Introduction
Osteoarthritis (OA) is a degenerative condition of
the joints that significantly affects the physical and mental
health of middle-aged and elderly people (1). There are >360 million people
suffering from OA all over the world, and the treatment of OA is an
economic burden on society (2).
The main pathological feature is the degeneration of articular
cartilage, which clinically manifests as frequently recurring joint
pain, joint dysfunction and other symptoms, such as joint swelling,
stiffness and deformity (3). The
main function of articular cartilage is to maintain the homeostasis
between extracellular matrix (ECM) synthesis and catabolism, and
reduce joint friction and impact (4). Chondrocytes synthesize and secrete
components of the ECM infrastructure, in addition to various
matrix-degrading enzymes, proteoglycan-degrading enzymes and other
hydrolytic enzymes; this facilitates the degradation and digestion
of denatured and dysfunctional ECM proteins, which alters the
structure of the ECM (5).
Therefore, apoptosis or abnormal physiological functioning of
chondrocytes markedly changes the dynamic balance of ECM
metabolism, and the integrity of the articular cartilage structure
and function; this is a common cause of OA (6).
Nuclear factor-erythroid 2-related factor 1 (Nrf1)
acts as a transcription factor belonging to the cap‘n’collar (CNC)
basic-region leucine zipper (bZIP) family, which serves as a
crucial integrator of nuclear and mitochondrial interactions,
modulating essential processes ranging from protein production to
mitochondrial biogenesis (7–9). It
also serves a prominent role in apoptosis (10,11),
suggesting that NRF1 may be an important target for mediating
chondrocytes apoptosis.
MicroRNAs (miRNAs) are non-coding RNAs of 19–25
nucleotides in length that downregulate the expression of specific
target genes through interacting with motifs found primarily in
their 3′-untranslatedregions (UTRs) (12,13).
miRNAs participate in the regulation of various physiological
activities, including cellular proliferation, development,
differentiation and apoptosis (14,15).
Previous studies have demonstrated that miRNAs regulate the
occurrence and development of OA through various mechanisms. miRNA
(miR)-26a was reported to suppress the activation of the NF-κB
signaling pathway to alleviate synovial inflammation and cartilage
injury in OA rats (16).
miR-10a-5p promoted chondrocyte apoptosis in OA by targeting
homeobox A1 (17). miR-93 targets
Toll-like receptor 4/NF-κB signaling to inhibit OA-associated
inflammation and chondrocyte apoptosis (18). TargetScan database analysis
predicted the existence of binding sites between NRF1 3′UTR and
miR-363-3p (19); therefore, the
present study investigated miR-363-3p. In addition, miR-363-3p is
involved in apoptosis regulation (20). However, the biological role of
miR-363-3p in chondrocyte apoptosis, and the relationship between
miR-363-3p and NRF1 have not been described. The present study
aimed to investigate whether miR-363-3p could target NRF1-regulated
chondrocyte apoptosis in OA model rats in vivo, and in
lipopolysaccharide (LPS)-treated chondrocytes in vitro.
Materials and methods
Animal studies
The Animal Care and Use Committee of Shandong
University (Shandong, China) approved all animal studies,
TargetScan database (release 7.2) (19) identified a putative miR-363-3p
target site within the NRF1-3′UTR which were conducted in a manner
consistent with the National Institutes of Health Guide for the
Care and Use of Laboratory Animals. A total of 36 male Wistar rats
(age, 6 weeks; weight, 180–200 g) were obtained from the Shandong
Center for Disease Control and housed at 24±2°C in 50±5% humidity,
under a 12-h light/dark cycle, with free access to food and water.
The rats were randomized into six groups: i) Untreated control
group (n=6); ii) untreated OA group (n=6); iii) OA +
agomir/antagomir miR-363-3p negative control (NC) group (n=6/6);
iv) OA + agomir/antagomir miR-363-3p group (n=6/6). The rats were
anaesthetized with 2% sodium phenobarbital (50 mg/kg; i.p.) and the
OA model was established by medial meniscectomy tear (MMT) surgery;
rats in the untreated control group were subjected to control
surgery. Then, 1 week after MMT surgery, rats in the OA +
agomir/antagomir miR-363-3p and the OA + agomir/antagomir
miR-363-3p NC groups were treated with intra-articular injections
of 5 nmol agomir/antagomir miR-363-3p or agomir/antagomir-NC
(Suzhou GenePharma Co., Ltd.), delivered using a medial
parapatellar approach. The rats which were subjected to MMT and
treated with agomir-NC as the negative control. Rats in control
group and OA group were normally bred, without any intervention.
Following 2 weeks, all rats were sacrificed for the articular
cartilages of the medial tibial plateau and the synovial fluid by
intraperitoneal injection with pentobarbital sodium overdose (150
mg/kg), which were collected and stored at −80°C for subsequent
analysis. The tissues were subsequently dehydrated in graded
concentrations of ethanol (50, 75, 85, 95 and 100%), cleared in
xylene, embedded in paraffin and sectioned into 4 µm tissue
sections.
Primary rat articular chondrocytes
isolation and culture
Intact tibial plateau cartilage tissue was cut into
2–3 mm slices with a scalpel blade, and then 20 slices were
digested in 0.25% trypsin (R&D Systems, Inc.) at 37°C for 30
min. Segments were washed twice using 1X PBS, followed by further
digestion using 3 mg/ml collagenase D (Thermo Fisher Scientific,
Inc.) in DMEM (Invitrogen; Thermo Fisher Scientific, Inc.) for 12 h
at 37°C and cartilage tissue were completely digested and
chondrocytes were clearly visible. The digested tissue was filtered
through a sterile 150 µM nylon mesh. At this point, collagen was
isolated and underwent centrifugation at 600 × g for 5 min at 4°C.
Subsequently, the pellet was suspended in DMEM and the undigested
cartilage was removed by 70 mm nylon mesh. Then, the chondrocytes
were cultured in a humidified atmosphere of 37°C and 5%
CO2 in 5 ml DMEM (Invitrogen; Thermo Fisher Scientific,
Inc.) supplemented with 10% FBS (Gibco; Thermo Fisher Scientific,
Inc.) and 1% penicillin-streptomycin. LPS-stimulation of
chondrocytes is an established cellular model for the study of OA
(21,22). In the present study, when the cells
reached 2×105/well, chondrocyte apoptosis was induced by
treatment with LPS (Sigma-Aldrich; Merck KGaA) at a final
concentration 5 µg/ml for 6 h (18).
Reverse transcription-quantitative PCR
(RT-qPCR)
TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) was used to isolate total RNA from
chondrocytes (5×106) and articular cartilage RNA (50
mg), according to the manufacturer's protocol. Total RNA was
quantified and subsequently reverse transcribed into cDNA using the
Revert Aid First Strand cDNA Synthesis kit (cat. no. K1622; Thermo
Fisher Scientific, Inc.), according to the manufacturer's protocol.
The thermocycling conditions of qPCR were 25°C for 5 min, 42°C for
60 min and 70°C for 10 min. qPCR was subsequently performed using
the Applied Biosystems SYBR®Green Master mix (Thermo
Fisher Scientific, Inc.) and an ABI StepOnePlus RT PCR platform
(Thermo Fisher Scientific, Inc.) was used following the
manufacturer's protocol. The following primers were used for the
qPCR: miR-363-3p, forward 5′-GGATGCGGATGGGCGAGAGC-3′, reverse
5′-TTAGCGGATGCGGAAAATC-3′; NRF1, forward
5′-TTACTCTGCTGTGGCTGATGG-3′, reverse 5′-CCTCTGATGCTTGCGTGGTCT-3′;
Bcl-2, forward 5′-GGGACGCGAAGTGCTATTGGT-3′, reverse
5′-CTCAGGCTGGAAGGAGAAGAT-3′; Bax, forward
5′-GGTTGCCCTCTTGTACTTTGC-3′, reverse 5′-TCTTCCAGATGGTGAGCGAG-3′;
p53, forward 5′-TTGCCGTCCCAAGCAATGGATGA-3′, reverse
5′-TCTGGGAAGGGACAGAAGATGAC-3′; cleaved caspase-3, forward
5′-ATGGAGAACAACAAAACCTCAGT-3′, reverse
5′-TTGCTCCCATGTATGGTCTTTAC-3′; β-actin, forward
5′-CCCGCCGCCAGCTCACCATGG-3′, reverse 5′-AAGGTCTCAAACATGATCTGGGTC-3′
(Invitrogen; Thermo Fisher Scientific, Inc.). Relative miR-363-3p
expression was normalized to U6, forward
5′-CGCTTCGGCAGCACATATACTA-3′ and reverse
5′-CGCTTCACGAATTTGCGTGTCA-3′. mRNA expressions were normalized to
the internal reference gene, β-actin; the expression levels were
quantified using the 2−ΔΔCq method (23).
Cell transfection
NRF1 siRNAs and control siRNA were purchased from
Invitrogen (Thermo Fisher Scientific, Inc.), and miR-363-3p agomir,
agomir NC, miR-363 antagomir and antagomir NC were purchased from
Suzhou GenePharma Co., Ltd. Once the chondrocytes reached 50%
confluency, agomir and antagomir (100 nM in vitro, 40
nmol/200 µl in vivo), and siRNA (100 nM) were transfected
with these constructs using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. Following 48 h of transfection at 37°C,
cells were harvested for further experimentation. siRNA
oligonucleotide sequences were as follows: NRF1 siRNA (si-NRF1)
forward, 5′-UUAAGCGCCAUAGUGACUG-3′; and control siRNA (si-NC):
forward 5′-UAUUUGGAUGUACCUGUGGACUUGG-3′.
