by positively regulating VEGF expression, which may be a critical
mechanism in repressing interstitial remodeling and maintaining
heart function. In addition, the activation of p38 induced by LV
pressure overload for 6 weeks could suppress the expression of
KLF15, and low expression of KLF15 lost its ability to inhibit CTGF
expression and angiogenesis by regulating VEGF expression,
resulting in occurrence of myocardial interstitial remodeling and
HF. Furthermore, the present study used KLF15-overexpressed TG mice
to identify that the downregulated expression of KLF15 was not a
consequence of myocardial interstitial remodeling, but it was a
crucial factor in regulating pathways of anti-fibrosis or
angiogenesis for preventing interstitial remodeling and HF
(28).
However, these findings also raised some questions.
First, AAC surgery was an acute coarctation process, which was
different from the chronic development of rheumatic aortic
stenosis. In the clinical practice, the compensatory period of
cardiac function in patients with LV pressure overload was much
longer than what was demonstrated in the present study. Additional
delayed response factors may be involved in the LV remodeling
process. Furthermore, it was still not clear whether the beneficial
changes of protecting heart function were regulated by KLF15 in
myocardiocytes independently or by a complex cross-talk between the
non-myocardiocytes and myocardiocytes. Future studies would clarify
this by establishing the co-culture system of myocardiocytes and
non-myocardiocytes in vitro. Overall, KLF15 may be helpful
in determining the optimum surgical timing and the evaluation of
prognosis. Exogenous administration of KLF15 may play an important
role in delaying heart failure in patients with LV overload disease
in clinical treatment.
HF induced by pressure overload was revealed to be
closely associated with myocardial interstitial remodeling, and
KLF15 was the critical factor regulating the expression of CTGF,
VEGF, p-p38 and p-Smad3 to effectively alleviate the progression of
HF through repression of interstitial remodeling and the promotion
of angiogenesis.
Acknowledgements
Not applicable.
Funding
The present study was supported by The National
Natural Science Foundation of China (grant no. 81170216).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SFZ performed most of the experiments and wrote the
manuscript. LC conceived the project. YBX and YP supervised the
experiments and made substantial contributions to conception and
design. YY, ZJ, YHJ, SC and FQT offered assistance with the data
analysis and interpretation and manuscript revision.
Ethics approval and consent to
participate
All procedures abided by the Guide for the Care and
Use of Laboratory Animals (Department of Health and Human Services
publication no. NIH 78-23,1996) and were approved by the Committee
on Animal Research of Army Military Medical University, Chongqing,
China.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Akinkuolie AO, Aleardi M, Ashaye AO,
Gaziano JM and Djousse L: Height and risk of heart failure in the
Physicians' Health Study. Am J Cardiol. 109:994–997. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xu R, Lin F, Zhang S, Chen X, Hu S and
Zheng Z: Signal pathways involved in reverse remodeling of the
hypertrophic rat heart after pressure unloading. Int J Cardiol.
143:414–423. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Christia P, Bujak M, Gonzalez-Quesada C,
Chen W, Dobaczewski M, Reddy A and Frangogiannis NG: Systematic
characterization of myocardial inflammation, repair, and remodeling
in a mouse model of reperfused myocardial infarction. J Histochem
Cytochem. 61:555–570. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ruwhof C and van der Laarse A: Mechanical
stress-induced cardiac hypertrophy: Mechanisms and signal
transduction pathways. Cardiovasc Res. 47:23–37. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bernardo BC, Weeks KL, Pretorius L and
McMullen JR: Molecular distinction between physiological and
pathological cardiac hypertrophy: Experimental findings and
therapeutic strategies. Pharmacol Ther. 128:191–227. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Berk BC, Fujiwara K and Lehoux S: ECM
remodeling in hypertensive heart disease. J Clin Invest.
117:568–575. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Baudino TA, Carver W, Giles W and Borg TK:
Cardiac fibroblasts: Friend or foe? Am J Physiol Heart Circ
Physiol. 291:H1015–H1026. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Leenders JJ, Wijnen WJ, van der Made I,
Hiller M, Swinnen M, Vandendriessche T, Chuah M, Pinto YM and
Creemers EE: Repression of cardiac hypertrophy by KLF15: Underlying
mechanisms and therapeutic implications. PLoS One. 7:e367542012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yu Y, Ma J, Xiao Y, Yang Q, Kang H, Zhen
J, Yu L and Chen L: KLF15 is an essential negative regulatory
factor for the cardiac remodeling response to pressure overload.
Cardiology. 130:143–152. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Oka T, Akazawa H, Naito AT and Komuro I:
Angiogenesis and cardiac hypertrophy: Maintenance of cardiac
function and causative roles in heart failure. Circ Res.
114:565–571. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fisch S, Gray S, Heymans S, Haldar SM,
Wang B, Pfister O, Cui L, Kumar A, Lin Z, Sen-Banerjee S, et al:
Kruppel-like factor 15 is a regulator of cardiomyocyte hypertrophy.
