Introduction
Myocardial ischemia is a serious threat to patients
with cardiovascular disease. Even the process of restoring the
blood supply to the myocardium after the treatment of
cardiovascular disease tends to have a serious effect on the
recovery of the heart, as a result of the sudden recovery of
coronary blood supply. This is known as myocardial ischemia
reperfusion injury (MIRI) (1).
This is especially true for patients after heart surgery. MIRI is a
cause of the increasing rate of myocardial infarction, heart
failure and mortality (2).
Therefore, the reduction of MIRI in patients is key to alleviating
myocardial infarction rates, heart failure and mortality, and to
improving quality of life.
Research into alleviating MIRI has demonstrated
that, numerous methods, such as ischemic pre-conditioning, ischemic
post-conditioning, drug pre-conditioning and drug post-conditioning
can play a role in myocardial protection (3–7). At
the same time, a variety of myocardial protection mechanisms are
involved. This includes the hypoxia inducible factor 1/hypoxia
response element pathway (HIF-1/HRE) and the mitochondrial
ATP-sensitive potassium channels (mitoKATP channels)
(8,9).
The study of Zhao et al (10) in vivo showed that ischemic
post-conditioning (IPO) could increase the expression of inducible
nitric oxide synthase (iNOS) by activating the HIF-1α pathway and
reduce the infarct size of the myocardium. In myocardial cells and
isolated heart perfusion experiments, drug post-conditioning can
increase HIF-1α, and then activate iNOS to reduce MIRI. This
process can be reversed by HIF-1α small interfering (si)RNA or
2-methoxyestradiol (2ME2; a HIF-1α subunit blocker) (11). All these studies suggest that both
IPO and drug post-conditioning can alleviate MIRI by activating the
HIF-1/HRE pathway.
A study by Jin et al (12) has also shown that IPO can open
mitoKATP channels to play a role in myocardial
protection. Diazoxide (a specific mitoKATP channel
opener) post-conditioning (DPO) can also alleviate MIRI (13). MitoKATP channels are
potassium channels in the mitochondrial membrane, which are
composed of Kir and SUR subunits. The regulation of
mitoKATP channels is primarily related to the regulatory
subunit SUR, which is mainly controlled by ATP. Other metabolites,
such as protein kinase A, protein kinase C and PIP2, can also
regulate mitoKATP channels (14). As signaling molecules, the reactive
oxygen species (ROS) produced by mitoKATP channels can
activate downstream signaling pathways and ultimately reduce MIRI
by reducing calcium overload. Thus, whether the downstream
signaling pathways activated by mitoKATP channels
include the HIF-1/HRE pathway in IPO and whether DPO can also
activate the HIF-1/HRE pathway through mitoKATP channel
opening merits investigation.
In order to study the above problems, a rat heart
perfusion model was established using a Langendorff experiment
device (15). Using cardioplegia,
cardiac arrest was simulated in clinical cardiopulmonary bypass and
the myocardial protective effect of IPO/DPO was observed. The
present study aimed to assess whether the HIF-1/HRE pathway
participates in the myocardial protection mechanism of DPO. Also,
5-hydroxydecanoic acid (5HD; a specific mitoKATP
channels blocker) and 2ME2 (a HIF-1α subunit blocker) were used to
observe the expression changes in the HIF-1/HRE pathway, and
whether or not IPO/DPO could open mitoKATP channels and
then activate the HIF-1/HRE pathway. The present study will provide
a theoretical basis for the clinical application of diazoxide to
the treatment of MIRI.
Materials and methods
Materials
The experimental animals used were 80 healthy male
Sprague Dawley (SD) rats (weight, 250–300 g; 16–20 weeks old),
which were provided by the laboratory animal center of DaPing
Hospital, Chongqing. Before the experiment, the rats were housed in
groups of three or four for at least 1 week (12-h light/dark cycle,
free access to food and water, temperature 20–25°C, humidity
50–65%).
Diazoxide and 5HD were purchased from Sigma-Aldrich;
Merck KGaA. 2ME2 was purchased from Selleck Chemicals. The VEGF
antibody (cat. no. NB100-664) and HIF-1α antibody (cat. no.
NB100-105) were purchased from Novus Biologicals, LLC. The HO-1
antibody (cat. no. ab13248) and iNOS antibody (cat. no. ab49999)
were purchased from Abcam. β-actin antibody (cat. no. 66009-1-Ig)
was purchased from Proteintech Group, Inc. IRDye 800CW secondary
antibodies (cat. no. 926-32210) was purchased from LI-COR
Biosciences. The Sensiscript RT kit and Real-Time amplification kit
were purchased from Takara Bio, Inc. The primers were purchased
from Shanghai Generay Biotech Co., Ltd.
Methods
Establishment of rat heart perfusion
model in vitro
SD rats were anesthetized by intraperitoneal
injection of 1% pentobarbital sodium (60 mg/kg) and heparin (500
U/kg), and then a thoracotomy was performed. The aorta was removed
from the heart quickly and completely. After that, the heart was
fixed on the Langendorff system via the aorta and low flow
retrograde perfusion of K-H solution (oxygenated with 95%
O2 and 5% CO2; 37°C) was performed in the
aortic root. A small incision was made on the left atrial appendage
and the piezometric tube was inserted into the left ventricle
through the small opening. The left ventricular end diastolic
pressure was adjusted to 2–5 mmHg using the PowerLab physiological
experiment system. The K-H solution and the ambient temperature of
the heart are controlled at 37°C. Thereafter, the flow rate of
aortic perfusion was regulated and the perfusion pressure increased
slowly and stabilized at ~70 mmHg. After the heart was perfused for
20 min, the left ventricular developed pressure (LVDP), heart rate
(HR) and arrhythmia were observed. At LVDP >80 mmHg, HR >250
times/min and arrhythmia <2/min, follow-up experiments were
carried out.
