Introduction
Bromodomain (BD) and BD extra-terminal domain (BET)
protein family members, including BD-containing 2 (BRD2), BRD3,
BRD4 and BD testis-associated (BRDT), specifically recognize
acetylated lysine acid on histones mainly through two conserved
N-terminal regions (BD1 and BD2) (1,2). BET
proteins bind to the acetylation site on the chromosome and recruit
other transcriptional regulatory complexes to regulate gene
expression. Their ability to bind to a single acetylation site on
histones is generally weak, whereas their ability to bind to
multiple acetylation sites is strong (3).
BRD4 is the most well-studied and important
functional member of the BET protein family. Its binding
characteristics and vital functions in transcriptional regulation
have been extensively reported. BRD4 interacts with multiple
protein complexes in the active promoter and enhancer regions,
including Mediator, positive transcription elongation factor b
(P-TEFb) and jumonji domain containing 6 (Jmjd6), and serves
multiple roles in transcriptional regulation (4,5).
BRD4 was first discovered during the isolation and purification of
human Mediator complexes (6).
Subsequent studies have indicated that BRD4 and Mediator interact
in multiple cell types and have similar binding distributions
across the chromosome. This co-localization helps them bind more
stably to DNA (6). The Mediator
complex is able to link the transcription factor (TF) activation
effect to RNA polymerase II (RNAPII), while the interaction of BRD4
with Mediator suggests that it serves an important role in
TF-mediated transcriptional activation. During RNA transcription,
the transcriptional initiator RNAPII binds to the transcriptional
arrest-associated factors DRB sensitivity-inducing factor (DSIF)
and negative elongation factor (NELF), halting transcription at a
position located 20–60 nt downstream of the transcription
initiation site. P-TEFb is a transcription elongation factor that
phosphorylates DSIF and NELF. Once recruited to the promoter
region, P-TEFb promotes RNAPII elongation by phosphorylating DSIF
and NELF (7). Previous studies
have indicated that BRD4 recognizes the acetylation site of the
activated promoter region, recruits P-TEFb to the vicinity of the
gene promoter and links the active chromatin region in the
acetylated state to RNAPII for extension (4). It has also been reported that BRD4
forms a protein complex with the histone demethylase Jmjd6 in the
distal enhancer region to promote RNAPII-mediated elongation
(8). These distal enhancers are
known as anti-pause enhancers, which also suggests that the
complexes formed by BRD4, Jmjd6 and P-TEFb bind to chromatin
through long-distance interactions, as they are close to the distal
and deep promoters of the gene in three-dimensional space, thus
regulating transcriptional activation and elongation of genes
(8–10). The majority of studies
investigating BRD4 are limited to specific cell types and are based
on in vitro biochemical experiments. Therefore, at the
genome-wide level, the integration of multi-level chromatin
immunoprecipitation-sequencing (ChIP-seq) data from multiple cell
lines is of great significance for comprehensive and in-depth
analyses of the binding characteristics of BRD4.
The BET inhibitors JQ1 and I-BET were developed by
two independent research groups and reported in 2010 (11,12).
The inhibitors compete with acetylation sites for histone binding
to the BD of the BET protein, making it impossible for the BET
protein to interact with the histones and resulting in dissociation
of the BET protein from the chromatin. The discovery of BET
inhibitors has made BRD4 a potential target for the treatment of a
variety of diseases, particularly cancer. The BRD4-NUT (nuclear
protein in testis) fusion protein is a driver TF for midline
cancer. JQ1 may cause detachment of BRD4-NUT from the chromatin,
which promotes terminal differentiation and apoptosis of cancer
cells. Recently, a large number of studies have indicated that BET
inhibitors exhibit good therapeutic effects on hematopoietic
malignancies, including acute myeloid leukemia (AML) (13,14),
multiple myeloma (MM) (15),
Burkitt's lymphoma (BL) (16),
diffuse large B-cell lymphoma (DLBCL) (17) and T-cell acute lymphoblastic
leukemia (T-ALL) (18). In
addition to hematological tumors, BET inhibitors also have good
effects on a range of solid tumor types. It has been reported that
neuroblastoma (19,20) and medulloblastoma (20) are triggered by amplification of
N-MYC and MYC, respectively, which are sensitive to JQ1 treatment,
and their proliferation is negatively influenced by inhibition of
N-MYC or MYC by JQ1. In a human lung adenocarcinoma cell line, JQ1
inhibited proliferation by reducing the expression of the
proto-oncogene FOS-like 1, AP-1 TF subunit (FOSL1) and its
downstream target genes (21). In
addition, in activated endothelial cells and macrophages, JQ1
eliminated the inflammatory response mediated by super-enhancers,
thereby exerting an anti-inflammatory effect. The majority of
previous studies have linked the anti-cancer effect of BET
inhibitors to their inhibition of important proto-oncogenes,
including MYC and its downstream target genes. Subsequent studies
have reported that a similar therapeutic effect to that of BET
inhibitors may also be achieved by inhibiting other
proto-oncogenes, including B-cell lymphoma 2 (BCL2) and E2F1, or
key TFs, including FOSL1 and androgen receptor (AR) (22). Notably, overexpression of MYC or
BCL2 in BET inhibitor-treated cancer cells has been reported to
only partially attenuate the anti-cancer effect of the BET
inhibitors (15,16,19).
This finding may indicate that the target of the BET inhibitor is
not a single gene, but more likely includes multiple genes involved
in the regulation of cancer. Based on this, it may be speculated
that BET inhibitors have significant therapeutic effects on a
variety of cancer types of different origins and pathogeneses,
possibly by inhibiting certain key proto-oncogenes or a core gene
set associated with cancer development to thereby interfere with
proliferation, induce cell cycle arrest and trigger apoptosis.
BRD4 is an important member of the RNAPII
transcriptional complex and is a co-activator of transcriptional
regulation. However, the mechanisms of BET inhibitors exhibit
distinct heterogeneity among different cell types. A super-enhancer
is a small group of classic enhancer regions with super-enrichment
of the co-activator, mediator complex subunit 1 (MED1) (23–25).
The genes associated with super-enhancers are mostly cell-specific
genes that determine cell identity. BRD4 is highly enriched in
these super-enhancer regions and actively regulates the expression
of this small group of important genes (26). BET inhibitors result in the
dissociation of BRD4 from these regions, thereby selectively
inhibiting super-enhancer-associated genes. However, a large number
of studies have reported that BRD4 directly interacts with multiple
TFs through BD recognition of acetylated lysines in TFs, including
nuclear factor-κB, GATA-binding protein 1 and AR (22), or by undetermined means, which may
involve P53, YY1 TF and MYC/MYC-associated factor X (MAX) (27). BET inhibitors not only directly
inhibit the expression of key TFs, but also disrupt the binding of
BRD4 to TFs in order to inhibit the activation of downstream target
genes. The binding characteristics of BRD4 and the mechanism of
action of BET inhibitors have been reported in specific cell types;
however, the integration of multi-omics data from multiple cell
lines to investigate the general mechanism in various types of
cancer has not been previously performed. Such an analysis will
provide a more unified theoretical basis for cancer treatment.
In the present study, the binding characteristics of
BRD4 were systematically evaluated in multiple cell lines by
integrating a set of ChIP-seq datasets for BRD4 and its associated
factors in six different cancer cell types, as well as the
expression profiles of the cells following JQ1 treatment (data
provided in Tables SI and
SII). The cell lines investigated
in the current study included the human small cell lung cancer
(SCLC) cell line H2171, the glioblastoma multiforme cell line U-87
MG, the MM cell line MM.1S, the DLBCL cell line cLy1, the T-ALL
cell line KOPT-K1 and the tumor necrosis factor (TNF)-α-activated
human endothelial cell line HUVEC-C (28). The results of the current study
revealed the general mechanism of BET inhibitors in the treatment
of cancer. Notably, a core gene set that is regulated by JQ1 and is
important for the progression of various cancer types was
identified, which provides potential targets for cancer therapy, as
well as a broader and more uniform perspective for understanding
the mechanism underlying the action of BET inhibitors.
Materials and methods
Data integration
For comprehensive analysis of BRD4 binding across
the genome, ChIP-seq data were collected for multiple factors,
including BRD4, H3K27Ac, RNAPII, MED1, MYC, MAX, E2F1 and histone
H3 trimethylated at lysine 4 (H3K4me3), in six tumor cell lines
(namely H2171, U-87 MG, MM.1S, Ly1, KOPT-K1 and HUVEC-C). H2171
(ATCC® CRL-5929™) used in the present study was a human
SCLC cell line. The U-87 MG (ATCC® HTB-14™) cell line
was also used, which is most probably a glioblastoma cell line of
unknown origin. The MM cell line MM.1S (ATCC® CRL-2974™)
was also examined, as well as the DLBCL cell line Ly1, which has
previously been used in the study of Chapuy et al (17). In addition, the T-ALL cell line
KOPT-K1 was used, which was kindly gifted by Dr A.T. Look from
Dana-Farber Cancer Institute (Boston, MA, USA) (16). The TNF-α-activated human
endothelial cell line HUVEC-C (ATCC® CRL-1730™) was also
used in the current study, which has previously been investigated
in the study of Brown et al (28).