Cell viability assay
Chondrocyte viability was detected using the MTT
assay. A total of 2×105 cells/well were seeded and
transfected for 48 h of transfection. Following this incubation, 20
µl MTT (Sigma-Aldrich, Merck KGaA) was added to each well for 6 h.
The purple formazan was dissolved in 200 µl DMSO and the viability
was analyzed using a Multiskan Spectrum microplate reader (Thermo
Fisher Scientific, Inc.) at a wavelength of 450 nm.
Flow cytometric analysis of
apoptosis
Following 48 h of transfection, chondrocytes
(1×106) were collected after centrifugation at 500 × g
for 20 min at room temperature, to assess the apoptotic status.
After washing the cells using 1X PBS, the chondrocytes were stained
with Annexin V at 4°C for 15 mins, followed by an additional 5 min
stain with propidium iodide from the Annexin V-FITC Apoptosis
Detection kit (BD Biosciences), according to the manufacturer's
protocol. Apoptotic cells were subsequently analyzed using flow
cytometry, which was performed using the BD FACS Calibur Flow
Cytometry Machine (BD Biosciences), and the results were analyzed
using the FlowJo.7.6.1 software (BD Biosciences).
ELISA
Tumor necrosis factor (TNF)-α, interleukin (IL)-1β
and IL-6 expression levels in synovial fluid (100 µl/well), which
was extracted by joint puncture, were analyzed using ELISA kits
(TNF-α, cat. no. RTA00; IL-1β, cat. no. RLB00; IL-6, cat. no.
R6000B; R&D Systems, Inc.), according to the manufacturer's
protocols.
Immunohistochemistry (IHC)
NRF1, Bax, Bcl-2 and caspase-3 levels were measured
by IHC. Briefly, tissues were fixed in 4% paraformaldehyde and
subsequently paraffin embedded and cut into 4-µm-thick sections.
After deparaffinized, the tissue sections were hydrated in graded
ethanol, and then blocked with 3% hydrogen peroxide for 20 min at
room temperature. Subsequently, sections were treated with 10%
normal goat serum (cat. no. 16210072; Gibco; Thermo Fisher
Scientific, Inc.) for 30 min at 37°C. After that, the sections were
incubated overnight at 4°C with the following primary antibodies
(Abcam): Rabbit monoclonal anti-NRF1 (1:100; cat. no. ab175932),
rabbit polyclonal anti-Bax (1:200; cat. no. ab53154), rabbit
polyclonal anti-Bcl-2 (1:200; cat. no. ab196495), rabbit monoclonal
anti-cleaved caspase 3 (1:200; cat. no. ab2302) and anti-β-actin
(1:500; cat. no. ab8224). Following primary antibody incubation,
sections were washed three times using 1X PBS, and incubated with
horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG
secondary antibodies (1:400; cat. no. G-21234; Thermo Fisher
Scientific, Inc.) for 2 h at room temperature. The slides were
subsequently stained with 1 mg/ml 3,3′-diaminobenzidine and
hematoxylin counterstain for 10 min at room temperature. A light
microscope (Olympus; model BX51) was used to capture the images
(magnification, ×400).
Immunofluorescence staining
Chondrocytes were fixed with 4% paraformaldehyde for
30 min at 4°C and subsequently permeabilized with 0.1% Triton X-100
for 10 min at 4°C. Following blocking in 10% donkey serum
(Sigma-Aldrich; Merck KGaA) for 20 min at room temperature, the
cells were incubated with primary antibodies (Abcam) targeting NRF1
(1:200; cat. no. ab175932), p53 (1:200; cat. no. ab131442), Bcl-2
(1:200; cat. no. ab196495) and cleaved caspase-3 (1:150; cat. no.
ab2302) overnight at 4°C. The samples were subsequently incubated
with goat anti-rabbit IgG (H+L) highly cross-adsorbed secondary
antibodies (1:400; cat. no. A32731; Invitrogen; Thermo Fisher
Scientific, Inc.) at room temperature for 30 min. Nuclear staining
was achieved using DAPI [Roche Diagnostics (Shanghai) Co., Ltd.]. A
fluorescent microscope (Leica) was used to capture the images
(magnification, ×200 or ×400).
TUNEL assay
Articular cartilage tissue sections were fixed in 4%
paraformaldehyde for 24 h at room temperature and embedded in
paraffin. Then, 4 µm-thick paraffin sections were deparaffinized
using 100% xylene, rehydrated for 5 min twice at room temperature
and washed with H2O. This was followed by rehydrated
with ethanol at graded concentrations (100% 5 min, 100% 3 min, 95%
3 min, 85% 3 min, 70% 3 min, 50% 3 min) at room temperature. Then,
the tissues sections were treated with 100 µl proteinase K [20
µg/ml; Roche Diagnostics (Shanghai) Co., Ltd.)] for 20 min at room
temperature, and washed 1X PBS. Subsequently, chondrocyte apoptosis
in the articular cartilage was measured using a In situ Cell
Death Detection kit (cat. no. 11684817910; Roche Diagnostics Co.,
Ltd.), according to the manufacturer's protocols. Cells with brown
nuclei were deemed TUNEL-positive and were counted by a microscope
using three fields of view/section. A light microscope (Olympus
Corporation; model BX51) was used to capture the images
(magnification, ×200).
Safranin O staining
Sections of knee cartilage tissue (4 µm) were fixed
with 4% formaldehyde at 20°C for 30 mins, paraffin embedded and
serially separated into 4 µm sections. Sections were then stained
with 0.1% Weigert's iron hematoxylin at room temperature for 5 min
and washed by water for 1 min at room temperature. The sections
were differentiated for 30 sec in 1% ethylic acid solution,
incubated in 0.2% fast green solution (Thermo Fisher Scientific,
Inc.) for 1 min at room temperature and then rinsed with distilled
water for 1 min. The sections were incubated in 0.1% safranin O
solution (Thermo Fisher Scientific, Inc.) for 2 min at room
temperature. The Safranin O solution (0.1–0.5 mg/ml; prepared in
H2O) was used as a counterstain and staining occurred at
room temperature for 5 min. A light microscope (Olympus
Corporation; model BX51) was used to capture the images
(magnification ×100). After the Safranin O staining, the degree of
articular cartilage lesions was scored by three independent
observers according to the modified Mankin scoring principle
(24). The score range was 0–14,
and the higher the score, the more severe the joint injury.
Dual-luciferase reporter assay
TargetScan database (release 7.2) (19) identified a putative miR-363-3p
target site within the NRF1-3′ untranslated region (UTR). Wild-type
(WT) and mutant (MT) versions of the NRF1 candidate miR-363-3p
target sequences were generated and cloned into the pGL3 vector
bearing a firefly luciferase reporter element (Promega
Corporation), yielding wild-type (wt-pGL3-NRF1-3′UTR) or mutant
(mut-pGL3-NRF1-3′UTR) constructs. 293T cells (American Type Culture
Collection) were plated into 24-well plates (2×105/well)
and subsequently transfected with the wt-pGL3-NRF1-3′UTR or
mut-pGL3-NRF1-3′UTR constructs using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. Following incubation for 48 h, cells were
collected and the luciferase activity was detected using a
Dual-Luciferase Reporter assay system (Promega Corporation).
Scramble miRNA was used as a negative control, and the
Renilla luciferase intensity as normal control, according to
the manufacturer's protocol.
Western blotting
Intact tibial plateau cartilage tissue or
chondrocytes (5×106) protein were extracted on ice using
a lysis buffer supplemented with protease inhibitors [Roche
Diagnostics (Shanghai) Co., Ltd.] for 50 min at 4°C and centrifuged
for 15 min at 12,000 × g. Total protein levels were quantified
using a bicinchoninic acid assay kit (Thermo Fisher Scientific,
Inc.) The protein samples were diluted in 5X sample buffer (ratio,
4:1; cat. No. MB01015; GenScript) and incubated for 5 min. A total
of 40 µg of proteins was separated by 10% SDS-PAGE gel. The PVDF
membrane was blocked with TBS containing 5% non-fat milk and 0.1%
Tween for 1 h at room temperature. Following blocking, membranes
were incubated overnight at 4°C with primary antibodies (Abcam)
against NRF1 (1:100; cat. no. ab175932), p53 (1:200; cat. no.
ab131442), cleaved caspase 3 (1:200; cat. no. ab2302) and β-actin
(1:500; cat. no. ab8224). Following the primary incubation,
membranes were incubated at room temperature for 1 h with
HRP-conjugated secondary antibodies (1:500; cat. no. A20207;
Invitrogen; Thermo Fisher Scientific, Inc.). Protein bands were
visualized and detected using Odyssey Infrared Imaging system Model
9120 (LI-COR Biotechnology) with Quantity One software (version
2.4; Bio-Rad Laboratories, Inc.). β-actin was used for
normalization.
Statistical analysis
Statistical analysis was performed using the SPSS
version 22.0 (IBM Corp.) software, and data are presented as the
mean ± SD. All experiments were performed ≥3 times. Statistical
significance between groups was determined using Student's t-test
or one-way ANOVA with Tukey's post hoc test. Simple correlations
were assessed by using the Pearson correlation. The target genes of
NRF1 were predicted with TargetScan. P<0.05 was considered to
indicate a statistically significant difference.