A total of 80 hearts were randomly divided into the
following 10 groups (n=8, each group): Group N, group I/R, group
IPO, group IPO+2ME2, group IPO+5HD, group DPO, group DPO+2ME2,
group DPO+5HD, group I/R+2ME2 and group I/R+5HD (Fig. 1). Group N was aerobically perfused
with K-H solution for 120 min. Group I/R, after being aerobically
perfused for 20 min, was subjected to 40 min hypoxia and 60 min
reperfusion. Group IPO was treated the same as group I/R, but 10
sec hypoxia plus 10 sec reperfusion was performed six times prior
to reperfusion. Group IPO+5HD, was treated the same as group IPO,
but perfused with 100 µM 5HD for 5 min before ischemic
post-conditioning (13). Group
IPO+2ME2 was treated the same as group IPO, but perfused with 2 µM
2-methoxyestradiol for 10 min before ischemic post-conditioning
(11). Group DPO was treated the
same as group I/R, but perfused with 50 µM diazoxide for 5 min
before reperfusion (13). Group
DPO+5HD was treated the same as group DPO, but perfused with 100 µM
5HD for 5 min before diazoxide post-conditioning. Group DPO+2ME2
was treated the same as group DPO, but perfused with 2 µM 2ME2 for
10 min before diazoxide post-conditioning. Group I/R+5HD was
treated the same as group I/R, but the heart was perfused with 100
µM 5HD for 5 min at 35 min after stopping the perfusion. Group
I/R+2ME2 was treated the same as group I/R, but the heart was
perfused with 2 µM 2ME2 for 10 min at 30 min after stopping the
perfusion.
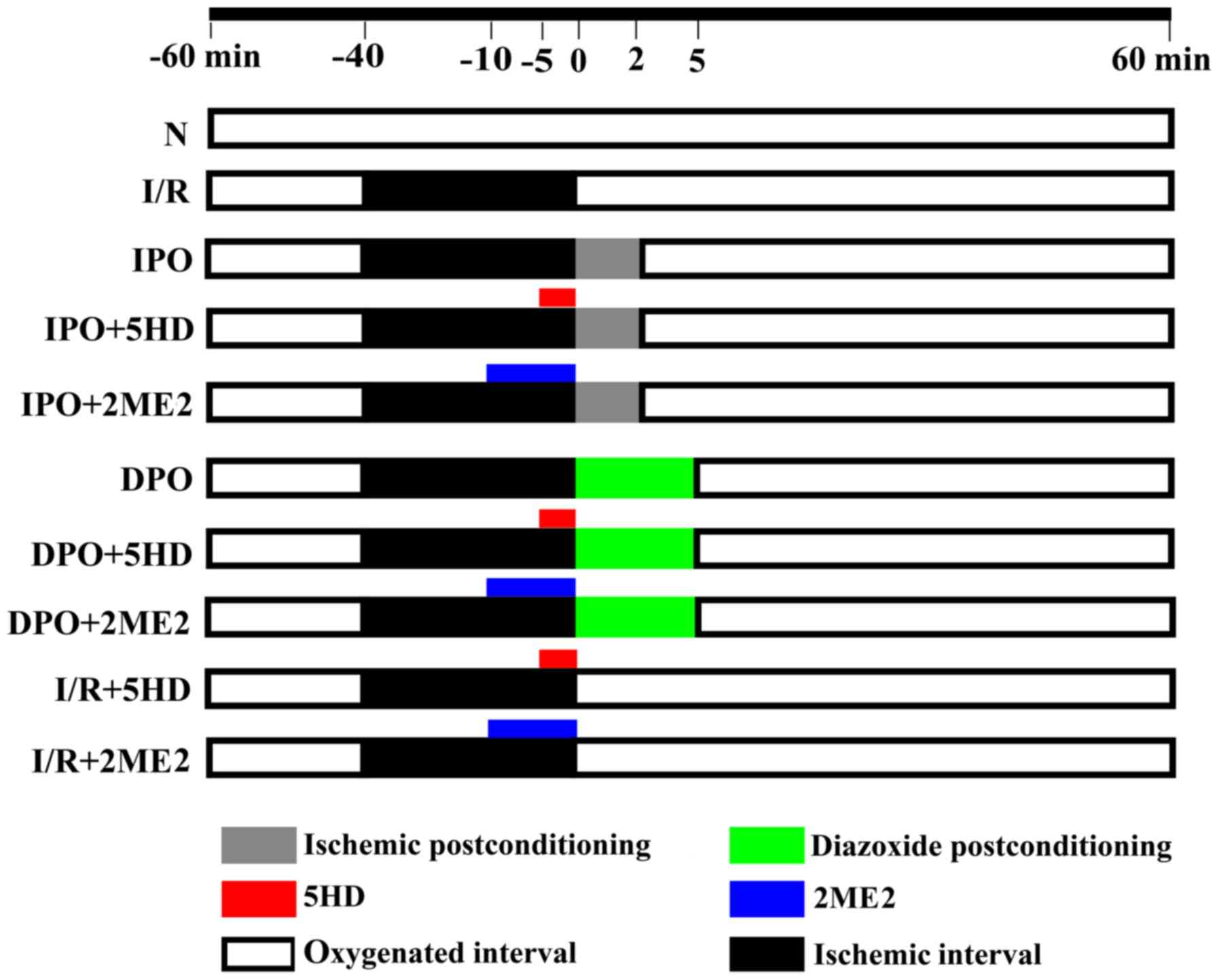 | Figure 1.Perfusion of the isolated heart.
Using a Langendorff perfusion device and K-H solution, the rat
hearts were randomly divided into the following 10 groups
(n=8/group): Group N, group I/R, group IPO, group IPO+2ME2, group
IPO+5HD, group DPO, group DPO+2ME2, group DPO+5HD, group I/R+2ME2
and group I/R +5HD. The K-H solution contained (in g/l) 6.8959
NaCl, 2.1799 D-glucose, 2.1003 NaHCO3, 0.2952
MgSO4 7H2O, 0.3504 KCl, 0.1633
KH2PO4 and 0.22198 CaCl2. The St.
Thomas cardioplegic solution contained (in g/l) 6.4284 NaCl, 1.6264
MgCl2 6H2O, 1.1928 KCl, 0.8401
NaHCO3 and 0.1332 CaCl2. IPO, ischemic
post-conditioning; DPO, diazoxide post-conditioning; I/R,
ischemia/reperfusion; 5HD, 5-hydroxydecanoic acid; 2ME2,
2-methoxyestradiol. |
Except for in group N, perfusion of the K-H solution
in all other groups was stopped after 20 min and cardiac arrest was
attained by immediately perfusing the St. Thomas cardioplegic
solution (4°C). After that, the hearts were maintained in a 32°C
environment for 40 min.