Raw ChIP-seq data were downloaded from the Sequence
Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) of the National
Center for Biotechnology Information (NCBI). According to the
unified processing analysis pipeline, the detailed information and
analysis results are provided in Table SI. For analysis of the effects of
JQ1 on gene expression in the corresponding cell lines, the gene
expression profiles of multiple cell lines prior to and following
JQ1 treatment were also integrated. Raw data for all cell lines
were downloaded from the NCBI Gene Expression Omnibus (GEO;
http://www.ncbi.nlm.nih.gov/geo/)
database. The accession numbers and microarray data are provided in
Table SII. The collected gene
expression profiles were mainly divided into three categories, as
follows: Gene expression profiles treated with JQ1 matched with the
corresponding ChIP-seq data served as the training set; Expression
profiles treated with JQ1 without corresponding ChIP-seq data
served as the test set; and a class of normal gene expression data
without JQ1 treatment for all the six cell lines under the same
platform served as the expression set.
Bioinformatics analysis
All ChIP data were analyzed and processed using a
unified procedure to eliminate data processing bias. Reads were
filtered with Trimmomatic (version 0.32) (29) and then mapped to the hg19 genome
using Bowtie (version 2.2.4) (30). Subsequent to normalization to the
indicated counts, tdf files were generated to visualize the genomic
coverage using IGVtools (version 2.3.52) (31). Only one mapped read to each unique
region of the genome that was >175 bp was kept and used in peak
calling. Peak calling and motif analyses were then performed using
MACS (version 1.4.2) (32) and
HOMER (version 4.1) (33) with
default parameters. Motifs with lengths of 8, 10 and 12 bases were
used for de novo motif analysis. A simple script was
developed to calculate the normalized read density of the ChIP-seq
data in any given region. Each given site was extended to 2 kb from
the center of the vertex, and the 4-kb region was divided into 40
bins in units of 100 bp. Subsequently, the density of reads per bin
(100 bp) was calculated. In order to allow for comparisons among
multiple groups, the density of the reads was normalized to the
total mapped reads to produce a signal in units of reads per 10
million mapped reads per 100 bp. Super-enhancers were defined and
calculated as previously described, with slight modifications
(24,26).
The gene expression profile platforms included
Affymetrix (http://www.affymetrix.com/site/mainPage.affx) and
PrimeView (http://www.primeviewglobal.com/), and data processing
was performed with R software (version 3.3.4) using the affy
packages (34). The data on gene
transcript platforms, including HuEx-1_0-st and HuGene-1_0-st, were
analyzed using the oligo package (version 1.48.0, http://www.bioconductor.org/packages/release/bioc/html/oligo.html).
The original data were normalized using the Robust Multi-array
Average method (35,36), whereas differentially regulated
genes were identified using the limma package (37). A false discovery rate (FDR) of
<0.01 was set as the screening standard. Gene set enrichment
analysis (GSEA) of the core gene set regulated by JQ1 was also
performed among the test set (38). Enrichment analysis was performed
with Gene Ontology (GO) and the Encyclopedia of Genes Genomes
(KEGG) pathway (https://david.ncifcrf.gov/).
Statistical analysis
One-way analysis of variance based on the function
‘aov’ in R software (version 3.4.5) was performed to analyze the
overall differences Tukey and Student-Newman-Keuls tests based on
functions of ‘TurkeyHSD’ and ‘SNK.test’ in package agricolae
(version 1.3–1) in R software (version 3.4.5) were conducted to
assess multiple comparisons between different groups. A
statistically significant difference was indicated by
P<0.05.
Results
Most BRD4 sites bind to cell
type-specific enhancer regions, while a small portion of
significantly enriched sites bind to the promoter regions of all
cell lines
In order to investigate the distribution of BRD4
genome-wide binding in multiple cell lines, the BRD4 sites from the
six cell lines examined in the present study were merged into one
collection. Two or more binding sites with overlapping positions
(≥1 bp) on the genome were merged into one binding interval, and
this interval was set as being shared by the cell line to which the
binding site belonged. Thus, the binding sites of the six cell
lines, including H2171 (n=24,273), HUVEC-C (n=10,173), KOPT-K1
(n=17,975), Ly1 (n=18,696), MM.1S (n=20,768) and U-87 (n=29,216),
were merged into 76,946 BRD4 binding intervals (Table SI). In theory, each final merged
BRD4 binding intervals will be yes or no when considering whether
or not it belongs to the binding site of BRD4 in the certain cell
line of the six cell lines. The number of all combinations in all
the six cell lines is theoretically 26−1 except that the
group S000000, which represents binding sites, does not belong to
any of the six cell lines. Finally, the total number of binding
sites for all cell lines was theoretically divided into 63 groups
(calculated from 26−1) according to the cell lines in
which each BRD4 binding interval was located.
To evaluate the statistical significance of the
number of binding sites in each group, an independent random
sampling method was used to calculate a background number for each
group. The BRD4 binding sites of the six cell lines were integrated
into one collection (without merging between different binding) and
the same number sites for each cell was randomly extracted from the
collection as BRD4 binding sites in each corresponding cell line.
Then, the six random extracted groups were merged into one
collection. By repeating the aforementioned steps 1,000 times, a
random background for the number of sites in each group was
constructed. Assuming that the background had a normal
distribution, the statistical z-score value was calculated.
According to the order of the number of binding sites in each group
from large to small (for numbers <250), it was revealed that the
number of BRD4 binding sites that were cell line-specific and only
occurred in one cell line was the highest. A total of 96.68% of the
BRD4 binding events were cell line-specific. As presented in
Table I, the cell line-specific
BRD4 binding sites in the U-87 and H2171 cell lines accounted for
29.67 and 27.72% of the total number of BRD4 binding sites,
respectively, and represented 60.16 and 67.65% of the BRD4 binding
sites in the respective cell lines. These results indicated that
the genome-wide distribution of BRD4 binding sites differs greatly