Results
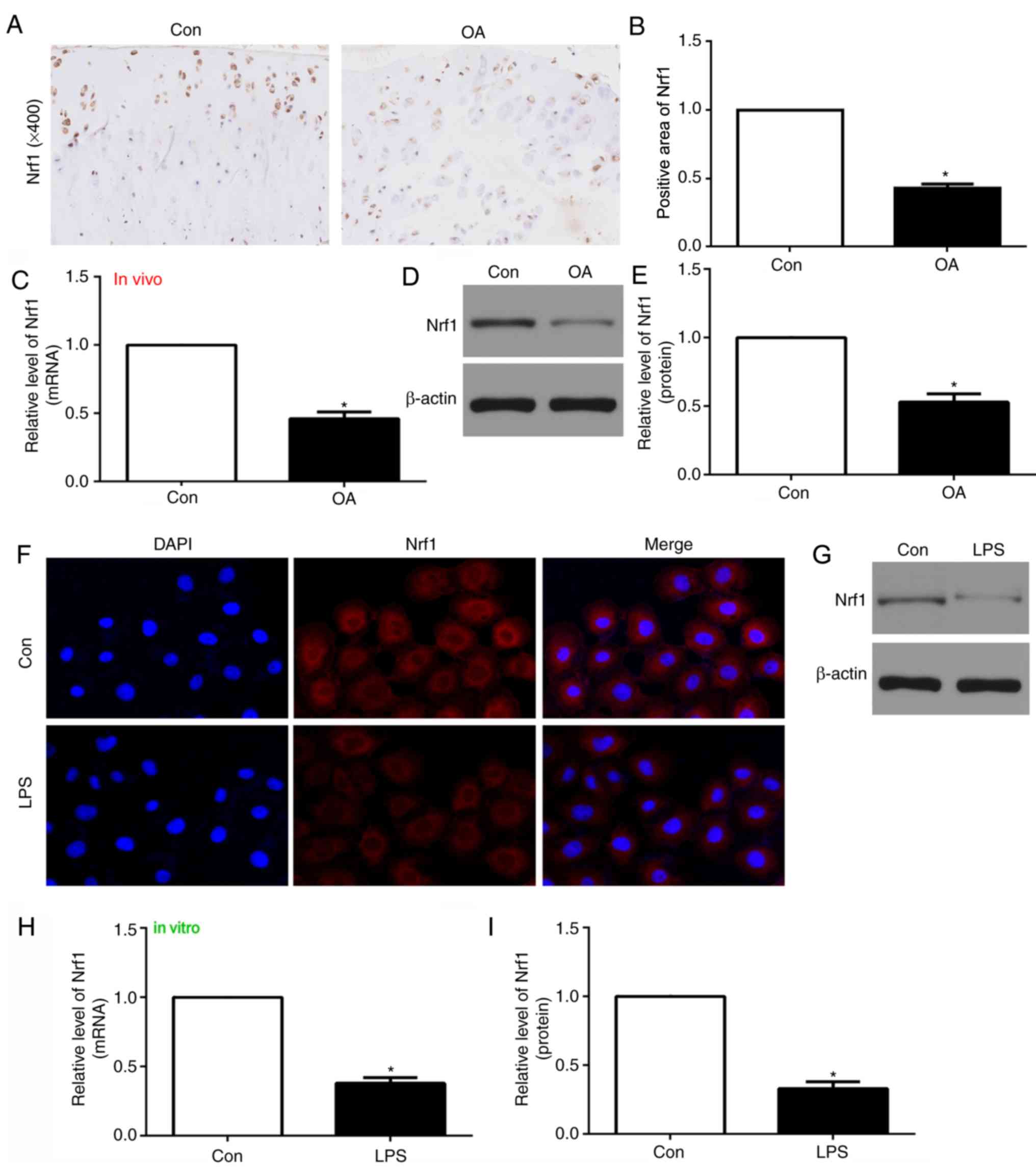
NRF1 expression is reduced in the
articular cartilage of OA rats in vivo and LPS-treated chondrocytes
in vitro
To investigate the potential involvement of NRF1 in
OA, the gene and protein expression levels were investigated in
vivo by RT-qPCR and western blotting, respectively, in addition
to IHC in vitro. The mRNA and protein expression levels of
NRF1 were revealed to be downregulated within the articular
cartilage of OA model rats compared with the control group
(Fig. 1A-E). Consistent with this,
LPS-treated chondrocytes exhibited reduced NRF1 mRNA and protein
expression compared with untreated (control) chondrocytes (Fig. 1F-I). These data demonstrated that
NRF1 may be important in OA.
Influence of NRF1 on chondrocyte
apoptosis in vitro
To investigate how NRF1 affected LPS-induced
chondrocyte injury, siRNA-NRF1 was used to knockdown NRF1 gene
expression levels in the chondrocytes. siRNA-NRF1 significantly
decreased NRF1 gene expression levels in chondrocytes relative to
the si-NC group, demonstrating an effective transfection efficiency
(Fig. 2A). In addition, NRF1
significantly decreased the mRNA and protein expression levels in
LPS-treated chondrocytes compared with the NCs (Fig. 2B-D). Flow cytometric analysis
confirmed that LPS induced apoptosis in chondrocytes and that
chondrocytes transfected with si-NRF1 further promoted LPS-induced
early apoptosis rather than late apoptosis (Fig. 2E). This indicated that NRF1 may
prevent apoptosis in vitro.
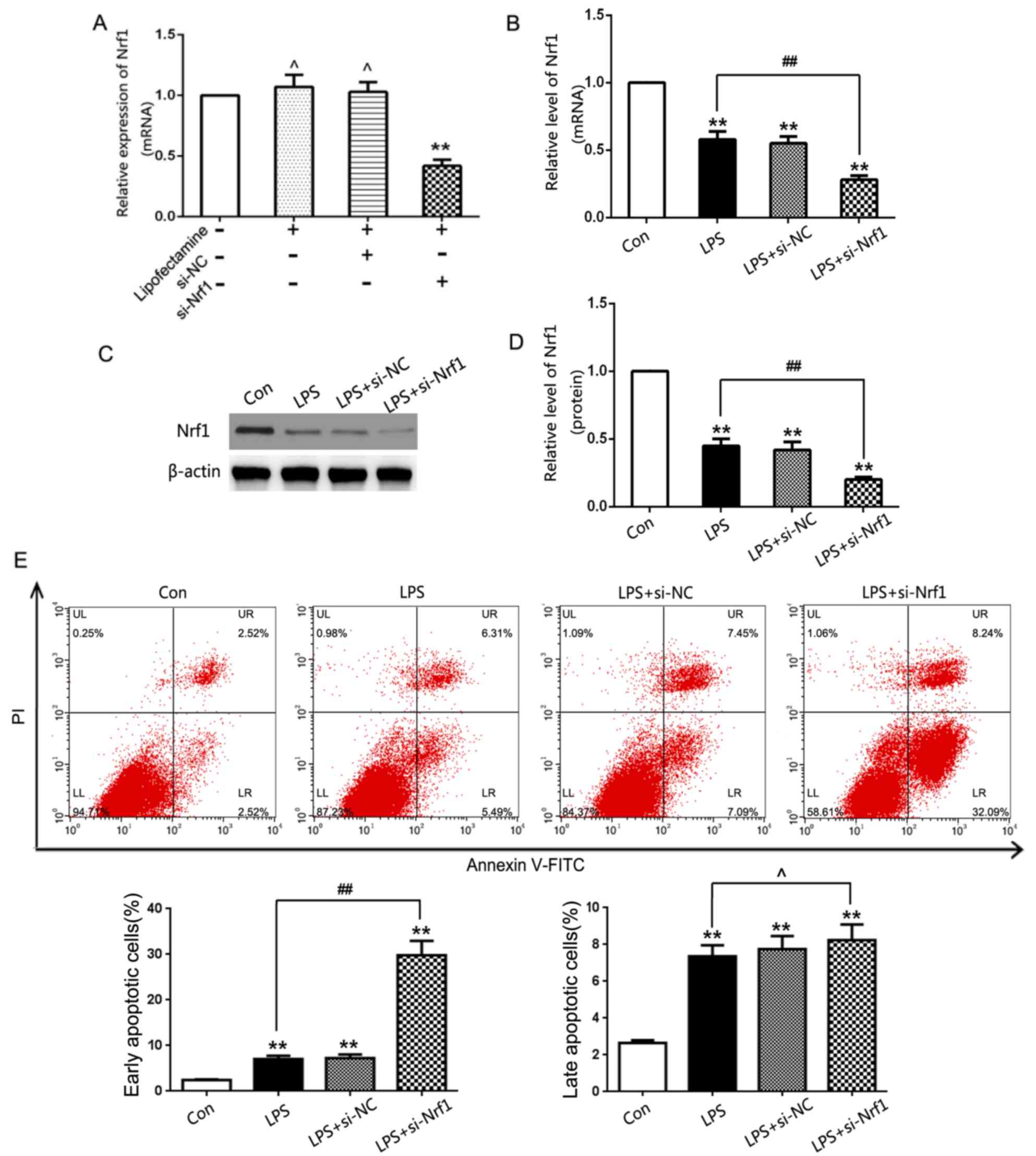 | Figure 2.Influence of NRF1 on LPS-induced
chondrocyte apoptosis in vitro. (A) Relative mRNA expression
levels of NRF1 were determined by RT-qPCR analysis in cultured
chondrocytes following the genetic knockdown of NRF1 with siRNA
(n=6/group). (B) mRNA expression levels of NRF1 were assessed in
LPS-treated chondrocytes by RT-qPCR. (C) Western blot analysis and
(D) quantification following the genetic knockdown of NRF1 with
siRNA (E) Flow cytometry analysis was performed to determine
cellular apoptosis following the genetic knockdown of NRF1 with
siRNA in LPS-treated chondrocytes. **P<0.01 vs. con,
##P<0.01, ,^P>0.05 vs. LPS group. RT-qPCR, reverse
transcription-quantitative PCR; LPS, lipopolysaccharide; OA,
osteoarthritis; NRF1, nuclear respiratory factor 1; siRNA, small
interfering RNA; NC, negative control; si/siRNA, small interfering
RNA; Con, untreated control; PI, propidium iodide. |
miR-363-3p and NRF1 expression are
negatively correlated in vivo, and miR-363-3p targets NRF1 in
vitro
miR-363-3p expression levels in vivo were
significantly increased in the OA group compared with the control
group, and in the LPS-treated chondrocytes compared with the
control chondrocytes in vitro (Fig. 3A). The correlation between
miR-363-3p and NRF1 expression was assessed using Pearson's
correlation analysis, which revealed that NRF1 mRNA expression was
negatively correlated with miR-363-3p expression in vivo
(Fig. 3B). TargetScan database
analysis identified a putative miR-363 target site within the
NRF1-3′UTR (Fig. 3C). A
dual-luciferase reporter approach was used to confirm this
predicted binding site, using luciferase reporter constructs
bearing wt-pGL3-NRF1-3′UTR or mut-pGL3-NRF1-3′UTR constructs.