Cardiac function monitoring
HR, LVDP, left ventricular end diastolic pressure
(LVEDP) and maximal left ventricular pressure (+dp/dtmax) were
measured using the PowerLab system at 20 min and 2 h.
Detection of infarct size in the
heart
First, 1% TTC dye solution was prepared for
incubation in the dark at 37°C for ~25 min.
The heart was quickly removed and placed into a
−80°C refrigerator for 7 min. Along the transverse section of the
heart, it was cut into five thick circular slices (0.1–0.3 cm).
After that, the cardiac slices were immersed in the above 1% TTC
dye solution and incubated at 37°C in the dark for 25 min. After 25
min, the cardiac slices were placed in 10% formaldehyde for 7 days
at 20–25°C. The cardiac slices were arranged from big to small in
order and images were captured using a Sony camera. Finally, the
infarct size was calculated using ImageJ software (version 1.46R;
National Institutes of Health).
Detection of myocardium under
transmission electron microscope
At the end of perfusion, several 0.1 cm3
pieces of myocardial tissue were cut rapidly from the left
ventricle of the heart. The myocardial tissues were fixed with 4°C
precooled 2.5% glutaraldehyde fixative (for no more than 2 weeks).
They were rinsed in phosphate buffer (washed once at intervals of 2
h, three times) and then fixed with 4°C precooled osmic acid (1%)
for 2 h. After successive gradient acetone dehydration, epoxy resin
embedding polymerization (45°C for 12 h and 60°C for 48 h),
UltracutE ultrathin sectioning (50–70 nm each slice), double
staining with acetic acid uranium dioxide (20–25°C for 20 min) and
lead citrate (20–25°C for 10 min), the ultrastructure of the
cardiomyocytes was observed under a Hitachi H7500 transmission
electron microscope.
The mitochondrial Flameng score criteria under
transmission electron microscope is as follows (16). The greater the myocardial
mitochondria damage, the higher the score and injury is scored as
0–4 points: 0, the structure of mitochondria is normal and they are
full of particles; 1, the structure of mitochondria is essentially
normal, but the matrix particles are lost; 2, mitochondrial
swelling and matrix transparency are apparent; 3, rupture of
mitochondrial cristae with matrix transparency and concentration;
4, the mitochondrial cristae are split, the integrity of the
mitochondria inside and outside the membrane has been lost, and
they appear vacuolated.
In each electron microscopy experiment, 100
mitochondria were observed and 20 mitochondria were randomly
selected from each field. A total of five visual fields were
selected randomly. The mitochondrial Flameng score was calculated
as the mean of the 100 mitochondrial total scores.
Reverse transcription-quantitative PCR
(RT-qPCR)
At the end of the experiment, the left ventricular
myocardium was placed in an enzyme-free cryopreservation tube (2
ml) and frozen in liquid nitrogen (−196°C). The frozen myocardium
was transferred to a −80°C refrigerator.
RNA isoPlus solution, chloroform, isopropanol and
ethanol were used to extract the RNA from the myocardium. The
concentration and purity of the RNA were determined using a
Varioskan Flash (Thermo Fisher Scientific, Inc.). After that, cDNA
was synthesized using a reverse transcription kit (Takara Bio,
Inc.) and 2400 PCR instrument (Bio-Rad Laboratories, Inc.). The
reverse transcriptase temperature profile was 37°C for 15 min, 85°C
for 5 min and maintenance at 4°C. To evaluate the target genes, the
cDNA was amplified using a Takara Bio, Inc., qPCR kit and CFX
Connect instrument (Bio-Rad, Laboratories, Inc.). The amplification
reaction temperature profile was 95°C for 3 min, followed by 95°C
for 10 min and 61.5°C for 30 min for a total of 40 cycles. The Cq
values of samples were taken to analyze and calculate the relative
expression of the target genes (17).
The primer sequence for each gene were as follows:
HIF-1α (forward, 5′-CCCATTCCTCATCCATCAAACATT-3′ and reverse,
5′-CTTCTGGCTCATAACCCATCAACTC-3′); HO-1 (forward,
5′-ATGAGGAACTTTCAGAAGGGTC-3′ and reverse,
5′-GGAAGTAGAGTGGGGCATAGAC-3′); VEGF (forward,
5′-CCTCTCCCTACCCCACTTCCT-3′ and reverse,
5′-CACTTTCTCTTTTCTCTGCCTCCAT-3′); iNOS (forward,
5′-TCCTCAGGCTTGGGTCTTGTTAG-3′ and reverse,
5′-GGGTTTTCTCCACGTTGTTGTT-3′); β-actin (forward,
5′-CTGAACCCTAAGGCCAACCG-3′ and reverse,
5′-GACCAGAGGCATACAGGGACAA-3′).
Western blotting
Myocardial proteins were extracted using RIPA lysis
buffer (Beijing Solarbio Science & Technology Co., Ltd.). After
the protein concentration was determined by bicinchoninic acid,
various target proteins were assessed by western blot analysis.
Equivalent amounts of protein (40 µg/5 µl) from the experimental
groups were analyzed by SDS-PAGE (the gel percentages were 10% for
separating gel and 4% for stacking gel) and the proteins were
transferred to PVDF membranes.. Then, membranes were blocked using
western blocking buffer (Beijing Solarbio Science & Technology
Co., Ltd.) at room temperature for 2 h. After incubation with the
primary (4°C, 12 h) and secondary (20–25°C, 2 h) antibodies in
succession, the PVDF membranes were scanned and detected by the
Odyssey Infrared Imaging System (LI-COR Biosciences). The dilution
ratio of each antibody was 1:500 (HIF-1 α), 1:1,000 (VEGF), 1:250
(HO-1), 1:1,000 (iNOS), 1:5,000 (β-actin) and 1:10,000 (IRDye 800cw
secondary antibody).