among the different cell lines. In addition, it was revealed that
only 1,509 BRD4 (S111111) sites were shared by all cell lines;
however, their significance was high when compared to the
background distribution generated by the number of randomly
sampling group of S111111 with significant z-score, indicating that
BRD4 binding in all cell lines was not a random event (Table I). In order to study the BRD4
binding sites shared by multiple cell lines, BRD4 binding sites
that co-existed in at least five of the cell lines were defined as
a common BRD4 set.
 | Table I.Number of each group in the overall
union of BRD4 binding sites. |
Table I.
Number of each group in the overall
union of BRD4 binding sites.
| Group | Peak numbers | Percentage (%) | bg counts | zscore |
|---|
| S000001 | 17,576 | 29.67 | 7,134 | 1,236.56 |
| S100000 | 16,420 | 27.72 | 5,590 | 1,364.17 |
| S000100 | 7,009 | 11.83 | 4,040 | 408.01 |
| S000010 | 6,740 | 11.38 | 4,589 | 281.63 |
| S001000 | 5,413 | 9.14 | 3,852 | 218.53 |
| S010000 | 4,119 | 6.95 | 2,014 | 328.88 |
| S010001 | 1,642 | 2.77 | 803 | 161.96 |
| S111111 | 1,509 | 2.55 | 293 | 304.88 |
| S001010 | 1,476 | 2.49 | 986 | 89.23 |
| S001110 | 1,411 | 2.38 | 306 | 271.45 |
| S001111 | 1,288 | 2.17 | 338 | 228.33 |
| S000110 | 1,146 | 1.93 | 1,032 | 20.82 |
| S101111 | 1,135 | 1.92 | 516 | 138.58 |
| S001100 | 994 | 1.68 | 864 | 24.77 |
| S100001 | 813 | 1.37 | 2,282 | −228.46 |
| S011111 | 764 | 1.29 | 138 | 185.07 |
| S000011 | 697 | 1.18 | 1,862 | −185.14 |
| S001011 | 587 | 0.99 | 579 | 1.54 |
| S100010 | 586 | 0.99 | 1,445 | −145.26 |
| S101110 | 510 | 0.86 | 242 | 68.97 |
| S100100 | 429 | 0.72 | 1,264 | −143.34 |
| S000101 | 341 | 0.58 | 1,634 | −212.80 |
| S101000 | 313 | 0.53 | 1,207 | −160.35 |
| S001001 | 303 | 0.51 | 1,554 | −210.82 |
| S101010 | 271 | 0.46 | 437 | −37.15 |
| S101011 | 256 | 0.43 | 508 | −53.59 |
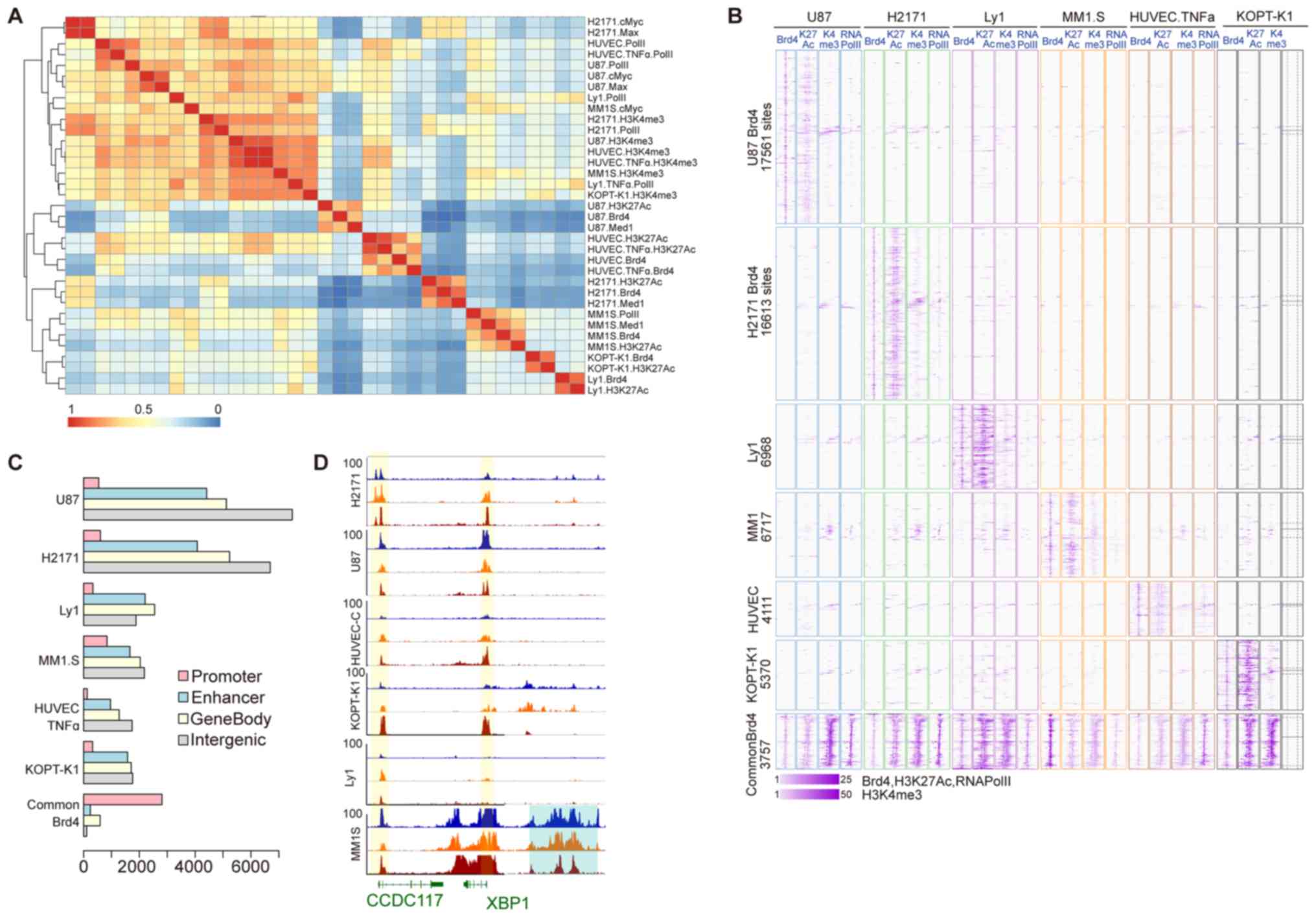
To further analyze the distribution characteristics
of BRD4 in the functional region and its interaction with various
associated factors (10), a number
of histone modifications and coactivators, namely H3K27Ac, H3K4me3,
MED1, MYC, MAX and RNAPII, were integrated relative to the BRD4
function. The binding intervals of BRD4 that are presented in
Table I were used as potential
BRD4 binding sites in all cell lines, and their binding densities
were then extracted. Thus, it was possible to quantitatively
analyze the correlation between BRD4 and associated factors
surrounding the potential BRD4 binding sites. Based on the
correlation analysis of various factors, it was indicated that all
factors could be divided into two categories. The first category is
a cell line-independent class that mainly includes the histone
modification H3K4me3, the TFs MYC and MAX, and RNAPII (Fig. 1A). In general, transcription start
sites and promoter regions of actively transcribed genes are marked
by H3K4me3, H3K27Ac and RNAPII (39), while MYC/MAX dimers have also been
reported to be mainly incorporated in the promoter regions of genes
(40). This observation suggests
that several of the aforementioned factors may be associated with
the BRD4 sites due to their significant enrichment in multiple cell
lines and distribution in the gene promoter region. The second
category is a cell line-specific class, in which each subset
comprises BRD4, H3K27Ac and/or MED1, which indicates that the
majority of the BRD4 sites in the genome bind by recognition of the
acetyl group of H3K27Ac and are co-occupied by MED1. Active
enhancers may be identified by enrichment of monomethylated H3K4
and H3K27Ac (41), and MED1 is a
co-activated complex consisting of a linker enhancer and a
promoter. Therefore, it may be concluded that the BRD4 binding site
is mostly cell line-specific and is highly correlated with the
enhancer region. In addition, according to the clustering distance,
the distribution of BRD4 in the MM.1S, KOPT-K1 and Ly1 cells was
closer, and their common binding sites (S001110) were also
significant (z-score=271.45); by contrast, U-87 was closer to the
distribution in HUVEC-Cs, and the z-score of their common binding
site (S010001) was 162.
Next, a heatmap of BRD4 and its co-binding factors
at each BRD4 binding interval was generated (Fig. 1B). The results revealed that, at
cell line-specific BRD4 binding sites, only BRD4 and H3K27Ac of the
corresponding cell line have binding signals with a low density for
H3K4me3 and RNAPII. In the BRD4 binding sites shared by multiple
cell lines, BRD4, H3K27Ac, H3K4me3 and RNAPII were all enriched
among all the cell lines examined. The binding sites of each group
were further mapped to RefSeq genes to study the gene distributions
(Fig. 1C). It was observed that
the cell line-specific BRD4 binding sites are located distant from
the gene transcription initiation site, and are mainly distributed
in the enhancer and intergenic regions. By contrast, the BRD4
binding sites shared by multiple cell lines are mainly distributed
in promoter regions, which was consistent with the results
presented in Fig. 1A. X-box
binding protein 1 (XBP1) has been reported to be highly expressed
in MM and to have an important role in the genesis of this tumor
(42). The binding signals of
BRD4, H3K27Ac and RNAPII around the XBP1 gene in the six cell lines
are presented in Fig. 1D. In the
XBP1 upstream enhancer region, only the MM.1S cell line had a high
density of BRD4 and H3K27Ac, and a relatively low density of
RNAPII. BRD4 and RNAPII were found to bind to the promoter regions
of XBP1 and CCDC117 in multiple cell lines. Based on these results,
the distribution of BRD4-binding sites in all the investigated cell
lines was obtained. The majority of the BRD4 binding sites were
found to only bind to specific cell lines and to co-localize with
H3K27Ac and MED1 in enhancer regions. By contrast, a certain
portion of BRD4 binding sites were observed to bind to all cell
lines and to mainly co-bind with RNAPII, H3K4me3 and MYC/MAX in
promoter regions.
Cell type-common BRD4 binding sites
co-localize with oncogenic TFs, including E2F1 and MYC, in promoter
regions
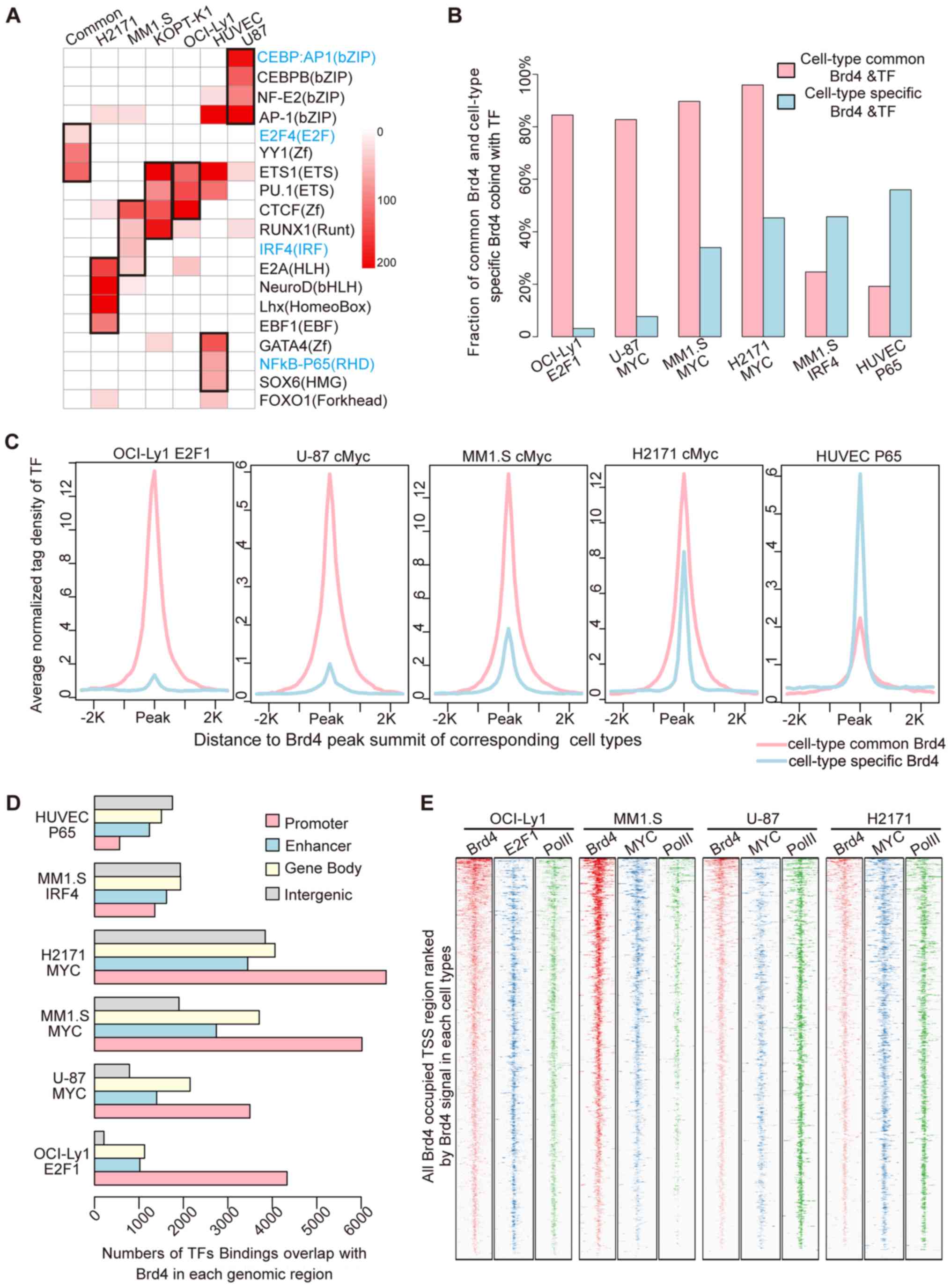
To regulate transcription, BRD4 binds to a complex
involved in transcription and interacts with a variety of TFs. To
examine the interactions of cell type-specific and cell type-common
BRD4 binding sites with specific TFs, a de novo motif
analysis of these two categories of BRD4 sites was performed to
obtain potential TFs that may bind in proximity to the two
categories of BRD4 binding. It was observed that the cell
line-specific TFs were significantly enriched at the cell
line-specific BRD4 binding sites (Fig.