miR-363-3p overexpression led to a significant decrease in
wt-pGL3-NRF1-3′UTR reporter activity, but not in
mut-pGL3-NRF1-3′UTR activity, which demonstrated similar activity
to the groups of scramble miRNA (Fig.
3D). The present results suggested that NRF1 may be a direct
target of miR-363-3p.
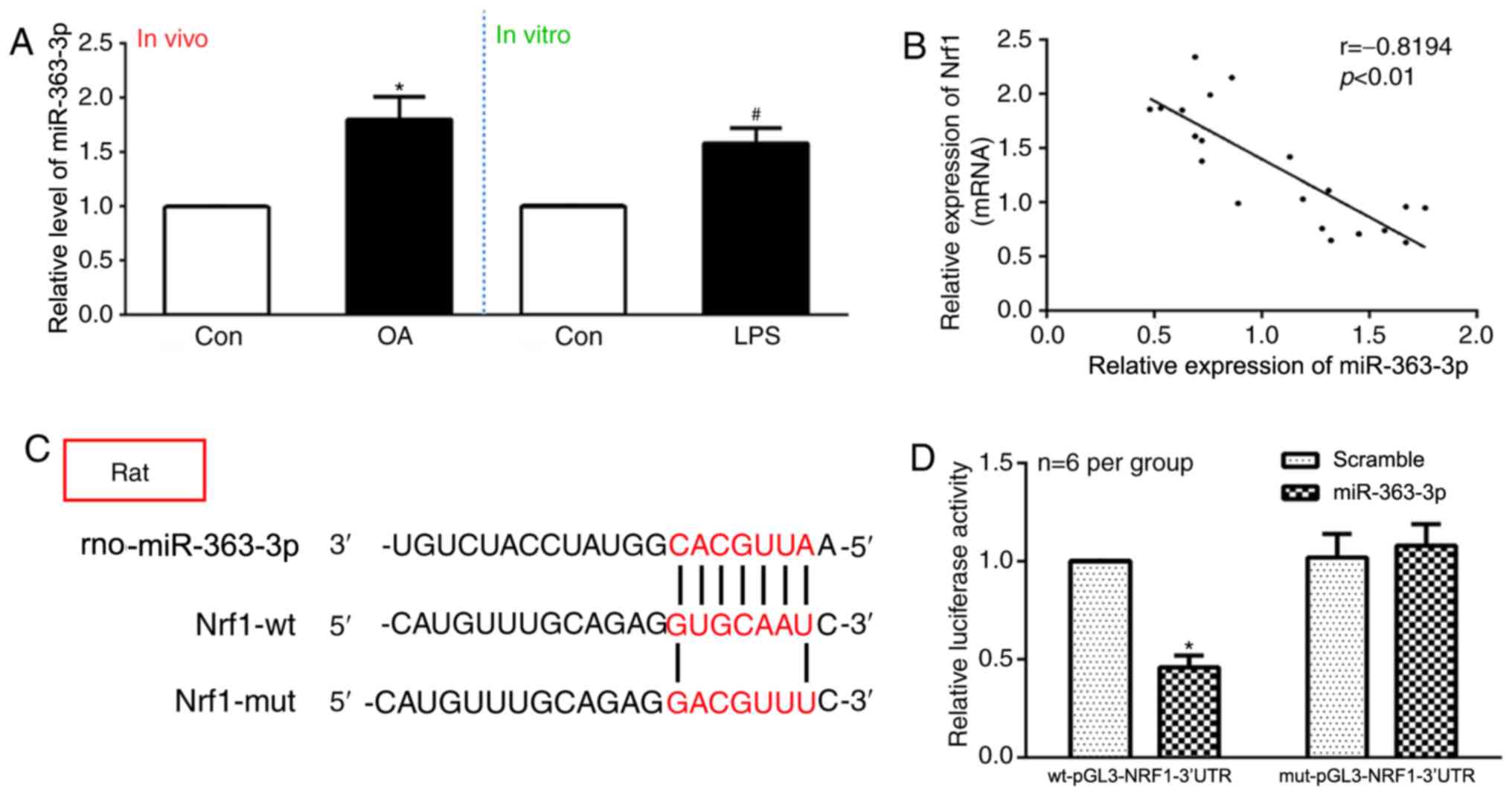 | Figure 3.NRF1 is a direct target of
miR-363-3p. (A) Expression levels of miR-363-3p were evaluated by
RT-qPCR in OA rats compared with the control group (in
vivo), and LPS-treated chondrocytes compared with control
chondrocytes (in vitro) (n=rats 6/group). (B) Pearson's
correlation analysis revealed that the mRNA expression of
miR-363-3p was inversely correlated with the expression of NRF1
(P<0.01). (C) TargetScan database predicted the existence of
binding sites between NRF13′UTR and miR-363-3p. The mutation was
generated on the NRF1 3′UTR sequence in the complementary site for
the binding region of miR-363-3p. (D) Dual-reporter luciferase
assay of miR-363-3p expression in wt-pGL3-NRF1-3′UTR or
mut-pGL3-NRF1-3′UTR constructs. *P<0.05 vs. control group, Wt,
wild-type; mut, mutant; miR, microRNA; OA, osteoarthritis; NRF1,
nuclear respiratory factor 1; UTR, untranslated region; RT-qPCR,
reverse transcription-quantitative PCR; NC, negative control; LPS,
lipopolysaccharide; Scramble, scramble-miRNA. |
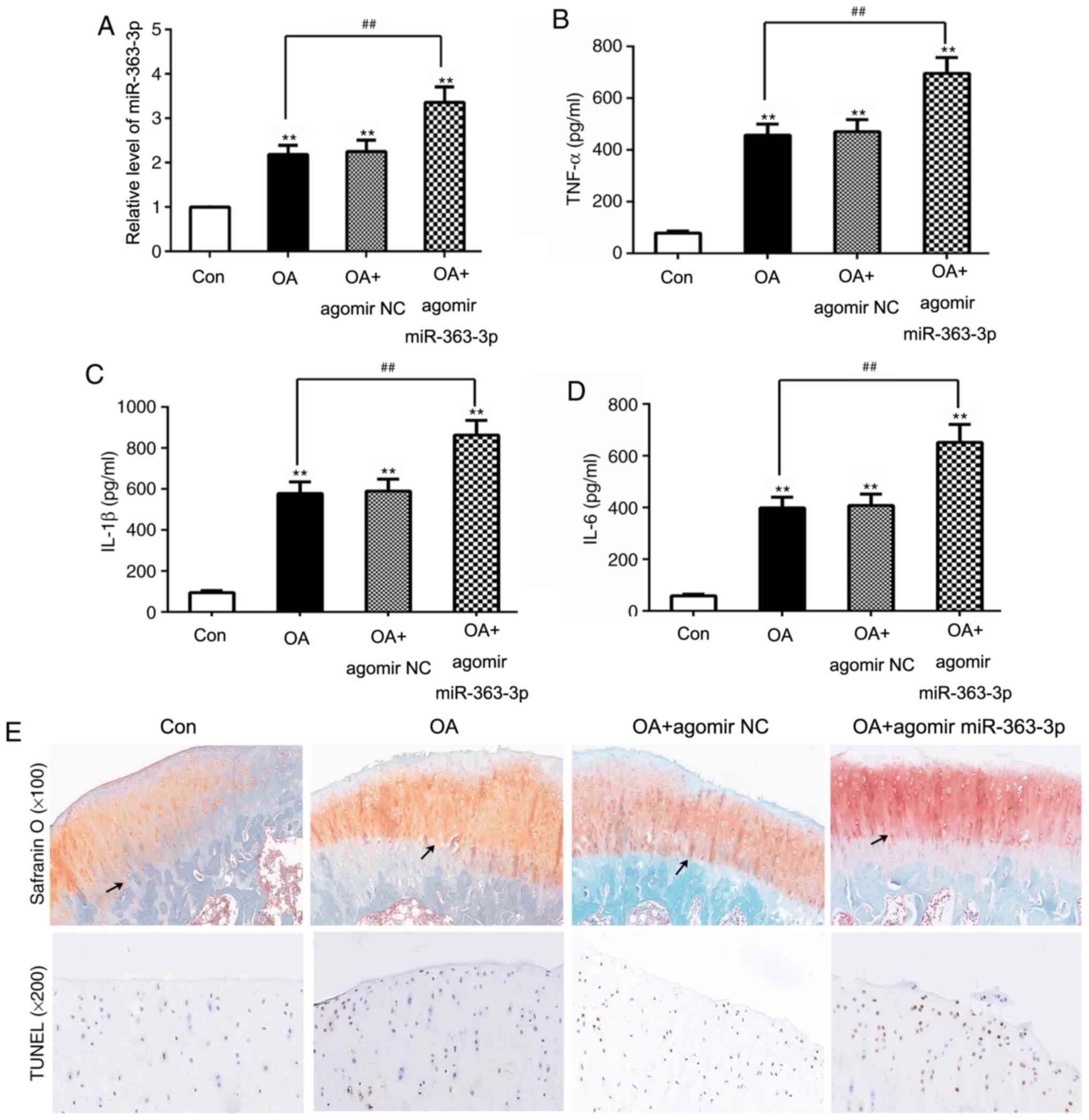
miR-363-3p promotes apoptosis in OA
model rats
To assess the functional relevance of miR-363-3p in
OA development, OA model rats were intra-articularly administered
agomir miR-363-3p. Expression of miR-363-3p in OA rats was
significantly increased compared with the control groups.