Statistical methods
All the data are presented as the mean ± standard
error of the mean and were analyzed using SPSS 17.0 statistical
software (SPSS, Inc.) One-way ANOVA followed by Dunnetts's T3 test
were used for intergroup comparison. P<0.05 was considered to
indicate a statistically significant difference.
Results
Cardiac function
After the heart was aerobically perfused for 20 min,
there were no significant differences in HR, LVDP, LVEDP and
+dp/dtmax among the groups (P>0.05; Table I). At the end of the experiment,
there were no significant differences in HR, LVDP, LVEDP and
+dp/dtmax among the groups (P>0.05; Table II).
 | Table I.Cardiac function indexes after
isolated heart aerobically perfused for 20 min. |
Table I.
Cardiac function indexes after
isolated heart aerobically perfused for 20 min.
| Groups | LVDP (mmHg) | HR (bpm) | LVEDP (mmHg) | +dp/dtmax
(mmHg/s) |
|---|
| N | 96.9±7.4 | 306±33 | 2.63±0.92 | 3,810±300 |
| I/R | 93.0±6.6 | 312±45 | 2.43±1.27 | 3,590±146 |
| IPO | 96.4±7.1 | 301±19 | 2.37±0.92 | 3,530±364 |
| IPO+5HD | 96.8±7.0 | 287±33 | 2.43±1.27 | 3,700±141 |
| IPO+2ME2 | 94.6±5.2 | 303±34 | 2.89±0.60 | 3,550±360 |
| DPO | 96.8±8.8 | 306±26 | 2.38±0.74 | 3,550±267 |
| DPO+5HD | 93.3±8.5 | 309±28 | 2.38±0.92 | 3,810±294 |
| DPO+2ME2 | 93.9±5.2 | 308±41 | 2.22±0.67 | 3,810±457 |
| I/R+5HD | 90.8±6.2 | 298±22 | 2.33±0.52 | 3,590±370 |
| I/R+2ME2 | 96.7±5.7 | 284±17 | 2.14±1.07 | 3,510±408 |
| Table II.Cardiac function indexes at the end
of the experiment. |
Table II.
Cardiac function indexes at the end
of the experiment.
| Groups | LVDP (mmHg) | HR (bpm) | LVEDP (mmHg) | +dp/dtmax
(mmHg/s) |
|---|
| N | 82.1±8.3 | 295±36 | 3.50±0.76 | 3,350±279 |
| I/R | 73.4±5.2 | 322±41 | 3.29±0.48 | 3,220±539 |
| IPO | 84.6±7.5 | 297±19 | 3.38±0.52 | 3,410±387 |
| IPO+5HD | 76.3±4.8 | 297±16 | 3.14±0.90 | 3,110±153 |
| IPO+2ME2 | 76.0±8.2 | 320±26 | 3.67±0.71 | 3,380±431 |
| DPO | 80.6±5.3 | 317±30 | 3.13±0.99 | 3,140±405 |
| DPO+5HD | 77.6±8.1 | 321±20 | 3.25±1.16 | 3,400±313 |
| DPO+2ME2 | 74.0±6.3 | 322±18 | 3.11±0.78 | 3,200±435 |
| I/R+5HD | 73.2±6.4 | 322±51 | 3.00±0.63 | 3,150±625 |
| I/R+2ME2 | 77.9±6.8 | 304±20 | 3.14±1.07 | 3,330±539 |
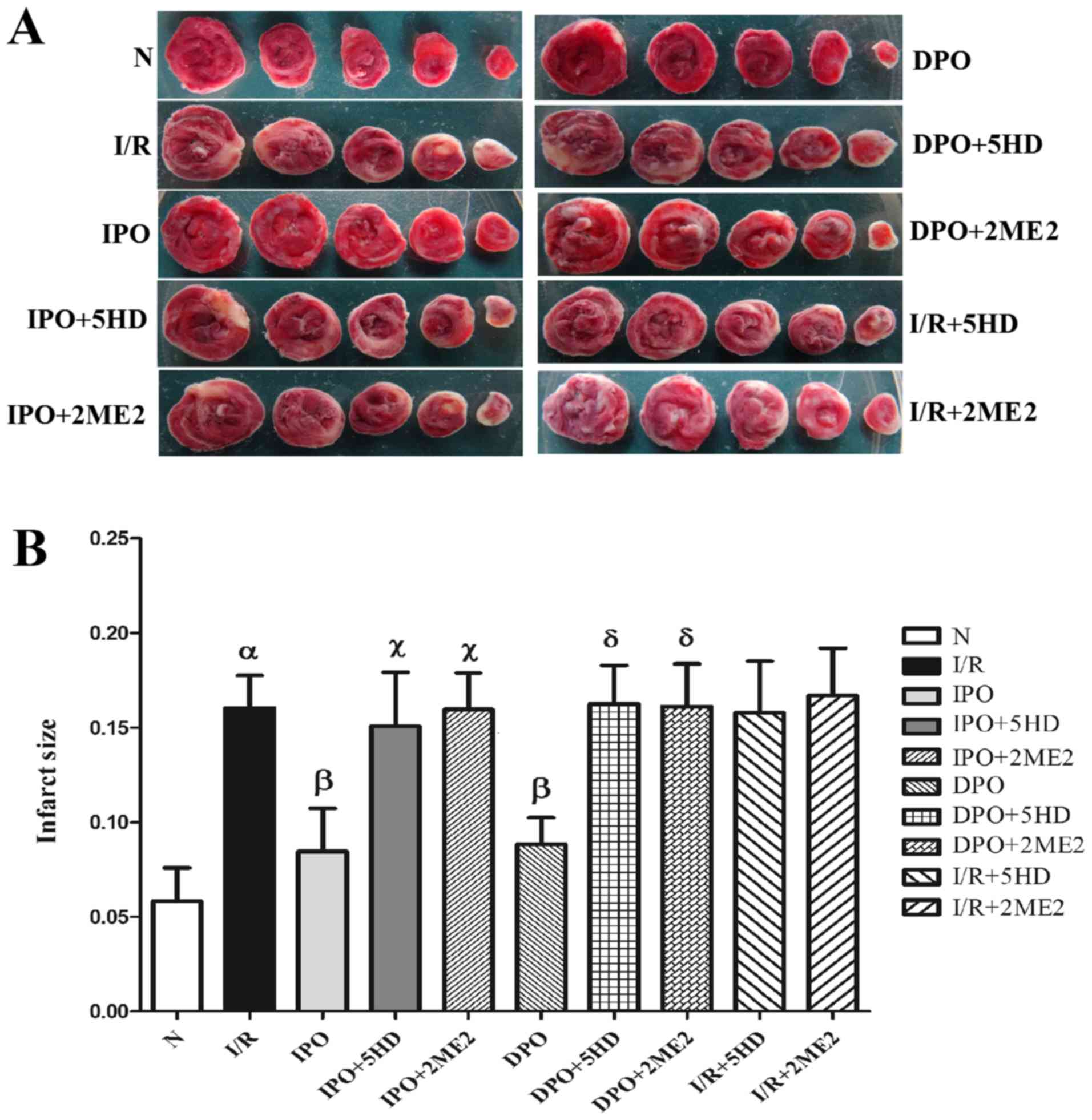
Infarct area of the myocardium
The infarct area of group N was 5.83±1.77%. In
comparison, the infarct size of group I/R increased significantly
(P<0.05; Fig. 2). The infarct
areas in groups IPO and DPO were significantly decreased compared
with group I/R (P<0.05), and the infarct areas in groups IPO+5HD
and IPO+2ME2 were significantly increased compared with group IPO
(P<0.05; Fig. 2). The infarct
sizes in groups DPO+5HD and DPO+2ME2 were significantly increased
compared with group DPO (P<0.05; Fig. 2). Compared with the I/R group,
there were no significant differences in infarct size between
groups I/R+5HD and I/R+2ME2 (P>0.05; Fig. 2B).