Proc Natl Acad Sci USA. 104:7074–7079. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shindo T, Manabe I, Fukushima Y, Tobe K,
Aizawa K, Miyamoto S, Kawai-Kowase K, Moriyama N, Imai Y, Kawakami
H, et al: Kruppel-like zinc-finger transcription factor KLF5/BTEB2
is a target for angiotensin II signaling and an essential regulator
of cardiovascular remodeling. Nat Med. 8:856–863. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chin MT: KLF15 and cardiac fibrosis: The
heart thickens. J Mol Cell Cardiol. 45:165–167. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Haldar SM, Lu Y, Jeyaraj D, Kawanami D,
Cui Y, Eapen SJ, Hao C, Li Y, Doughman YQ, Watanabe M, et al: Klf15
deficiency is a molecular link between heart failure and aortic
aneurysm formation. Sci Transl Med. 2:26ra262010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu Y, Zou S, Yang Q, Xiao Y, Yan Y, Chen
H, Wu H, Luo Y, Yu P and Chen L: KLF15 is a positive regulatory
factor in the process of myocardial remodeling and angiogenesis
induced by pressure overload. Int J Clin Exp Med. 9:13394–13406.
2016.
|
|
16
|
Simoes PA, Zamarioli A, Bloes P, Mazzocato
FC, Pereira LH, Volpon JB and Shimano AC: Effect of treadmill
exercise on lumbar vertebrae in ovariectomized rats:
Anthropometrical and mechanical analyses. Acta Bioeng Biomech.
10:39–41. 2008.PubMed/NCBI
|
|
17
|
Karamanolis G, Delladetsima I, Kouloulias
V, Papaxoinis K, Panayiotides I, Haldeopoulos D, Triantafyllou K,
Kelekis N and Ladas SD: Increased expression of VEGF and CD31 in
postradiation rectal tissue: Implications for radiation proctitis.
Mediators Inflamm. 2013:5150482013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yan L, Wei X, Tang QZ, Feng J, Zhang Y,
Liu C, Bian ZY, Zhang LF, Chen M, Bai X, et al: Cardiac-specific
mindin overexpression attenuates cardiac hypertrophy via blocking
AKT/GSK3β and TGF-β1-Smad signalling. Cardiovasc Res. 92:85–94.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wynn TA and Ramalingam TR: Mechanisms of
fibrosis: Therapeutic translation for fibrotic disease. Nat Med.
18:1028–1040. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Koitabashi N, Danner T, Zaiman AL, Pinto
YM, Rowell J, Mankowski J, Zhang D, Nakamura T, Takimoto E and Kass
DA: Pivotal role of cardiomyocyte TGF-β signaling in the murine
pathological response to sustained pressure overload. J Clin
Invest. 121:2301–2312. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Teekakirikul P, Eminaga S, Toka O, Alcalai
R, Wang L, Wakimoto H, Nayor M, Konno T, Gorham JM, Wolf CM, et al:
Cardiac fibrosis in mice with hypertrophic cardiomyopathy is
mediated by non-myocyte proliferation and requires Tgf-β. J Clin
Invest. 120:3520–3529. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Holzem KM, Marmerstein JT, Madden EJ and
Efimov IR: Diet-induced obesity promotes altered remodeling and
exacerbated cardiac hypertrophy following pressure overload.
Physiol Rep. 3(pii): e124892015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pelouch V, Dixon IM, Golfman L, Beamish RE
and Dhalla NS: Role of extracellular matrix proteins in heart
function. Mol Cell Biochem. 129:101–120. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cosaceanu D, Budiu RA, Carapancea M,
Castro J, Lewensohn R and Dricu A: Ionizing radiation activates
IGF-1R triggering a cytoprotective signaling by interfering with
Ku-DNA binding and by modulating Ku86 expression via a p38
kinase-dependent mechanism. Oncogene. 26:2423–2434. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Leenders JJ, Wijnen WJ, Hiller M, van der
Made I, Lentink V, van Leeuwen RE, Herias V, Pokharel S, Heymans S,
de Windt LJ, et al: Regulation of cardiac gene expression by KLF15,
a repressor of myocardin activity. J Biol Chem. 285:27449–27456.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Small EM, Thatcher JE, Sutherland LB,
Kinoshita H, Gerard RD, Richardson JA, Dimaio JM, Sadek H, Kuwahara
K and Olson EN: Myocardin-related transcription factor-a controls
myofibroblast activation and fibrosis in response to myocardial
infarction. Circ Res. 107:294–304. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Flanders KC, Major CD, Arabshahi A,
Aburime EE, Okada MH, Fujii M, Blalock TD, Schultz GS, Sowers A,
Anzano MA, et al: Interference with transforming growth
factor-beta/Smad3 signaling results in accelerated healing of
wounds in previously irradiated skin. Am J Pathol. 163:2247–2257.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tian Y and Morrisey EE: Importance of
myocyte-nonmyocyte interactions in cardiac development and disease.
Circ Res. 110:1023–1034. 2012. View Article : Google Scholar : PubMed/NCBI
|