Significantly elevated miR-363-3p expression levels were
demonstrated in the cartilage of OA + agomir miR-363-3p rats
compared with the OA groups (Fig.
4A). Levels of pro-inflammatory cytokines in these rats were
determined by ELISA, which revealed that TNF-α, IL-1β and IL-6
levels in OA rats was significantly increased compared to the
control groups, and significantly increased in the agomir
miR-363-3p OA group compared with OA and control groups (Fig. 4B-D). Furthermore, the effect of
miR-363 on the articular cartilage of OA rats was analyzed by
Safranin O and TUNEL staining. The OA group treated with agomir
miR-363-3p exhibited a significantly damaged surface cartilage
layer, and the tide line was distorted and moved forward compared
with the OA group, as revealed by Safranin O staining (Fig. 4E). In addition, compared with the
OA group, agomir miR-363-3p increased the number of positive
dark-brown stained cells, which were bigger compared with those in
the OA group (Fig. 4E). Mankin's
score of articular cartilage was previously evaluated by light
microscopy, as previously described; compared with the control
group (0.33±0.52; n=6), significant pathological changes of
articular cartilage in the OA group (4.50±1.05; n=6) and OA+agomir
NC group (5.00±0.89; n=6) were observed. Agomir miR-363-3p
(8.50±1.05; n=6) significantly increased the Mankin's score of OA
articular cartilage. In addition, compared with the OA group,
agomir miR-363-3p increased the number of positive dark-brown
stained cells, which were bigger compared with those in the OA
group (Fig. 4E). Taken together,
these data suggested that miR-363-3p aggravated LPS-stimulated
apoptosis and proinflammatory cytokine production.
miR-363-3p targets NRF1 and promotes
LPS-induced chondrocyte apoptosis via the upregulation of p53 in
vitro
First of all, the present study detected target gene
expression by RT-qPCR, which showed the transfections of agomir and
antagomir miR-363-3p were effective (Fig. 5A and B). To further investigate
whether miR-363-3p regulates chondrocyte apoptosis, the
chondrocytes transfected with agomirmiR-363-3p were stimulated with
LPS, and the overexpression of miR-363-3p in the chondrocytes was
evaluated using RT-qPCR analysis (Fig.
5A). The present results suggested that the expression level of
miR-363-3p was significantly increased in the LPS group compared
with the control group. In addition, agomir miR-363-3p further
upregulated miR-363-3p expression in LPS-induced chondrocytes
(Fig. 5C). Subsequently, the MTT
assay to assess chondrocyte viability revealed that miR-363
overexpression significantly reduced chondrocyte viability compared
with the control groups (Fig. 5D).
Furthermore, miR-363-3p overexpression significantly downregulated
NRF1 mRNA and protein expression levels compared with the agomir
NC, whereas chondrocytes transfected with a miR-363-3p antagomir
significantly increased NRF1 mRNA and protein expression compared
with the antagomir NC (Fig. 5E-G).
Expression levels of p53, cleaved caspase-3 and Bcl-2, were
assessed through immunofluorescence, and p53 expression was also
assessed through western blotting and RT-qPCR (Fig. 5E, H and I). The overexpression of
miR-363-3p in the chondrocytes significantly increased mRNA and
protein expression levels of p53 and cleaved caspase-3, and reduced
the expression of Bcl-2 compared with the respective NC
chondrocytes (Fig. 5J). Antagomir
miR-363 reduced expression levels of p53 and cleaved caspase-3, and
increased Bcl-2 expression levels in chondrocytes (Fig. 5E, H-J). The present results
suggested that targeting NRF1 promoted LPS-induced chondrocyte
apoptosis in vitro.
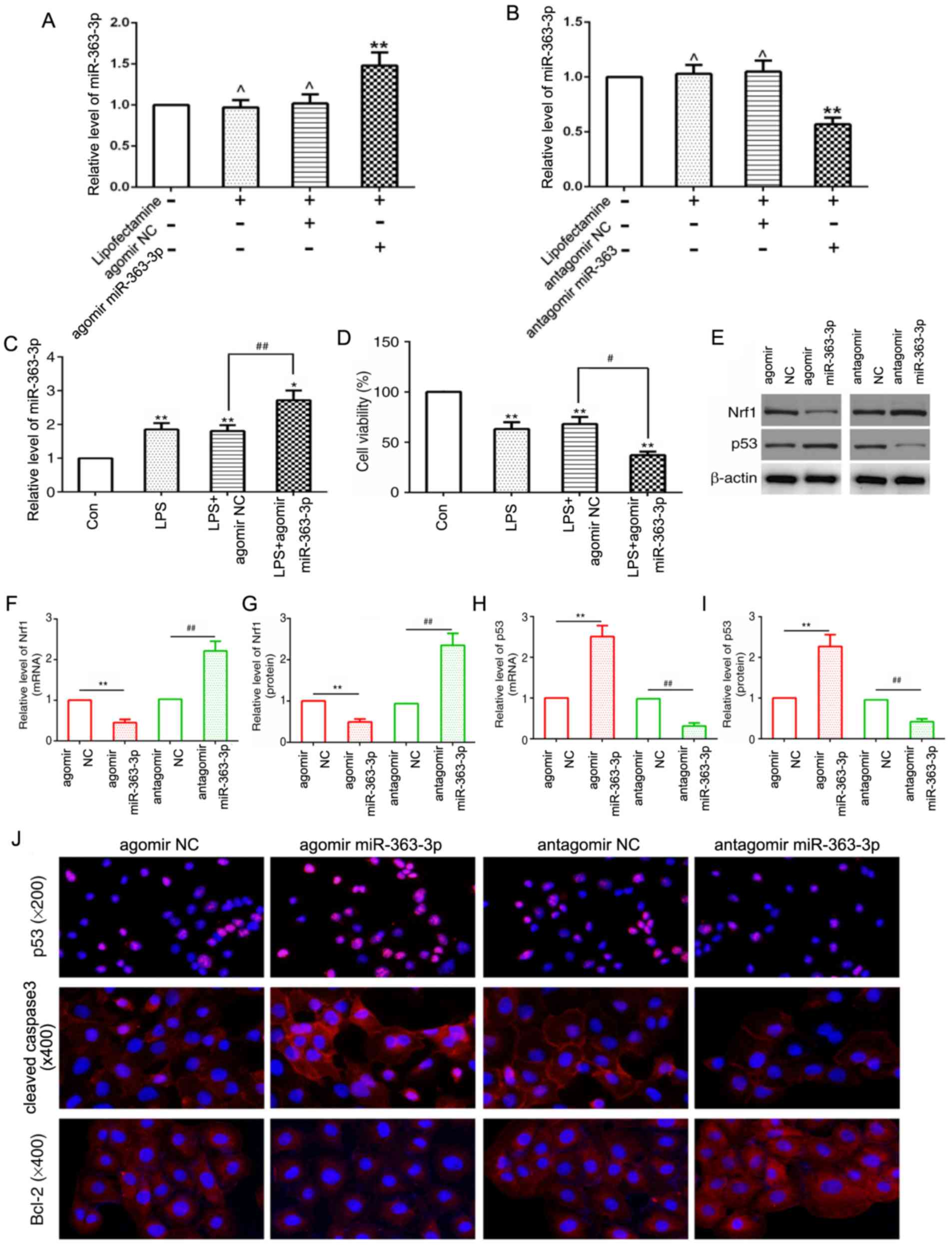 | Figure 5.miR-363-3p enhances LPS-induced
chondrocyte apoptosis and promotes the expression of p53 in
vitro. (A and B) Transfection efficiency of agomir and
antagomir miR-363-3p, was determined in cultured chondrocytes. (C)
Relative expression levels of miR-363-3p were determined by RT-qPCR
analysis in agomir miR-363-3p transfected chondrocytes stimulated
with LPS compared with control groups. (D) Cell viability was
measured in agomir miR-363-3p transfected chondrocytes stimulated
with LPS compared with control groups using an MTT assay. (E) NRF1
and p53 protein expression was assessed by western blot analysis in
the agomir or antagomir miR-363-3p transfected chondrocytes
compared with their respective NCs. (F) mRNA and (G) protein
expression level of NRF1, and (H) mRNA and (I) protein expression
level of p53 following transfection of chondrocytes with agomir or
antagomir miR-363-3p compared with their respective NCs. (J)
Protein expression levels of p53 (magnification, ×200), cleaved
caspase-3 (magnification, ×400) and Bcl-2 (magnification, ×400),
were assessed by immunofluorescence in chondrocytes transfected
with agomir or antagomir miR-363-3p, compared with their respective
NCs. **P<0.01, ,*P<0.05, ^P>0.05 vs. control
group, ##P<0.01 vs. LPS + miR-363-3p group,
#P<0.05 vs. LPS+miR-363 NC group. miR, microRNA; LPS,
lipopolysaccharide; RT-qPCR, reverse transcription-quantitative
PCR; NRF1, nuclear respiratory factor 1; NC, negative control; Con,
control. |
miR-363-3p targeting of NRF1 promotes
chondrocyte apoptosis via the upregulation of p53 in OA model
rats
To further confirm that miR-363-3p regulates
chondrocyte apoptosis in OA, in vivo experiments were
conducted. miR-363-3p overexpression in OA model rats resulted in a
significant increase of Bax and cleaved caspase-3 expression, and a
decrease of Bcl-2 expression (Fig.