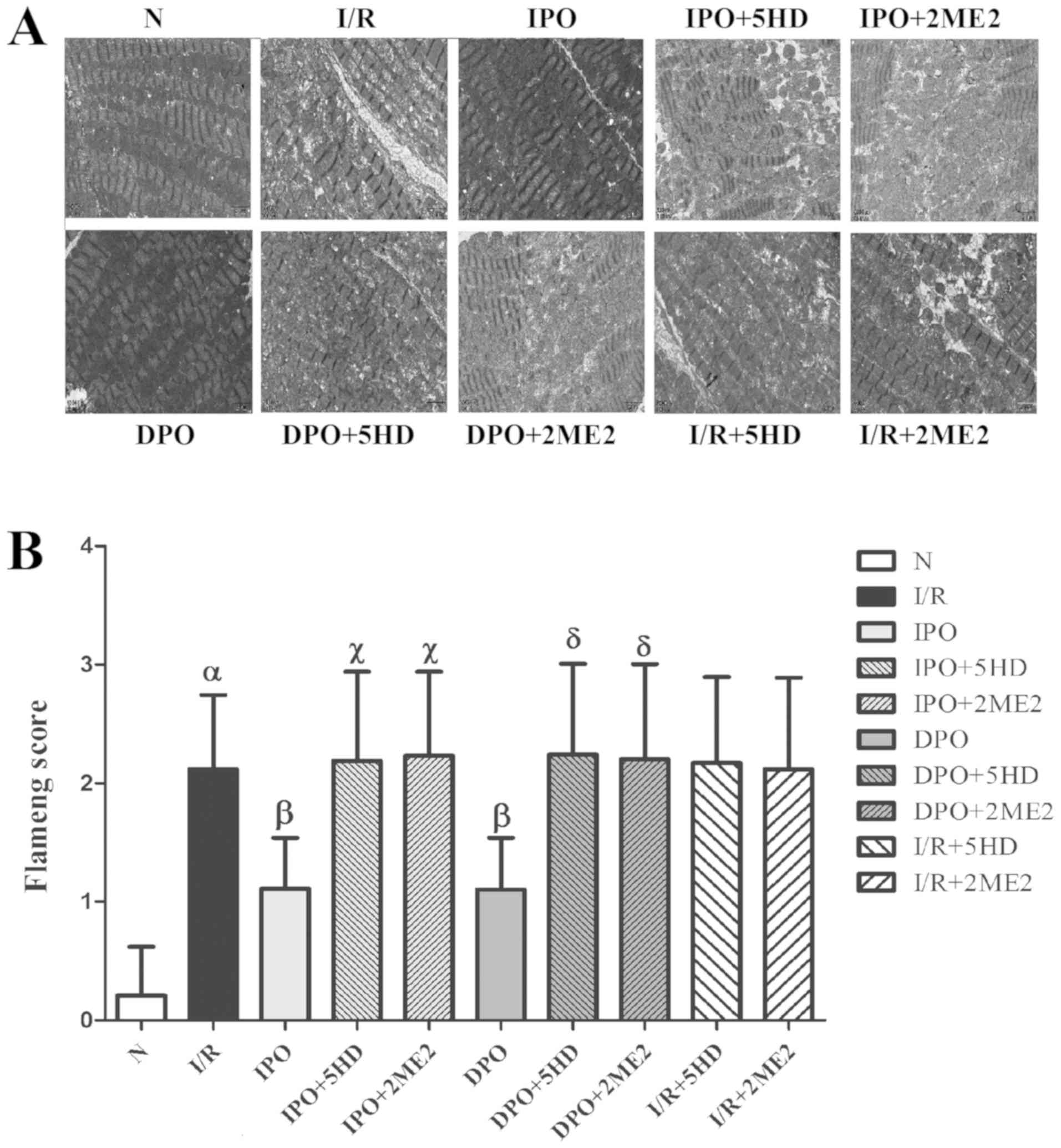
Myocardial morphology
Using the transmission electron microscope, the
myocardial morphology in each group was assessed. The mitochondrial
structure of group N was essentially normal and the mitochondrial
score was 0.21±0.409. The mitochondrial score in group I/R was
significantly increased compared with group N (P<0.05; Fig. 3). The mitochondrial scores of
groups IPO and DPO were significantly decreased compared with the
I/R group (P<0.05), and the mitochondrial score of groups
IPO+5HD and IPO+2ME2 was significantly increased compared with
group IPO (P<0.05). The mitochondrial scores in groups DPO+5HD
and DPO+2ME2 were significantly increased compared with group DPO
(P<0.05). There were no significant differences in the
mitochondrial scores among groups I/R+5HD, I/R+2ME2 and I/R
(P>0.05; Fig. 3).