2A). The interferon-regulatory factor (IRF) family was
specifically enriched in MM.1S cells, nuclear factor κB (NFκB)-P65
was specifically enriched in TNF-α-activated HUVEC-Cs, and the
CCAAT enhancer-binding protein family was specifically enriched in
U-87 cells. In addition, the hematopoietic-specific TFs PU.1 and
runt-related transcription factor 1 were enriched in the blood
cancer cell lines MM.1S, KOPT-K1 and Ly1. Furthermore, the E2F, YY1
and ETS motifs were enriched in the common BRD4 binding sites
shared among multiple cell lines.
Previous studies have demonstrated that E2F, YY1 and
ETS1 are mainly involved in gene promoter regions and are
associated with fundamental functions of cell lines and
tumorigenesis (43). Furthermore,
the BRD4 inhibitor JQ1 inhibits the proliferation of cancer cells
mostly by depressing the expression of the proto-oncogenes MYC or
N-MYC, which are also the major binding sites in the gene promoter
region. Thus, it was hypothesized that the cell type-common BRD4
binding sites may co-localize with cancer-associated TFs, including
E2F1 and MYC, in the promoter region. To validate this hypothesis,
ChIP-seq data for E2F1 and MYC in multiple cell lines were
collected. First, the co-localization of TF binding events with the
cell type-common and cell type-specific BRD4 binding sites was
assessed (Fig. 2B). The results
indicated that the majority of the cell type-common BRD4 binding
sites were bound by E2F1 and MYC, and that the co-localization
ratio exceeded 80%. The co-binding ratio of the TFs IRF4 and
NFκB-P65 in the cell type-specific BRD4 binding sites was only 20%.
However, at the cell line-specific BRD4 binding sites, binding of
E2F1 and MYC was low, with only 8% of the cell line-specific BRD4
contained in E2F1 binding sites in the Ly1 cell line.
To further illustrate the interaction of the cell
type-common BRD4 binding sites with E2F1 and MYC, the average
binding signal of each TF in the two groups of BRD4 binding sites
was evaluated (Fig. 2C). The
results revealed that the binding signals of E2F1 and MYC were
significantly higher in the cell type-common BRD4 binding sites as
compared with those in the cell line-specific sites. By contrast,
P65 exhibited significantly higher binding at the cell
line-specific BRD4 sites. Next, the distribution of sites co-bound
by TFs and BRD4 in specific cell lines across the whole genome was
assessed. The results indicated that E2F1 and MYC were mainly
distributed in the promoter region, whereas IRF4 and P65 were
mainly distributed in non-promoter regions (Fig. 2D). Finally, to further analyze the
genome-wide interaction of BRD4 with E2F1 and MYC, the present
study then focused on binding near transcription start sites
(TSSs). As indicated in the binding profiles in Fig. 2E, BRD4 and E2F1 or MYC exhibited
significant co-localization at TSSs with RNAPII binding, and it was
suggested that BRD4 co-localized with E2F1 or MYC in the promoter
regions in the whole genome, particularly at cell type-common BRD4
binding sites.
In summary, using motif scanning combined with
analysis of TF binding profiles, it was indicated that cell
type-common BRD4 binding sites co-localized with the oncogenic TFs
E2F1 and MYC at promoter regions with subsequent expression of
downstream target genes, resulting in the development of cancer.
These findings also suggest a potential mechanism by which BRD4
inhibitors effectively inhibit the proliferation of various cancer
types.
BRD4 super-enriched regions or genes
in each cell line comprise cell type-specific and cell type-common
gene sets
Recent studies have indicated that most BRD4 binding
sites are super-enriched in certain regions that are termed as
super-enhancers (9,21), which actively regulate the
expression of surrounding genes. However, the definition of
super-enhancers in previous studies is limited to super-enrichment
of co-activators, such as MED1, in non-promoter regions. Previous
studies by our and other groups have indicated that BRD4 and MED1
are co-localized throughout the whole genome (6), and thus, BRD4 may also serve as a
factor for defining super-activated regions. In addition, a
continuous distribution and ultra-high enrichment of BRD4 was
observed near gene promoters (Fig.
3B). Thus, the method used to scan for super-enhancers was
partially modified using the binding signal of BRD4. Promoter
regions containing a single BRD4 binding peak were removed to
retain only those in which multiple BRD4 binding peaks were
continuously distributed. This modified method was used to scan the
super-enhancer regions of all six cell lines and to align them to
the nearest RefSeq gene. Since the region scanned by this method
contained partial promoters, these regions were defined as BRD4
super-loaded regions (or super-enriched regions) instead of
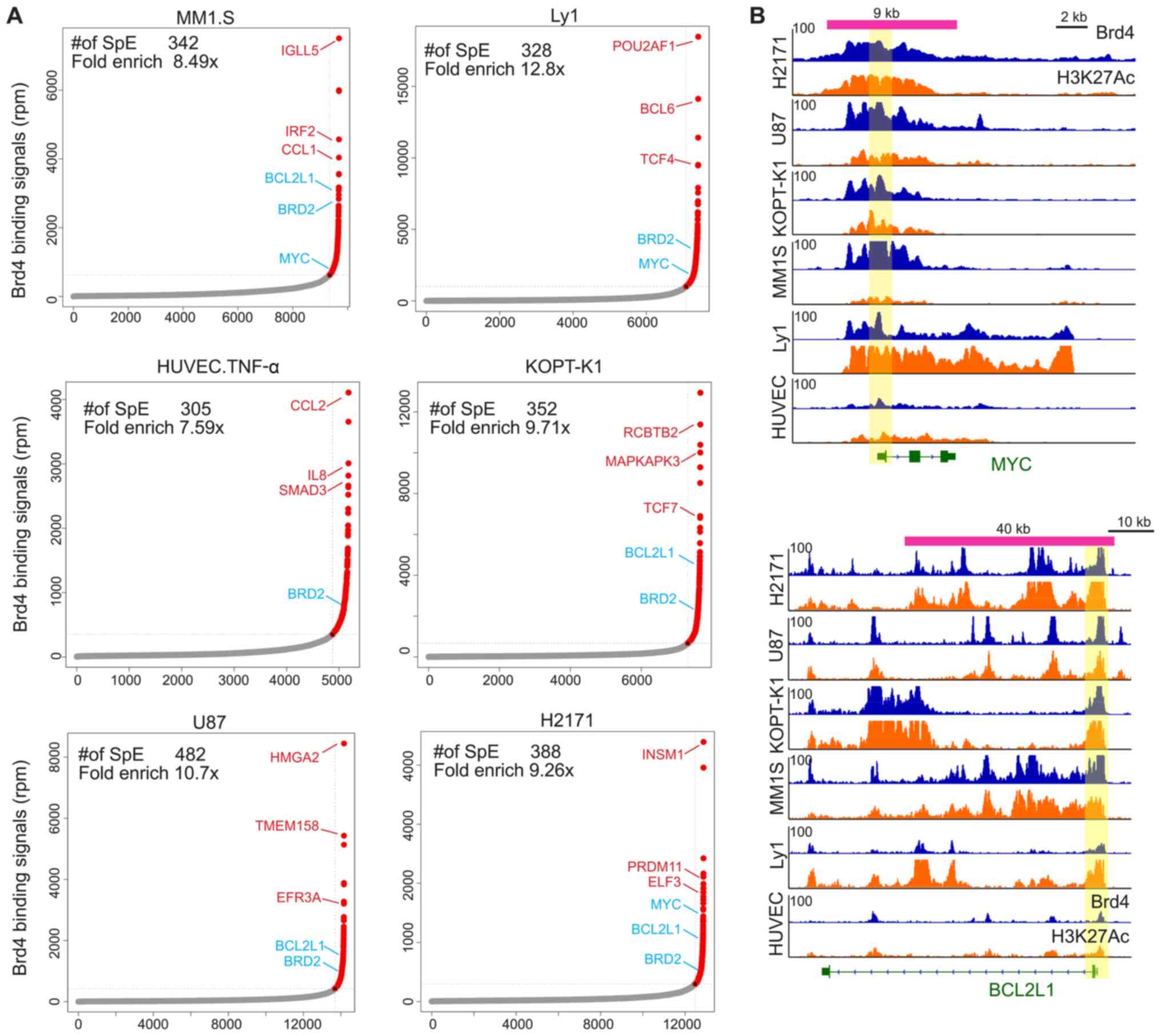
super-enhancers. A total of 305–482 BRD4 super-loaded regions were
scanned per cell line, as shown in Fig. 3A. The assignment of super-loaded
regions to genes in each cell type is provided in the supplementary
files (Tables SIII–SVIII for H2171, HUVEC-C, KOPT-K1, Ly1,
MM.1S and U-87, respectively). Subsequently, the BRD4
super-enriched genes were analyzed, and it was revealed that the
genes with the most abundant BRD4 binding sites were key TFs or
genes associated with cell-specific functions in the corresponding
cell lines. Namely, the gene IRF2 was identified in MM.1S cells,
POU class 2 homeobox-associating factor 1 and BCL6 transcriptional
repressor were identified in Ly1 cells, C-C motif chemokine ligand
2 and IL8 were identified in HUVEC-C cells, TCF7 was identified in
KOPT-K1 cells, HMGA2 was identified in U-87 cells, and INSM
transcriptional repressor 1 was identified in H2171 cells. These
detected TFs have previously been reported to serve important roles
in the corresponding cell lines (44–48).