6A-D), which suggested that miR-363-3p may promote chondrocyte
apoptosis in OA. Furthermore, NRF1 mRNA and protein levels were
decreased, and p53 mRNA and protein levels were increased following
the overexpression of miR-363-3p with agomir miR-363 (Fig. 6E-I). These results provided further
evidence that miR-363-3p targeting of NRF1 increased chondrocyte
apoptosis through the upregulation of p53 in vivo.
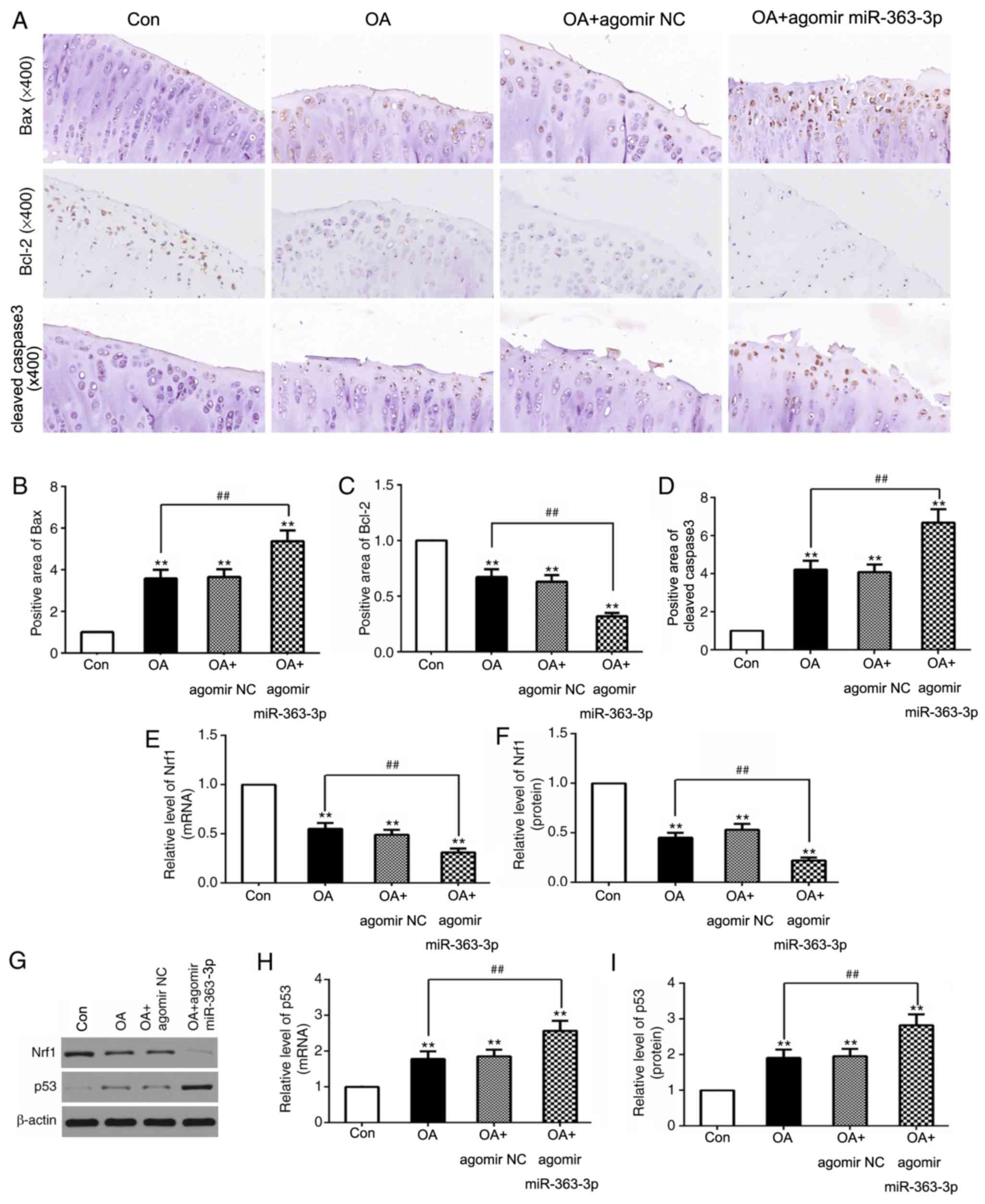 | Figure 6.miR-363-3p targeting of NRF1regulates
the expression of apoptotic index in chondrocytes of OA rats in
vivo. (A) Immunohistochemistry staining in chondrocytes and
quantification (brown area %; magnification, ×400) from the
sections of (B) Bax, (C) Bcl-2 and (D) cleaved caspase-3, following
injection of agomir miR-363-3p, or agomir NC (n=6 rats/group).
Reverse transcription-quantitative PCR was used to analyze the mRNA
levels of (E) NRF1 and (H) p53 following injection with agomir
miR-363-3p or its NC. (G) Protein expression levels of (F) NRF1 and
(I) p53 following injection with agomir miR-363-3p, or its NC, as
assessed by reverse transcription-quantitative PCR (n=6
rats/group). **P<0.01 vs. control, ##P<0.01 vs. OA
group. miR, microRNA; NRF1, nuclear respiratory factor 1; OA,
osteoarthritis; NC, negative control. |
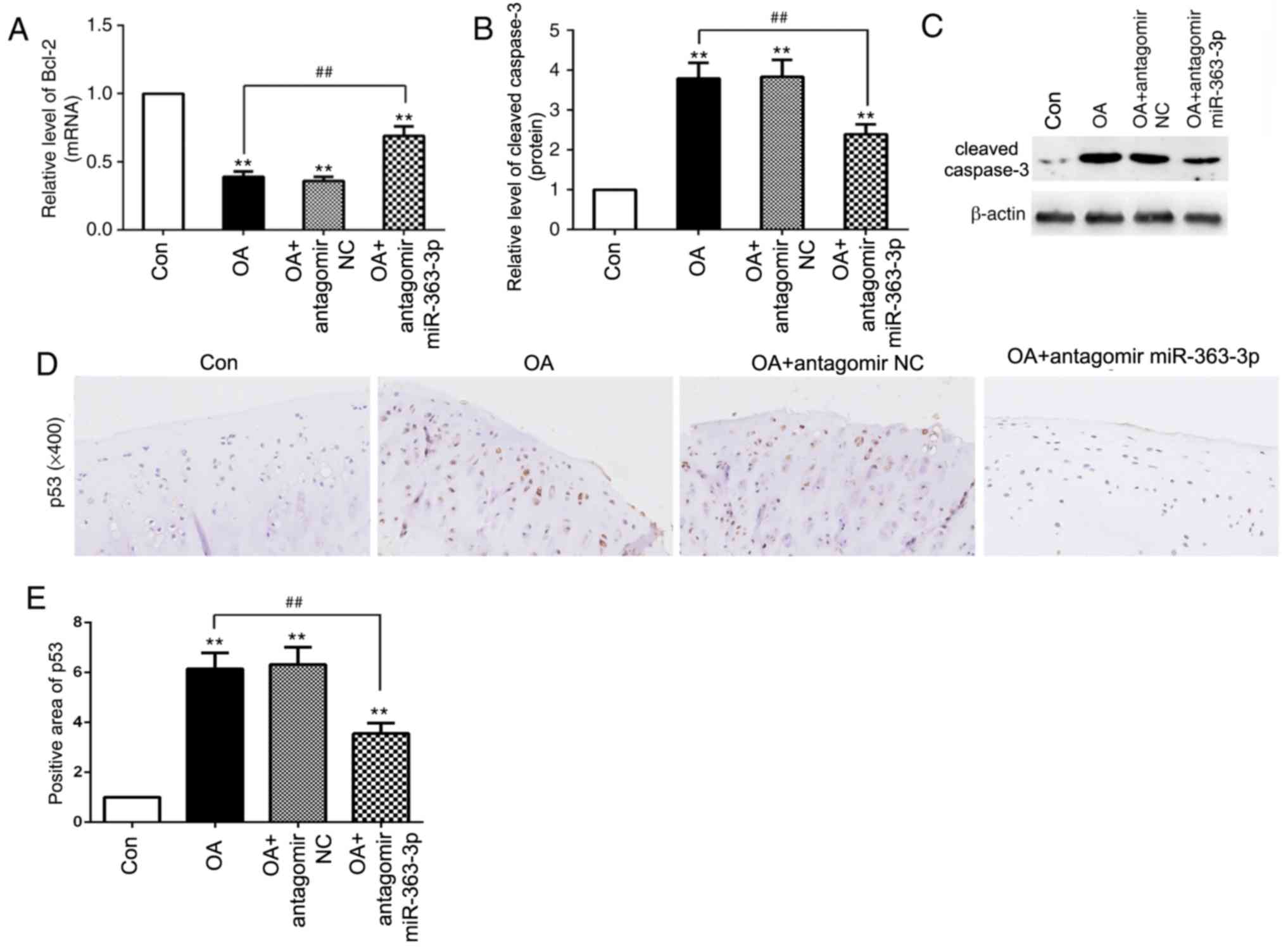
Inhibition of miR-363-3p reduces
chondrocyte apoptosis in OA rats
Based on the increased miR-363-3p expression levels
demonstrated in vivo, whether the inhibition of miR-363-3p
with antagomir miR-363 could reduce chondrocyte apoptosis was
investigated. Compared with the OA + antagomir NC group, the OA +
antagomir miR-363-3p demonstrated an increased mRNA expression of
Bcl-2 and decreased protein expression of cleaved caspase-3
(Fig. 7A-C), which suggested that
the inhibition of miR-36-3-3p may reduce chondrocyte apoptosis in
OA. Furthermore, compared with the OA group, antagomir miR-363-3p
decreased the expression level of p53 in OA rats (Fig. 7D and E). These results suggested
that inhibition of miR-363-3p may reduce chondrocyte apoptosis
through the downregulation of p53 in vivo.