Expression of HIF-1/HRE related
genes
There was no significant difference in HIF-1α mRNA
expression among the groups (P>0.05; Fig. 4A). The expression levels of HO-1,
iNOS and VEGF mRNA in group I/R were significantly increased
compared with group N (P<0.05). In groups IPO and DPO, the
expression levels (HO-1, iNOS and VEGF mRNA) were significantly
increased compared with group I/R (P<0.05). In groups IPO+5HD
and IPO+2ME2, the expression levels (HO-1, iNOS and VEGF mRNA) were
significantly decreased compared with group IPO (P<0.05). In
groups DPO+5HD and DPO+2ME2, the expression levels (HO-1, iNOS and
VEGF mRNA) were significantly decreased compared with group DPO
(P<0.05; Fig. 4).
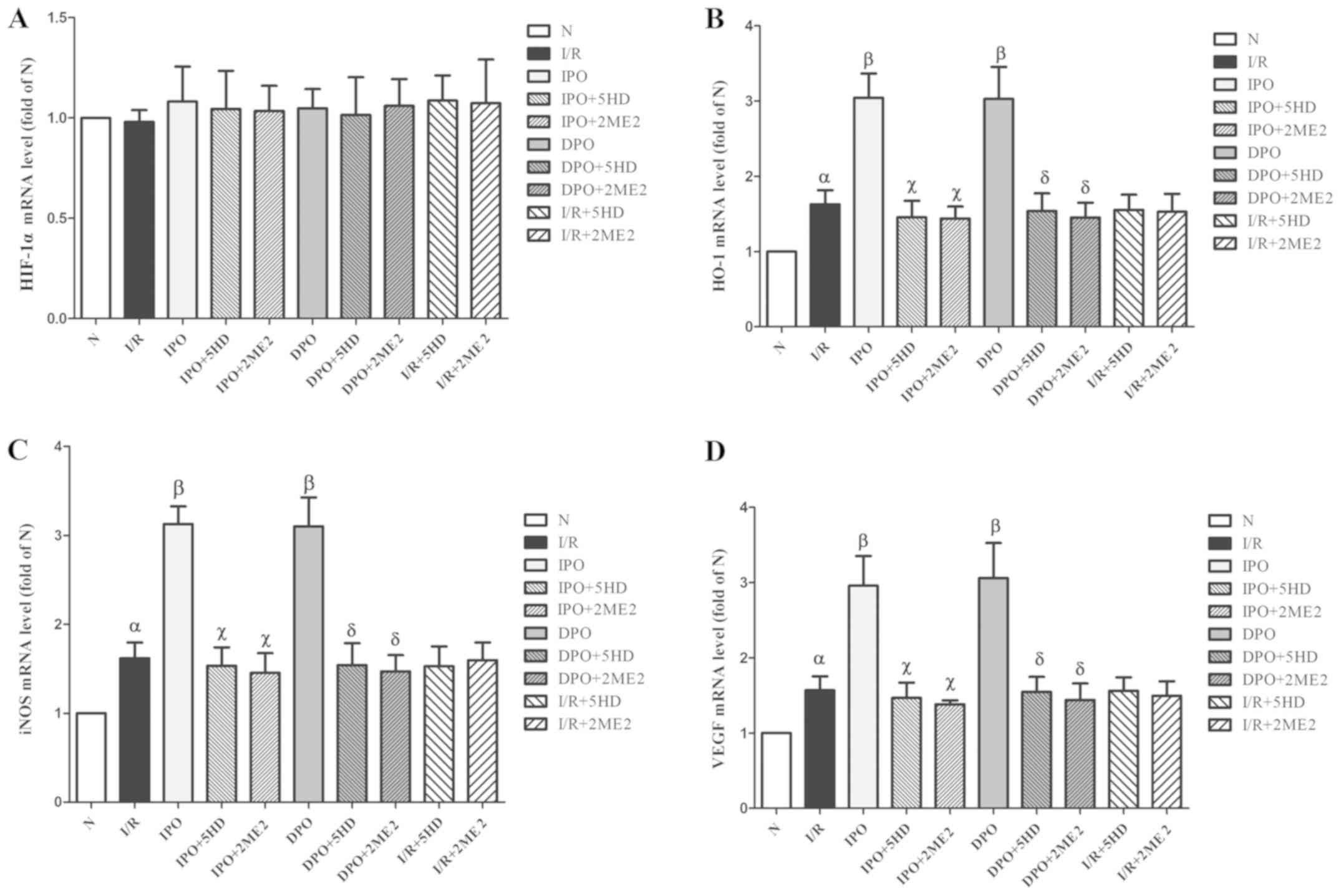 | Figure 4.Expression of HIF-1/hypoxic response
element-related genes in rats following myocardial
ischemia/diazoxide post-conditioning. (A) Expression level of
HIF-1α mRNA had no significant effect among the groups. The
expression levels of (B) HO-1, (C) iNOS and (D) VEGF mRNA
respectively. Compared with group I/R, group DPO/IPO significantly
increased the expression levels of HO-1, iNOS and VEGF mRNA; while
the blockers 5HD/2ME2 could reverse the above effects of DPO/IPO.
The results are expressed as the mean ± standard error of the mean,
n=6. αP<0.05 vs. group N; βP<0.05 vs.
group I/R; χP<0.05 vs. group IPO;
δP<0.05 vs. group DPO. IPO, ischemic
post-conditioning; DPO, diazoxide post-conditioning; I/R,
ischemia/reperfusion; 5HD, 5-hydroxydecanoic acid; 2ME2,
2-methoxyestradiol; HIF, hypoxia inducible factor; HO-1,
heme-oxygenase-1; iNOS, inducible nitric oxide; VEGF, vascular
endothelial growth factor. |
In groups I/R+5HD and I/R+2ME2, compared with group
I/R, there were no significant differences in the mRNA expression
of HO-1, iNOS and VEGF (P>0.05; Fig. 4B-D).