Notably, it was also observed that a small number of genes were
super-loaded with BRD4 in multiple cell lines, particularly MYC and
BCL2-related protein A1 (BCL2A1). A number of earlier studies have
indicated that MYC and BCL2A1 are involved in infinite
proliferation and cell cycle regulation during cancer development
(49,50). As presented in Fig. 3B, the MYC and BCL2A1 promoter
regions in multiple cell lines contained clusters of super-enriched
fragments of BRD4 and H3K27Ac, suggesting that MYC and BCL2A1 serve
an indispensable role in the development of different types of
cancer.
Cell type-common BRD4 super-loaded
genes are sensitive to BRD4 inhibitors and are highly associated
with tumorigenesis
The BRD4 super-loaded region contains most of the
BRD4 binding signals and actively regulates a small group of
functionally important genes. Therefore, identifying genes
associated with the BRD4 super-loaded region was an important aim
of subsequent analysis in the present study. A small number of
genes were found to be associated with BRD4 super-enriched regions
in multiple cell lines, suggesting their important functions in
multiple cell types. In order to further determine the
characteristics of these genes, the gene set was redefined. For
BRD4 hyper-enriched regions obtained in a specific cell line, it
was examined whether the region was bound by BRD4 in other cell
lines. If >2 BRD4 binding sites were detected in 5 or more cell
lines in the given region, this region was defined as a BRD4 cell
line-common super-enriched region. Otherwise, this region was
defined as a BRD4 super-enriched region specific to a particular
cell line. Subsequently, these two categories of BRD4
super-enriched regions were assigned to the nearest RefSeq gene,
and the common region-associated genes comprised the core gene set
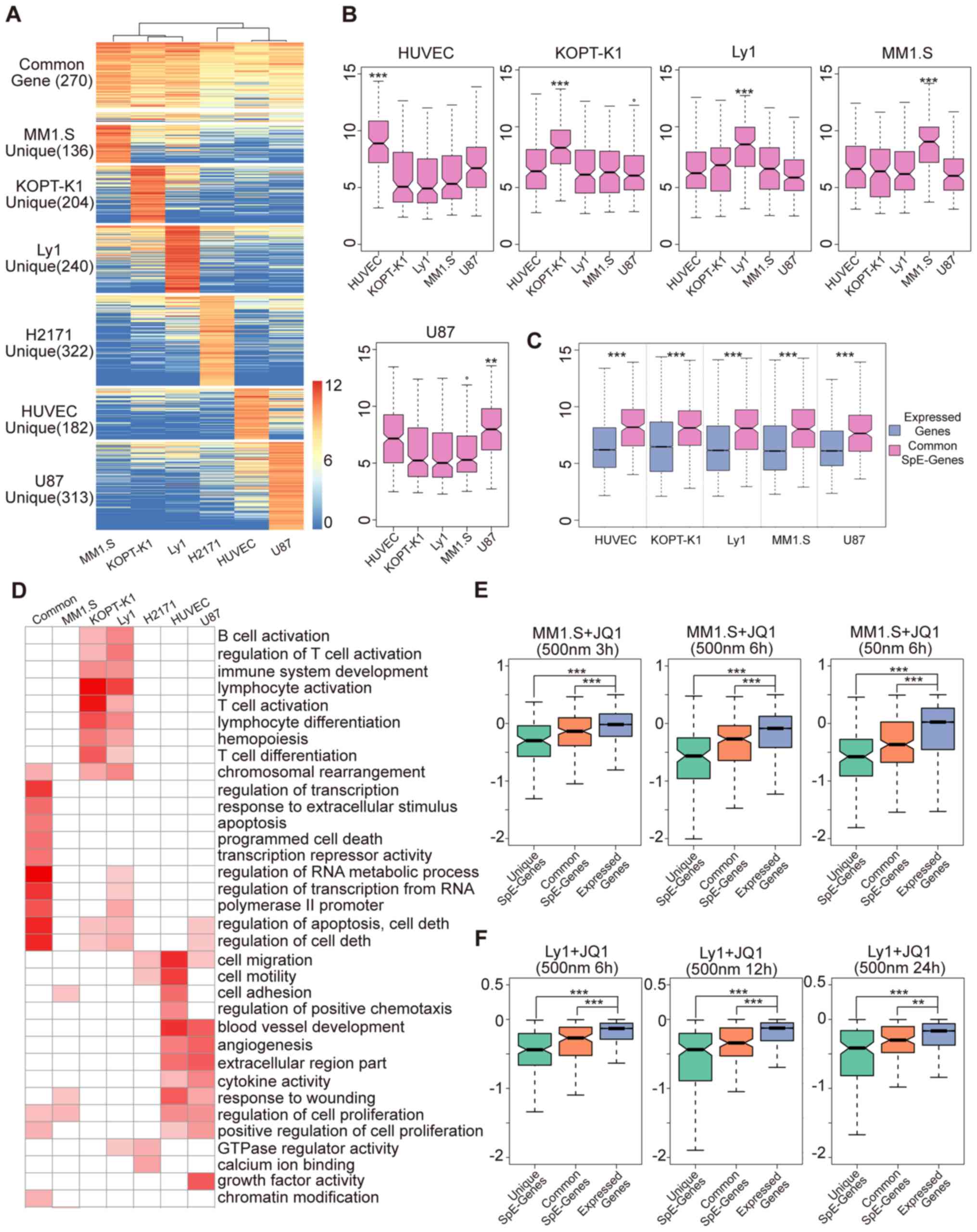
(Table SIX). The cell line-common
BRD4 super-enriched genes had evidently higher BRD4 signals in each
cell line (Fig. 4A), whereas a
strong BRD4 signal was only present in the particular cell line for
the cell line-specific BRD4 super-enriched genes. These results
indicated the regions of the two categories were reliably
determined using the aforementioned method.
BRD4 regulates gene expression through a variety of
mechanisms. To further study the expression of gene sets in each
cell line, the expression profiles were collected, and the results
were obtained by unified analysis (Fig. 4B and C). As indicated in the box
plots in Fig. 4B, the expression
of cell line-specific BRD4 super-enriched region-associated genes
was significantly higher in the corresponding cell line as compared
with that in the other cell lines. The box plots in Fig. 4C indicate that the cell line-common
BRD4 super-enriched genes were significantly overexpressed in each
cell line relative to all genes expressed in the genome. These
results indicated that the BRD4 super enrichment-associated genes
are actively transcribed in the genome, particularly the cell
line-common BRD4 super-enriched genes, which are actively expressed
in all cell lines. In addition, this potentially illustrates the
importance of this gene set in cellular biological processes.
To further study the functions of genes linked to
BRD4 super-enrichment, functional enrichment analysis among the
different gene groups was performed. As presented in Fig. 4D, the Gene Ontology (GO) functional
terms enriched by the cell line-specific gene groups were closely
associated with the function of specific cell types. In the KOPT-K1
and Ly1 lymphoid cell lines, the GO categories that were
specifically enriched by their genes are specific functions of
lymphocytes, including immune regulation and activation, and the
differentiation of B cells and T cells. HUVEC-Cs and U-87 cells are
epithelial-derived cell lines, and their corresponding genes were
significantly enriched in functions associated with cell adhesion,
cell migration, angiogenesis, extracellular matrix and epithelial
cells. Of note, the functional classes enriched by the cell
line-common BRD4 super-enriched region-associated genes represented
the basic functions during cellular biological processes, including
gene transcription regulation, RNA metabolism, apoptosis and
necrosis, and cell proliferation.
The BRD4 inhibitor JQ1 is able to suppress BRD4
target genes, thereby effectively inhibiting tumor cell
proliferation and promoting tumor cell apoptosis (9,11).
To test whether the BRD4 super-loaded target genes defined earlier
in the study were sensitive to BRD4 inhibitors, the expression
profiles of the MM.1S and Ly1 cell lines following JQ1 treatment
were obtained to assess JQ1 inhibition of the aforementioned gene
groups. The inhibitory effect of JQ1 on MM.1S-specific BRD4
super-loaded target genes was most evident at different
concentrations and treatment times (Fig. 4E). In contrast to all the expressed
genes, JQ1 also exerted a significant inhibitory effect on the
cell-common BRD4 super-enriched genes. The results obtained with
Ly1 cells were similar to those observed when the MM.1S cell line
was subjected to JQ1 treatment (Fig.
4F), suggesting the selective inhibition of super-enriched BRD4
genes by JQ1 in specific and multiple cell lines.