Discussion
OA is associated with articular cartilage
degeneration and cartilage extracellular matrix destruction
(25). The extrachondral matrix of
the articular cartilage is mainly secreted by chondrocytes
(26). In OA, a combination of
multiple factors is directly or indirectly involved in cartilage
matrix synthesis or metabolism, which ultimately results in
OA-associated degradation of the cartilage (27). In osteoarthritic cartilage, the
rate of chondrocyte loss is ~20% (28). This indicates that chondrocytes are
important for maintaining extrachondral matrix homeostasis and that
the abnormal behavior of chondrocytes leads to the degeneration of
the articular cartilage. An improved understanding of the molecular
mechanisms governing the apoptotic death of chondrocytes in OA will
therefore prove invaluable as a means of identifying novel
therapeutic strategies to treat this disease.
In addition to regulating mitochondrial
transcription and replication-associated gene expression, NRF1 is
involved in the regulation of apoptosis. The knockdown of NRF1 led
to increased apoptotic rates, which was likely associated with
mitochondrial cytochrome c release, and upregulated
expression levels of Bax, caspase-3 and caspase-9, which are linked
with apoptosis (29). The
overexpression of NRF1 inhibited palmitate-induced human cardiac
myocytes (HCMS) apoptosis (30).
In the present study, NRF1 was markedly reduced in the articular
cartilage of OA model rats, as well as in LPS-stimulated
chondrocytes, and NRF1 knockdown significantly enhanced chondrocyte
apoptosis in vitro. In consideration of the role of NRF1 in
apoptosis, the present study investigated the expression of NRF1,
and found that NRF1 was markedly reduced in the articular cartilage
of OA model rats, as well as in LPS-stimulated chondrocytes.
Therefore, the present results suggested that NRF1 may be a
potential regulator in chondrocytes apoptosis. The present study
constructed a siRNA-NRF1, found that NRF1 knockdown significantly
enhanced chondrocyte apoptosis in vitro.
miRNA expression is closely associated with
apoptotic death in different pathological progressions (31). miRNA-410-3p protected
hypoxia-induced proliferation suppression and apoptosis stimulation
in cardiomyocytes via targeting TRAF5 (32). In addition, miR-425 suppressed cell
apoptosis by targeting AMPH-1 in non-small-cell lung cancer
(33). miRNAs are important
factors for regulating the RNA network, with individual miRNAs
potentially being pro-apoptotic or anti-apoptotic in certain
situations (34). The present
study aimed to reveal miRNAs capable of binding to NRF1 and to
assess their regulatory importance in the context of chondrocyte
apoptosis in OA. In the present study, NRF1 was identified as a
target of miR-363-3p in chondrocytes. Previous research has
demonstrated that miR-363-3p is an important regulator of
apoptosis. miR-363 inhibition in cardiomyocytes reduced the
incidence of hypoxia-induced apoptotic death through enhancing
Notch signaling (20). The
upregulation of miR-363-3p accelerated apoptosis in laryngeal
cancer cells by targeting induced myeloid leukemia cell
differentiation protein 1 (35).
In addition, when expressed ectopically in HT29 and HCT116 cells,
miR-363-3p promoted apoptosis (36). Thus, the present study hypothesized
that miR-363 may represent a novel therapeutic target in OA.
IL-1β, IL-6 and TNF-α are crucial cytokines in
vivo (37–39). A previous study reported that the
initiation of apoptosis was due to the activation of the homologous
IL-1β-converting enzyme protease family; it converts the newly
synthesized precursor of IL-1β into active IL-1β, and results in
the breakdown of matrix proteins that maintain the structure and
function of cells, in addition to mediating apoptosis by activating
other members of the family (37).
IL-6 affects the growth, differentiation and gene expression of
numerous cell types, and its disrupted expression is closely
related apoptosis (38). TNF-α has
a potent apoptotic effect, and its apoptotic signal is mediated by
the TNF receptor 1 (TNFR1) (39).
In the present study, overexpression of miR-363-3p significantly
increased TNF-α, IL-1β and IL-6 expression levels in synovial
fluid. The present results suggested that inhibition of miR-363-3p
exerted its protective effects against OA by suppressing
proinflammatory cytokines induced apoptosis.
Bcl-2 is an oncogene identified by Tsujimoto et
al (40) in follicular
lymphoma. Bcl-2 family members are divided into two categories
according to their function; members that promote cell apoptosis,
such as Bax; and members that inhibit cellular apoptosis, such as
Bcl-2, and the balance between Bax/Bcl-2 controls the onset of
apoptosis (41). Caspase-3 exists
in the cytoplasm in an inactive format, where it coordinates
signals from multiple apoptotic pathways, and activation of
caspase-3 can initiate the degradation phase of apoptosis (42,43).
p53 is a recognized apoptotic gene vital to apoptosis, and it
contributes by both inhibiting cellular proliferation and inducing
cellular apoptosis (44,45). In vitro, the present results
suggested that miR-363-3p overexpression enhanced p53 and cleaved
caspase-3 expression, inhibited Bcl-2 expression, which indicated
the aggravated chondrocyte apoptosis, whereas inhibiting miR-363-3p
resulted reduced apoptosis. In addition, elevated miR-363-3p
expression increased apoptosis by downregulating NRF1 gene
expression in vivo. In order to further investigate the role
of miR-363-3p in OA chondrocyte apoptosis, supplementary
experiments were conducted. The present results suggested that
downregulation of miR-363-3p led to an increased expression of
Bcl-2, decreased expression of p53 and cleaved caspase-3, which
indicated that inhibition of miR-363-3p reduced chondrocyte
apoptosis. In conclusion, the present results suggested that
miR-363-3p inhibited NRF1, and this was linked with OA-associated
chondrocyte apoptosis.
Acknowledgements
Not applicable.
Funding
The present study was supported by The National
Natural Science Foundation of China (grant no. 81901413).
Availability of data and materials
All data generated and analyzed during the present
study are included in this article.
Authors' contributions
MZ, ZQW and BJL performed the experiments,
contributed to data analysis and wrote the manuscript. MZ, FYS and
AZC analyzed the data. MZG conceptualized the study design, and
contributed to data analysis and experimental materials. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The Animal Care and Use Committee of Shandong
University (Shandong, China) approved all animal studies, which
were conducted in a manner consistent with the National Institutes
of Health Guide for the Care and Use of Laboratory Animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Xie F, Kovic B, Jin X, He X, Wang M and
Silvestre C: Economic and humanistic burden of osteoarthritis: A
systematic review of large sample studies. Pharmacoeconomics.
34:1087–1100. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gu YT, Chen J, Meng ZL, Ge WY, Bian YY,
Cheng SW, Xing CK, Yao JL, Fu J and Peng L: Research progress on
osteoarthritis treatment mechanisms. Biomed Pharmacother.
93:1246–1252. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Barnett R: Osteoarthritis. Lancet.
391:19852018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ribitsch I, Mayer RL, Egerbacher M, Gabner
S, Kańduła MM, Rosser J, Haltmayer E, Auer U, Gültekin S, Huber J,
et al: Fetal articular cartilage regeneration versus adult
fibrocartilaginous repair: Secretome proteomics unravels molecular
mechanisms in an ovine model. Dis Model Mech. 11:dmm0330922018.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim JH, Jeon J, Shin M, Won Y, Lee M, Kwak
JS, Lee G, Rhee J, Ryu JH, Chun CH and Chun JS: Regulation of the
catabolic cascade in osteoarthritis by the zinc-ZIP8-MTF1 axis.