Expression of HIF-1/HRE related
proteins
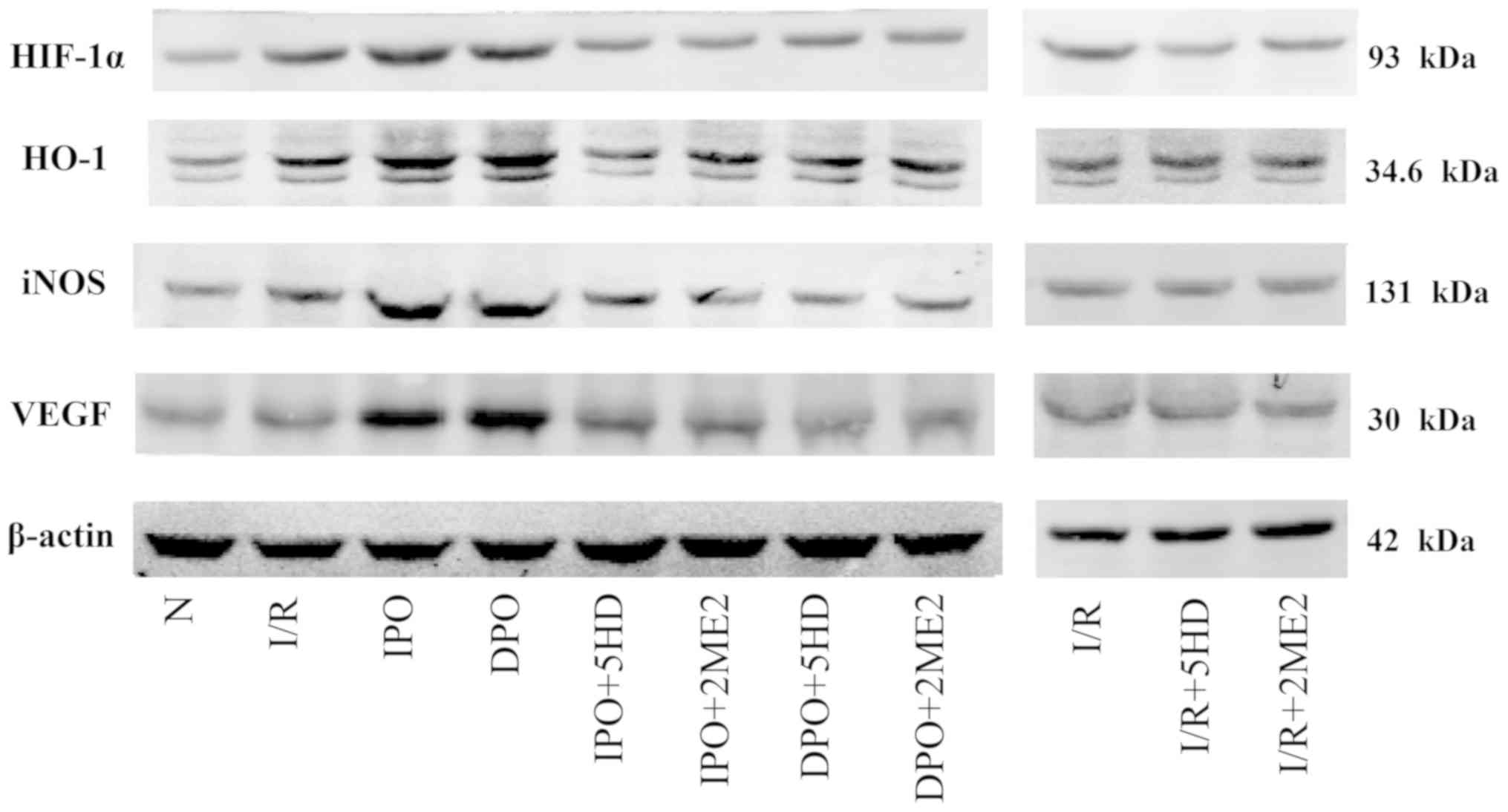
The expression of HIF-1α protein in group I/R was
significantly increased compared with group N (P<0.05; Figs. 5 and 6A). The expression levels of HIF-1, HO-1,
iNOS and VEGF in group IPO and DPO were significantly increased
compared with those in group I/R (P<0.05; Figs. 5 and 6B-D). In groups IPO+5HD and IPO+2ME2, the
expression levels (HIF-1, HO-1, iNOS and VEGF) were significantly
decreased compared with those in group IPO (P<0.05; Figs. 5 and 6). In groups DPO+5HD and DPO+2ME2, the
expression levels (HIF-1, HO-1, iNOS and VEGF) were significantly
decreased compared with those in group DPO (P<0.05; Figs. 5 and 6).