Taken together, the cell line-specific and cell
line-common BRD4 super-loaded regions were defined, and in-depth
research on the expression, function and sensitivity of BRD4
hyper-accumulation-associated genes was performed. It was revealed
that BRD4 hyper-accumulation-associated genes were actively
transcribed in cells and that JQ1 had a significant inhibitory
effect on their expression. The functional classes of cell
line-specific gene groups were mainly enriched in specific
functions of specific cell types, whereas the functional terms of
the multi-cell shared gene groups were the basic functions of
biological activities of cells, which were closely associated with
the occurrence of cancer.
JQ1-targeted core gene group shared by
multiple cell lines is significantly inhibited by JQ1 in different
tumor types
The BRD4 inhibitor JQ1 has a good therapeutic effect
on tumors of different origins (11,13,14,25).
It inhibits tumor proliferation and promotes apoptosis by
inhibiting genes with BRD4 hyper-enrichment, thus affecting tumors
with different sources (6,23). The results of the present study
suggested that super-enriched genes shared by multiple cells were
significantly associated with cancer occurrence, and that JQ1 had
an evident inhibitory effect on the expression of these genes
(Fig. 4E). Therefore, it was
hypothesized that JQ1 regulates a core gene group in different
types of cancer cell to inhibit cancer. To examine whether any core
gene group was regulated by JQ1, the 270 BRD4 super-enriched genes
shared by multiple cell types were screened. As presented in
Fig. 5A, genes that were
significantly regulated by JQ1 were screened in at least two of the
HUVEC-C, Ly1 and MM.1S cell lines, as the JQ1 regulatory core gene
group. The core regulatory genes of JQ1 contained a total of 67
genes (as indicated in Table
SIX), among which MYC, BCL2, TCF4 and ETV6 were previously
reported to have important roles in the development of various
cancer types (49,50). Notably, it was discovered that 8
genes were significantly upregulated by JQ1, particularly HEXIM
P-TEFb subunit 1 (HEXIM1). HEXIM1 is a transcriptional elongation
inhibitory factor that has a role opposite to that of BRD4 in the
regulation of transcriptional elongation and may thus have a
negative feedback effect on JQ1. Next, the current study examined
whether the 67 JQ1 core regulatory genes were significantly
inhibited by JQ1 in other cancer types. Expression data pre- and
post-JQ1 treatment generated independently by different
laboratories were collected as the test set, including for MM, BL,
neuroblastoma and medulloblastoma cell lines. Similar to the GSEA
results presented in Fig. 5B, the
JQ1 core regulatory gene group was significantly inhibited by JQ1
in other cancer cell lines (FDR<0.05). Subsequently, a
functional enrichment analysis was performed on these 67 genes,
indicating that their major functions were associated with
transcriptional regulation and cancer development. A total of 45%
of the genes in the core gene cluster (30/67) were TFs involved in
the transcriptional regulation process, with 13 and 11 genes
involved in apoptosis and cell proliferation, respectively,
accounting for 36% of the total genes. In addition, 6 genes were
involved in KEGG pathways, including MYC, BCL2L1, E2F3,
transforming growth factor-β receptor 2, TNF receptor-associated
factor 1 and baculoviral IAP repeat containing 3 (Fig. 5C). Overall, the JQ1 core regulatory
gene group was observed to serve an important role in a variety of
cancer types, which provides a novel perspective on the role of
BRD4 inhibitors in the treatment of cancer.
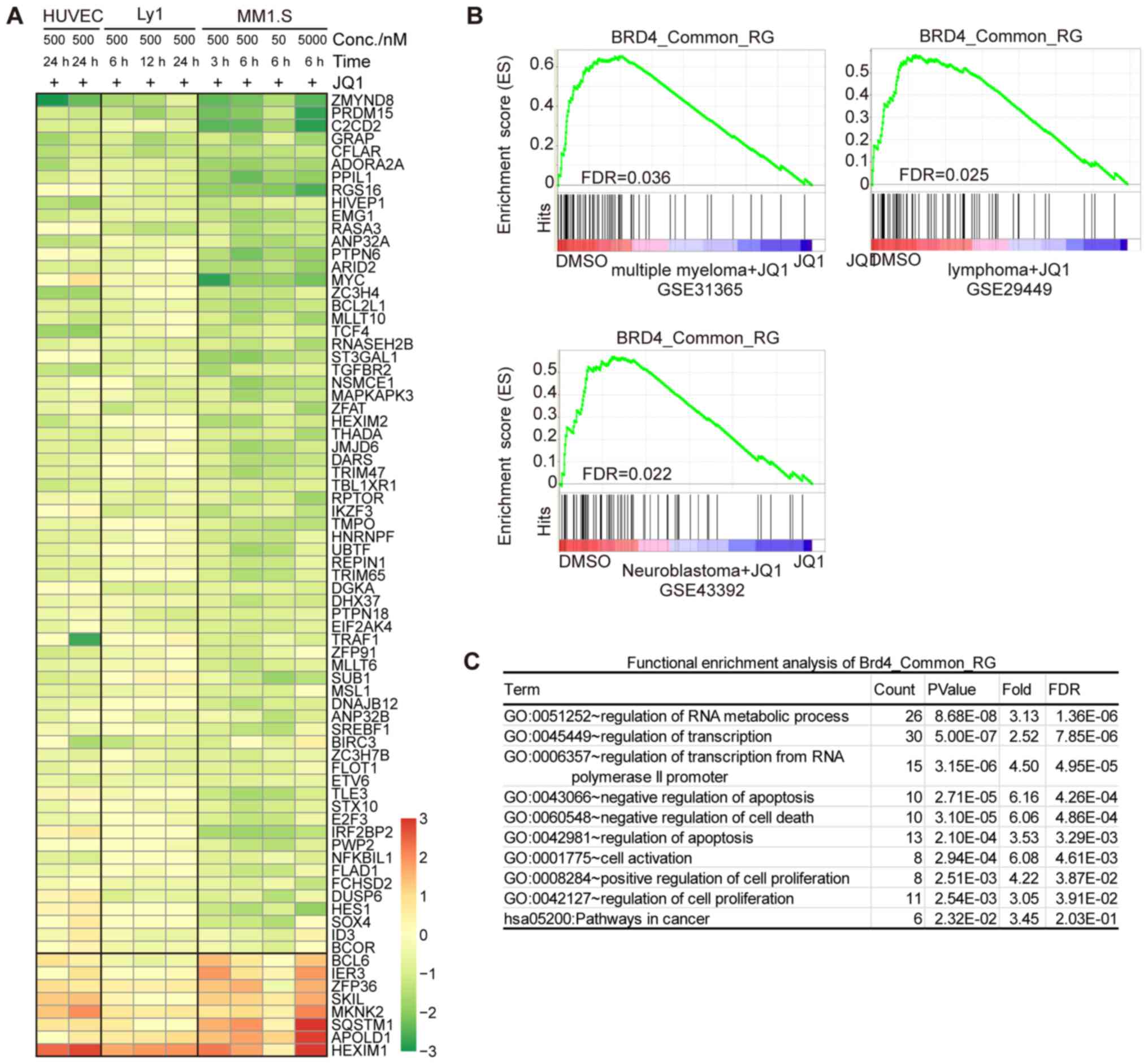 | Figure 5.Core set of common BRD4-regulated
genes concordantly sensitive to JQ1 in various cancer cell types.
(A) Heatmap of log2-expression changes in the core set
of common BRD4-regulated genes in HUVEC-C, Ly1 and MM.1S cells
treated with JQ1 under various conditions. HUVEC-CH UVEC-C common
BRD4 super-loaded genes that were significantly regulated [absolute
log2(fold change)>0.75 and FDR<0.01] by JQ1 in two
of the three cell types were selected. (B) Gene set enrichment
analysis plot of the core set of the common BRD4-regulated gene set
from gene expression profiling of different types of cancer cell
lines treated with JQ1, including multiple myeloma (GSE31365),
lymphoma (GSE29449) and neuroblastoma (GSE43392). (C) Functional
enrichment analysis (performed with the DAVID online tool) of the
core set of common BRD4-regulated genes. A count of >5 and
P-value of <0.01 were considered to indicate statistical
significance. BRD4, bromodomain containing 4; HUVEC-C, human
umbilical vein endothelial cell; FDR, false discovery rate; TNF-α,
tumor necrosis factor α; Conc., concentration; GO, Gene Ontology;
hsa, Homo sapiens. |
Discussion
BRD4 synergistically regulates the expression of
downstream target genes by interacting with histone modifications,
multiple co-activator complexes and TFs (51). The BRD4 inhibitor JQ1 competitively
binds to BRD4 to inhibit its transcriptional activation effect and
has been reported to have beneficial therapeutic effects in various
cancer types (11,13,15).
In the present study, the genome-wide binding characteristics of
BRD4 were systematically analyzed by integrating BRD4-associated
data from multiple cell lines to unveil a general mechanism by
which BRD4 inhibitors exert their anti-cancer effects. It was
identified that cell line-specific BRD4 binding sites were
co-localized with H3K27Ac and MED1 in enhancer regions, whereas
cell line-common BRD4 binding sites co-localized with the
proto-oncogene TFs MYC and E2F1 in gene promoter regions. Since
genes distributed in BRD4 super-enriched regions have vital roles
in cell biological processes, the present study also identified
these genes in each cell line. The results revealed that the cell
line-specific genes were associated with specific cell functions
and cell identity. Conversely, the cell line-common genes were
significantly enriched in fundamental cellular functions.