Cell. 156:730–743. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ma F, Li G, Yu Y, Xu J and Wu X:
MiR-33b-3p promotes chondrocyte proliferation and inhibits
chondrocyte apoptosis and cartilage ECM degradation by targeting
DNMT3A in osteoarthritis. Biochem Biophys Res Commun. 519:430–437.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Evans MJ and Scarpulla RC: NRF-1: A
trans-activator of nuclear-encoded respiratory genes in animal
cells. Genes Dev. 4:1023–1034. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Scarpulla RC: Nuclear respiratory factors
and the pathways of nuclear-mitochondrial interaction. Trends
Cardiovasc Med. 6:39–45. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang Y and Manning BD: mTORC1 signaling
activates NRF1 to increase cellular proteasome levels. Cell Cycle.
14:2011–2017. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang GM, Deng MT, Lei ZH, Wan YJ, Nie HT,
Wang ZY, Fan YX, Wang F and Zhang YL: Effects of NRF1 on
steroidogenesis and apoptosis in goat luteinized granulosa cells.
Reproduction. 154:111–122. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Niu N, Li Z, Zhu M, Sun H, Yang J, Xu S,
Zhao W and Song R: Effects of nuclear respiratory factor1 on
apoptosis and mitochondrial dysfunction induced by cobalt chloride
in H9C2 cells. Mol Med Rep. 19:2153–2163. 2019.PubMed/NCBI
|
|
12
|
Zhang H, Wu J, Keller JM, Yeung K, Keller
ET and Fu Z: Transcriptional regulation of RKIP expression by
androgen in prostate cells. Cell Physiol Biochem. 30:1340–1350.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Berezikov E, Guryev V, van de Belt J,
Wienholds E, Plasterk RH and Cuppen E: Phylogenetic shadowing and
computational identification of human microRNA genes. Cell.
120:21–24. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang S, Chen L, Jung EJ and Calin GA:
Targeting microRNAs with small molecules: From dream to reality.
Clin Pharmacol Ther. 87:754–758. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bao J, Li D, Wang L, Wu J, Hu Y, Wang Z,
Chen Y, Cao X, Jiang C, Yan W and Xu C: MicroRNA-449 and
microRNA-34b/c function redundantly in murine testes by targeting
E2F transcription factor-retinoblastoma protein (E2F-pRb) pathway.
J Biol Chem. 287:21686–21698. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhao Z, Dai XS, Wang ZY, Bao ZQ and Guan
JZ: MicroRNA-26a reduces synovial inflammation and cartilage injury
in osteoarthritis of knee joints through impairing the NF-κB
signaling pathway. Biosci Rep. 39:BSR201820252019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ma Y, Wu Y, Chen J, Huang K, Ji B, Chen Z,
Wang Q, Ma J, Shen S and Zhang J: miR-10a-5p promotes chondrocyte
apoptosis in osteoarthritis by targeting HOXA1. Mol Ther Nucleic
Acids. 14:398–409. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ding Y, Wang L, Zhao Q, Wu Z and Kong L:
MicroRNA93 inhibits chondrocyte apoptosis and inflammation in
osteoarthritis by targeting the TLR4/NF-κB signaling pathway. Int J
Mol Med. 43:779–790. 2019.PubMed/NCBI
|
|
19
|
Witkos TM, Koscianska E and Krzyzosiak WJ:
Practical aspects of microRNA target prediction. Curr Mol Med.
11:93–109. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Meng X, Ji Y, Wan Z, Zhao B, Feng C, Zhao
J, Li H and Song Y: Inhibition of miR-363 protects cardiomyocytes
against hypoxia-induced apoptosis through regulation of Notch
signaling. Biomed Pharmacother. 90:509–516. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wenzhao L, Jiangdong N, Deye S, Muliang D,
Junjie W, Xianzhe H, Mingming Y and Jun H: Dually regulatory roles
of HMGB1 in inflammatory reaction of chondrocyte cells and mice.
Cell Cycle. 18:2268–2280. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ni Z, Kuang L, Chen H, Xie Y, Zhang B,
Ouyang J, Wu J, Zhou S, Chen L, Su N, et al: The exosome-like
vesicles from osteoarthritic chondrocyte enhanced mature IL-1β
production of macrophages and aggravated synovitis in
osteoarthritis. Cell Death Dis. 10:5222019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mankin HJ: Biochemical and metabolic
abnormalities in osteoarthritic human cartilage. Fed Proc.
32:1478–1480. 1973.PubMed/NCBI
|
|
25
|
Pope JE, McCrea K, Stevens A and Ouimet
JM: The relationship between NSAID use and osteoarthritis (OA)
severity in patients with hip and knee OA: Results of a case
control study of NSAID use comparing those requiring hip and knee
replacements to those in whom surgery was not recommended. Med Sci
Monit. 14:CR604–CR610. 2008.PubMed/NCBI
|
|
26
|
Onuora S: Osteoarthritis: Cartilage matrix
stiffness regulates chondrocyte metabolism and OA pathogenesis. Nat
Rev Rheumatol. 11:5042015. View Article : Google Scholar
|
|
27
|
Yudoh K and Karasawa R: Statin prevents
chondrocyte aging and degeneration of articular cartilage in
osteoarthritis (OA). Aging (Albany NY). 2:990–998. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Héraud F, Héraud A and Harmand MF:
Apoptosis in normal and osteoarthritic human articular cartilage.
Ann Rheum Dis. 59:959–965. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sant KE, Hansen JM, Williams LM, Tran NL,
Goldstone JV, Stegeman JJ, Hahn ME and Timme-Laragy A: The role of
NRF1 and Nrf2 in the regulation of glutathione and redox dynamics
in the developing zebrafish embryo. Redox Biol. 13:207–218. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang J, Gu JY, Chen ZS, Xing KC and Sun
B: Astragalus polysaccharide suppresses palmitate-induced apoptosis
in human cardiac myocytes: The role of NRF1 and antioxidant
response. Int J Clin Exp Pathol. 8:2515–2524. 2015.PubMed/NCBI
|
|
31
|
Su Z, Yang Z, Xu Y, Chen Y and Yu Q:
MicroRNAs in apoptosis, autophagy and necroptosis. Oncotarget.
6:8474–8490. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Teng YL, Ren F, Xu H and Song HJ:
Overexpression of miRNA- 410-3p protects hypoxia-induced
cardiomyocyte injury via targeting TRAF5. Eur Rev Med Pharmacol
Sci. 23:9050–9057. 2019.PubMed/NCBI
|
|
33
|
Jiang L, Ge W and Geng J: miR-425
regulates cell proliferation, migration and apoptosis by targeting
AMPH-1 in non-small-cell lung cancer. Pathol Res Pract.
215:1527052019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wu G, Tan J, Li J, Sun X, Du L and Tao S:
miRNA-145-5p induces apoptosis after ischemia-reperfusion by
targeting dual specificity phosphatase 6. J Cell Physiol. Mar
18–2019.doi: 10.1002/jcp.28291 (Epub ahead of print).
|
|
35
|
Feng WT, Yao R, Xu LJ, Zhong XM, Liu H,
Sun Y and Zhou LL: Effect of miR-363 on the proliferation, invasion
and apoptosis of laryngeal cancer by targeting Mcl-1. Eur Rev Med
Pharmacol Sci. 22:4564–4572. 2018.PubMed/NCBI
|
|
36
|
Dong J, Geng J and Tan W: MiR-363-3p
suppresses tumor growth and metastasis of colorectal cancer via
targeting SphK2. Biomed Pharmacother. 105:922–931. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee KH and Kang TB: The molecular links
between cell death and inflammasome. Cells. 8:E10572019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhu Y, Saito K, Murakami Y, Asano M,
Iwakura Y and Seishima M: Early increase in mRNA levels of
pro-inflammatory cytokines and their interactions in the mouse
hippocampus after transient global ischemia. Neurosci Lett.
393:122–126. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu T, Bao YH, Wang Y and Jiang JY: The
role of necroptosis in neurosurgical diseases. Braz J Med Biol Res.
48:292–298. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tsujimoto Y, Cossman J, Jaffe E and Croce
CM: Involvement of the bcl-2 gene in human follicular lymphoma.
Science. 228:1440–1443. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu C, Shi Z, Fan L, Zhang C, Wang K and
Wang B: Resveratrol improves neuron protection and functional
recovery in rat model of spinal cord injury. Brain Res.
1374:100–109. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Clark WM, Rinker LG, Lessov NS, Hazel K,
Hill JK, Stenzel-Poore M and Eckenstein F: Lack of interleukin-6
expression is not protective against focal central nervous system
ischemia. Stroke. 31:1715–1720. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Glushakova OY, Glushakov AA, Wijesinghe
DS, Valadka AB, Hayes RL and Glushakov AV: Prospective clinical
biomarkers of caspase-mediated apoptosis associated with neuronal
and neurovascular damage following stroke and other severe brain
injuries: Implications for chronic neurodegeneration. Brain Circ.
3:87–108. 2017.PubMed/NCBI
|
|
44
|
Fridman JS and Lowe SW: Control of
apoptosis by p53. Oncogene. 22:9030–9040. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lu Q, Rau TF, Harris V, Johnsonm M,
Poulsen DJ and Black SM: Increased p38 mitogen-activated protein
kinase signaling is involved in the oxidative stress associated
with oxygen and glucose deprivation in neonatal hippocampal slice
cultures. Eur J Neurosci. 34:1093–1101. 2011. View Article : Google Scholar : PubMed/NCBI
|