Furthermore, these two categories were found to be sensitive to JQ1
treatment. In combination with the multiple expression profile data
generated following JQ1 treatment, 67 core regulatory genes were
finally screened from the BRD4 super-enriched gene set in various
cancer cell lines that were suppressed by JQ1. The core regulatory
genes mediated the general mechanism of BRD4 inhibitors, which
provides valuable gene targets for further in-depth studies on
transcriptional regulation and cancer treatment.
The ChIP-seq technique was also used to determine
the characteristics of the distribution of BRD4 in the whole
genome, providing a comprehensive perspective for the study of BRD4
in transcriptional regulation and tumor pathogenesis. It has
previously been reported that BRD4 binds to almost all activated
promoter regions across the genome, as well as most activated
enhancer regions in a variety of cell types (52). The overall binding characteristics
of BRD4 are similar to those of histone acetylation modifications,
including H3K9Ac and H3K27Ac, suggesting that it has the molecular
characteristic of specifically recognizing histone acetylation. In
the present study, ChIP-seq data were integrated from multiple cell
lines and types of binding factors to reveal that BRD4 co-localized
with a variety of factors in the promoter region, including
H3K4me3, RNAPII and H3K27Ac, while it also bound to H3K27Ac and
MED1 in the enhancer region. However, the genome-wide correlation
analysis suggested that BRD4 was more closely associated with
H3K27Ac and MED1 in all types of cell lines. In addition, most of
the BRD4 binding sites exhibited a cell line-specific distribution
and were localized in the enhancer region, whereas a small portion
of the BRD4 binding sites coincided with the promoter region in
multiple cell lines. These observations indicated that the binding
distribution of BRD4 in the promoter region of different cell lines
is more conservative, whereas the binding distribution in the
enhancer region is more extensive and the cell line specificity is
stronger.
BRD4 inhibitors were initially considered as
effective MYC inhibitors. In MM.1S cells, which is an MM cell line
that contains ectopic IgH/MYC, BRD4 binds in the distal IgH
super-enhancer region and activates high MYC expression through
long-term regulation (15). Recent
studies have indicated that BRD4 synergistically regulates MYC
expression by binding to the proximal promoter and distal enhancer
regions of the MYC gene in various cancer cell lines with abnormal
MYC expression (14,16,53).
In addition, BRD4 inhibitors inhibit tumorigenesis by reducing the
expression of several key proto-oncogenes and their downstream
target genes, such as MYC, BCL2 and E2F1 (17). In the present study, the results of
the ChIP-seq binding site analysis suggested that BRD4 binding
sites shared by multiple cell lines were co-localized with the key
proto-oncogenes MYC and E2F1 in partially activated promoter
regions. Gene expression profiling also revealed that genes
downregulated by BRD4 inhibitors in multiple cell lines were
significantly enriched in the regulatory sets of MYC and/or E2F1.
These results indicated that BRD4 synergizes with MYC and E2F1 to
activate its downstream target genes and promote cancer in a
variety of cell lines, partially revealing the general mechanism of
BRD4 inhibitors in various cell lines. However, overexpression of
MYC or BCL2 in cancer cells treated with JQ1 only partially
attenuated the anti-cancer effect of JQ1. This potentially suggests
the diversity of the target and the complexity of the mechanisms of
BRD4 inhibitors, which function not only through a key TF (such as
MYC or E2F1), but also through a synergistic regulatory network
that induces multiple TFs to exert an anti-cancer effect.
BRD4 is asymmetrically distributed in the genome and
is called the super-enhancer region in a small portion (<5% of
the BRD4 binding sites) of regions that are enriched by >50% of
the BRD4 binding signal. The super-enhancer is localized in the
transcription-activated enhancer region, which is distributed in
the transcriptional co-activator MED1 and cell line key TF binding
sites, and its associated genes are involved in cell identity and
cell-specific functions (26). In
the present study, it was revealed that the binding distribution of
BRD4 and MED1 in the whole genome was consistent, and that BRD4 may
be used to define the super-enhancer regions. In addition, a
consecutive distribution of ultra-high BRD4 binding signals was
observed around the promoter regions of numerous key genes. To
completely scan the BRD4 super-loaded region, all enhancer regions
and the promoter regions containing multiple BRD4 binding peaks
were included. BRD4 super-loaded regions were obtained by scanning
BRD4 signals. The present results indicated that the majority of
the BRD4 cell line-common super-loaded regions contained gene
promoter regions, and that the corresponding gene was actively
expressed. These results suggested that BRD4 was super-enriched not
only in a small part of the enhancer region, but also in the
promoter region, and that the associated genes were implicated in
fundamental cellular functions. Conversely, super-enhancer regions
were mostly cell-specific binding regions, and their associated
genes were involved in specific cell functions and cell identity
decisions.
Previous studies have reported that BRD4 inhibitors
have significant anti-cancer effects in inhibiting proliferation
and promoting apoptosis in various cancer cell lines, particularly
in hematological cancers (51).
Furthermore, JQ1 has exhibited a broad and potent inhibitory effect
in various human AML cell lines and patient samples. Subsequent
mechanistic studies have suggested that BRD4 inhibitors
significantly inhibit super-enhancer-associated genes (23). Therefore, the present study focused
on the genes associated with BRD4 super-enrichment regions, which
markedly narrowed the scope for subsequent research and refined its
focus. Through multi-level screening, a core regulatory gene set
around the BRD4 cell line-common super-loaded regions was obtained.
Most of these genes were TFs closely linked to cancer, including
the key proto-oncogene TFs MYC, BCL2L1 and TCF4. These findings
suggest that the gene regulatory network of various types of cancer
may contain a core gene group that is responsible for controlling
fundamental cellular functions, including transcription,
proliferation, apoptosis and the cell cycle. Finally, BRD4
inhibitors may exert their potent effect on various types of cancer
by inhibiting this core gene set.
In conclusion, the present study found that BRD4
binds to enhancers with H3K27Ac and mediator in a cell-type
specific manner and binds to the promoter region with oncogenic
transcription factors MYC or E2F1 in a group of cell-type common
binding sites. The genes with high BRD4 binding signal in cell-type
specific super-enhancers and cell-type common super-enhancers were
both sensitive to JQ1 treatment. The cell-type common ones across
six cell types are functionally important for tumorigenesis, and
the cell-type specific ones are functional enriched with cell
identity. The present study obtained a core set of JQ1 regulated
genes with cell-type common BRD4-super-loaded, which were
significantly inhibited by JQ1 in many other cancer cell lines, and
contributed to the cancer hallmarks. These results imply a common
mechanism underlying therapeutic effects of JQ1, and provide a
potential candidate for BRD4-mediated transcription regulation and
BET-inhibitors-related cancer therapy.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was funded by the Zhejiang
Provincial Department of Health Project (grant no. 2018KY515) and
the Key Research Project of Traditional Chinese Medicine of
Zhejiang Province of China (grant no. 2019ZZ015).
Availability of data and materials
The data of the present study are included in the
manuscript and supplementary materials. The raw gene expression
data are available from GEO (http://www.ncbi.nlm.nih.gov/geo/; accession nos. are
provided in the supplementary materials), and the raw ChIP-Seq data
are available on SRA (https://www.ncbi.nlm.nih.gov/sra/; accession nos. are
provided in the supplementary materials).
Authors' contributions
The study was conceived and designed by GJ, CW and
WD. WD, GJ and YL performed the experiments and analyzed the data,
including sequence alignment. GJ and YL contributed the materials
and analytical tools, as well as wrote and critically revised the
content of the manuscript. CW also provided funding and guidance.
All of the authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Dhalluin C, Carlson JE, Zeng L, He C,
Aggarwal AK and Zhou MM: Structure and ligand of a histone
acetyltransferase bromodomain. Nature. 399:491–496. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Belkina AC and Denis GV: BET domain
co-regulators in obesity, inflammation and cancer. Nat Rev Cancer.
12:465–477. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dey A, Chitsaz F, Abbasi A, Misteli T and
Ozato K: The double bromodomain protein Brd4 binds to acetylated
chromatin during interphase and mitosis. Proc Natl Acad Sci USA.
100:8758–8763. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jang MK, Mochizuki K, Zhou M, Jeong HS,
Brady JN and Ozato K: The bromodomain protein Brd4 is a positive
regulatory component of P-TEFb and stimulates RNA polymerase
II-dependent transcription. Mol Cell. 19:523–534. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jiang YW, Veschambre P, Erdjument-Bromage
H, Tempst P, Conaway JW, Conaway RC and Kornberg RD: Mammalian
mediator of transcriptional regulation and its possible role as an
end-point of signal transduction pathways. Proc Natl Acad Sci USA.
95:8538–8543. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Donner AJ, Ebmeier CC, Taatjes DJ and
Espinosa JM: CDK8 is a positive regulator of transcriptional
elongation within the serum response network. Nat Struct Mol Biol.
17:194–201. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Patel MC, Debrosse M, Smith M, Dey A,
Huynh W, Sarai N, Heightman TD, Tamura T and Ozato K: BRD4
coordinates recruitment of pause release Factor P-TEFb and the
pausing complex NELF/DSIF to regulate transcription elongation of
interferon-stimulated genes. Mol Cell Biol. 33:2497–2507. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu W, Ma Q, Wong K, Li W, Ohgi K, Zhang
J, Aggarwal A and Rosenfeld MG: Brd4 and JMJD6-associated
anti-pause enhancers in regulation of transcriptional pause
release. Cell. 155:1581–1595. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rahman S, Sowa ME, Ottinger M, Smith JA,
Shi Y, Harper JW and Howley PM: The Brd4 extraterminal domain
confers transcription activation independent of pTEFb by recruiting
multiple proteins, including NSD3. Mol Cell Biol. 31:2641–2652.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang Z, Yik JH, Chen R, He N, Jang MK,
Ozato K and Zhou Q: Recruitment of P-TEFb for stimulation of
transcriptional elongation by the bromodomain protein Brd4. Mol
Cell. 19:535–545. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Picaud S: Selective inhibition of BET
bromodomains. Nature. 468:1067–1073. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nicodeme E, Jeffrey KL, Schaefer U, Beinke
S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H,
et al: Suppression of inflammation by a synthetic histone mimic.
Nature. 468:1119–1123. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dawson MA, Prinjha RK, Dittmann A,
Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C,
Savitski MM, et al: Inhibition of BET recruitment to chromatin as
an effective treatment for MLL-fusion leukaemia. Nature.
478:529–533. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zuber J, Shi J, Wang E, Rappaport AR,
Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al:
RNAi screen identifies Brd4 as a therapeutic target in acute
myeloid leukaemia. Nature. 478:524–528. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Delmore JE, Issa GC, Lemieux ME, Rahl PB,
Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et
al: BET bromodomain inhibition as a therapeutic strategy to target
c-Myc. Cell. 146:904–917. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mertz JA, Conery AR, Bryant BM, Sandy P,
Balasubramanian S, Mele DA, Bergeron L and Sims RJ III: Targeting
MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl
Acad Sci USA. 108:16669–16674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chapuy B, McKeown MR, Lin CY, Monti S,
Roemer MG, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, et al:
Discovery and characterization of super-enhancer-associated
dependencies in diffuse large B cell lymphoma. Cancer Cell.
24:777–790. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Knoechel B, Roderick JE, Williamson KE,
Zhu J, Lohr JG, Cotton MJ, Gillespie SM, Fernandez D, Ku M, Wang H,
et al: An epigenetic mechanism of resistance to targeted therapy in
T cell acute lymphoblastic leukemia. Nat Genet. 46:364–370. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Puissant A, Frumm SM, Alexe G, Bassil CF,
Qi J, Chanthery YH, Nekritz EA, Zeid R, Gustafson WC, Greninger P,
et al: Targeting MYCN in neuroblastoma by BET bromodomain
inhibition. Cancer Discov. 3:308–323. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bandopadhayay P, Bergthold G, Nguyen B,
Schubert S, Gholamin S, Tang Y, Bolin S, Schumacher SE, Zeid R,
Masoud S, et al: BET bromodomain inhibition of MYC-amplified
medulloblastoma. Clin Cancer Res. 20:912–925. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lockwood WW, Zejnullahu K, Bradner JE and
Varmus H: Sensitivity of human lung adenocarcinoma cell lines to
targeted inhibition of BET epigenetic signaling proteins. Proc Natl
Acad Sci USA. 109:19408–19413. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Asangani IA, Dommeti VL, Wang X, Malik R,
Cieslik M, Yang R, Escara-Wilke J, Wilder-Romans K, Dhanireddy S,
Engelke C, et al: Therapeutic targeting of BET bromodomain proteins
in castration-resistant prostate cancer. Nature. 510:278–282. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lovén J, Hoke HA, Lin CY, Lau A, Orlando
DA, Vakoc CR, Bradner JE, Lee TI and Young RA: Selective inhibition
of tumor oncogenes by disruption of super-enhancers. Cell.
153:320–334. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Whyte WA, Orlando DA, Hnisz D, Abraham BJ,
Lin CY, Kagey MH, Rahl PB, Lee TI and Young RA: Master
transcription factors and mediator establish super-enhancers at key
cell identity genes. Cell. 153:307–319. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Di Micco R, Fontanals-Cirera B, Low V,
Ntziachristos P, Yuen SK, Lovell CD, Dolgalev I, Yonekubo Y, Zhang
G, Rusinova E, et al: Control of embryonic stem cell identity by
BRD4-dependent transcriptional elongation of
super-enhancer-associated pluripotency genes. Cell Rep. 9:234–247.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hnisz D, Abraham BJ, Lee TI, Lau A,
Saint-André V, Sigova AA, Hoke HA and Young RA: Super-enhancers in
the control of cell identity and disease. Cell. 155:934–947. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu SY, Lee AY, Lai HT, Zhang H and Chiang
CM: Phospho switch triggers Brd4 chromatin binding and activator
recruitment for gene-specific targeting. Mol Cell. 49:843–857.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Brown JD, Lin CY, Duan Q, Griffin G,
Federation A, Paranal RM, Bair S, Newton G, Lichtman A, Kung A, et
al: NF-κB directs dynamic super enhancer formation in inflammation
and atherogenesis. Mol Cell. 56:219–231. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bolger AM, Lohse M and Usadel B:
Trimmomatic: A flexible trimmer for Illumina sequence data.
Bioinformatics. 30:2114–2120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Langmead B, Trapnell C, Pop M and Salzberg
SL: Ultrafast and memory-efficient alignment of short DNA sequences
to the human genome. Genome Biol. 10:R252009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Thorvaldsdottir H, Robinson JT and Mesirov
JP: Integrative Genomics Viewer (IGV): High-performance genomics
data visualization and exploration. Brief Bioinform. 14:178–192.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang Y, Liu T, Meyer CA, Eeckhoute J,
Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W and
Liu XS: Model-based analysis of ChIP-Seq (MACS). Genome Biol.
9:R372008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Heinz S, Benner C, Spann N, Bertolino E,
Lin YC, Laslo P, Cheng JX, Murre C, Singh H and Glass CK: Simple
combinations of lineage-determining transcription factors
prime-regulatory elements required for macrophage and B cell
identities. Mol Cell. 38:576–589. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: affy-analysis of Affymetrix GeneChip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Irizarry RA, Bolstad BM, Collin F, Cope
LM, Hobbs B and Speed TP: Summaries of Affymetrix GeneChip probe
level data. Nucleic Acids Res. 31:e152003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Irizarry RA, Hobbs B, Collin F,
Beazer-Barclay YD, Antonellis KJ, Scherf U and Speed TP:
Exploration, normalization, and summaries of high density
oligonucleotide array probe level data. Biostatistics. 4:249–264.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gentleman RC, Carey VJ, Bates DM, Bolstad
B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al:
Bioconductor: Open software development for computational biology
and bioinformatics. Genome Biol. 5:R802004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Barski A, Cuddapah S, Cui K, Roh TY,
Schones DE, Wang Z, Wei G, Chepelev I and Zhao K: High-resolution
profiling of histone methylations in the human genome. Cell.
129:823–837. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Rahl PB and Young RA: MYC and
transcription elongation. Cold Spring Harb Perspect Med.
4:a0209902014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhou VW, Goren A and Bernstein BE:
Charting histone modifications and the functional organization of
mammalian genomes. Nat Rev Genet. 12:7–18. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Claudio JO, Masih-Khan E, Tang H,
Gonçalves J, Voralia M, Li ZH, Nadeem V, Cukerman E,
Francisco-Pabalan O, Liew CC, et al: A molecular compendium of
genes expressed in multiple myeloma. Blood. 100:2175–2186. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Whitfield TW, Wang J, Collins PJ,
Partridge EC, Aldred SF, Trinklein ND, Myers RM and Weng Z:
Functional analysis of transcription factor binding sites in human
promoters. Genome Biol. 13:R502012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jelinek DF, Aagaard-Tillery KM, Arendt BK,
Arora T, Tschumper RC and Westendorf JJ: Differential human
multiple myeloma cell line responsiveness to interferon-alpha.
Analysis of transcription factor activation and interleukin 6
receptor expression. J Clin Invest. 99:447–456. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Otsuki T, Yamada O, Sakaguchi H, Tomokuni
A, Wada H, Yawata Y and Ueki A: Human myeloma cell apoptosis
induced by interferon-alpha. Br J Haematol. 103:518–529. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mccoull W, Cheung T, Anderson E, Barton P,
Burgess J, Byth K, Cao Q, Castaldi MP, Chen H, Chiarparin E, et al:
Development of a novel B-cell lymphoma 6 (BCL6) PROTAC to provide
insight into small molecule targeting of BCL6. ACS Chem Biol.
13:3131–3141. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Rodriguez EF, Chowsilpa S and Maleki Z:
insulinoma-associated protein 1 immunostain: A diagnostic tool for
pulmonary small cell carcinoma in cytology. Acta Cytol. 62:333–338.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen C, Breslin MB and Lan MS: Sonic
hedgehog signaling pathway promotes INSM1 transcription factor in
neuroendocrine lung cancer. Cell Signal. 46:83–91. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gabay M, Li Y and Felsher DW: MYC
activation is a hallmark of cancer initiation and maintenance. Cold
Spring Harb Perspect Med. 4(pii): a0142412014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Vogler M: BCL2A1: The underdog in the BCL2
family. Cell Death Differ. 19:67–74. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Shi J and Vakoc CR: The mechanisms behind
the therapeutic activity of BET bromodomain inhibition. Mol Cell.
54:728–736. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Anand P, Brown JD, Lin CY, Qi J, Zhang R,
Artero PC, Alaiti MA, Bullard J, Alazem K, Margulies KB, et al: BET
bromodomains mediate transcriptional pause release in heart
failure. Cell. 154:569–582. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lamoureux F, Baud'huin M, Rodriguez
Calleja L, Jacques C, Berreur M, Rédini F, Lecanda F, Bradner JE,
Heymann D and Ory B: Selective inhibition of BET bromodomain
epigenetic signalling interferes with the bone-associated tumour
vicious cycle. Nat Commun. 5:35112014. View Article : Google Scholar : PubMed/NCBI
|