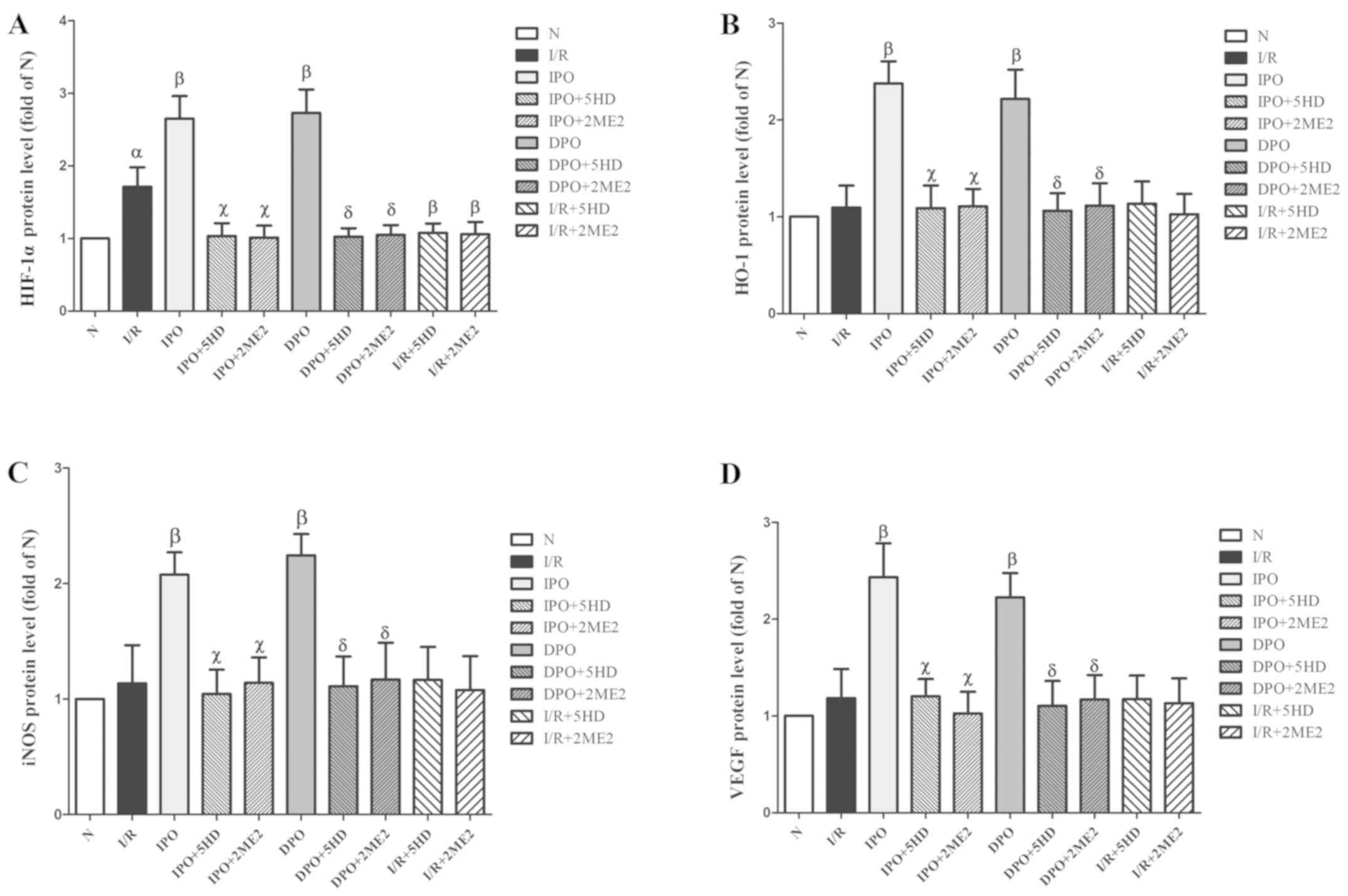 | Figure 6.Expression of HIF-1/hypoxic response
element-related proteins in rats following myocardial
ischemia/diazoxide post-conditioning. The expression of (A) HIF-1α,
(B) HO-1, (C) iNOS and (D) VEGF protein respectively. Compared with
group I/R, group DPO/IPO significantly increased the expression
levels of each protein; while the blockers 5HD/2ME2 could reverse
the above effects of DPO/IPO. The results are expressed as the mean
± standard error of the mean, n=6. αP<0.05 vs. group
N; βP<0.05 vs. group I/R; χP<0.05 vs.
group IPO; δP<0.05 vs. group DPO. IPO, ischemic
post-conditioning; DPO, diazoxide post-conditioning; I/R,
ischemia/reperfusion; 5HD, 5-hydroxydecanoic acid; 2ME2,
2-methoxyestradiol; HIF, hypoxia inducible factor; HO-1,
heme-oxygenase-1; iNOS, inducible nitric oxide; VEGF, vascular
endothelial growth factor. |
In groups I/R+5HD and I/R+2ME2, the protein
expression of HO-1, iNOS and VEGF was not significantly different
from that in group I/R (P>0.05; Figs. 5 and 6B-D), however, the expression of HIF-1α
protein in groups I/R+5HD and I/R+2ME2 was decreased compared with
group I/R (P<0.05; Figs. 5 and
6A).
Discussion
In the present study, the rat heart perfusion model
was established with the aid of the Langendorff experimental
device. The mechanism of action of IPO/DPO on MIRI was investigated
using cardioplegia to simulate cardioversion after cardiopulmonary
bypass.
In the present experiment, it was observed that
compared with group N, the infarct size of group I/R and the
mitochondrial Flameng score increased. Compared with group I/R,
both the infarct size and mitochondrial Flameng score decreased in
groups IPO and DPO. In addition, IPO/DPO increased the expression
(genes and proteins) of the HIF-1/HRE pathway (HO-1, iNOS and VEGF)
and HIF-1α protein. The myocardial protective effects of IPO/DPO
and their activation of the HIF-1/HRE pathway-related products
could be eliminated after the use of 5HD or 2ME2.
In addition, when groups I/R+5HD and I/R+2ME2 were
compared to group I/R, it was found that 5HD and 2ME2 had no effect
on MIRI. This was manifested by the size of the myocardial infarct
and the mitochondrial Flameng scores of myocardial cells. Although
HR, LVDP, LVEDP and +dp/dtmax directly reflect the state of cardiac
function, changes in cardiac function may take a certain period of
reperfusion to show. Therefore, there was no significant change in
cardiac functional indexes at the end of perfusion in this
experiment.
As an oxygen sensitive transcription factor, HIF can
make aerobic organisms adapt to anoxia. HIF-1 is one of the most
important factors in the HIF family and it is also an important
target for the study of myocardial protection (18–20).
HIF-1 is composed of α and β subunits. HIF-1α is HIF-1's regulatory
protein, which is very sensitive to changes in the oxygen
concentration and plays a key role in the regulation of HIF-1
function. HIF-1β is the basic expression protein and is not
regulated by oxygen (21). Under
normal oxygen supply, the proline and asparagine residues on HIF-1α
are hydroxylated. This promotes binding to the ligase complex pVHL-
ubiquitin E3 and rapid degradation. Under hypoxic conditions, the
hydroxylase activity is inhibited, thereby preventing the
degradation of HIF-1α. As a result, HIF-1α in the cytoplasm
increased and then transferred into the nucleus, combining with
HIF-1β. HIF in the nucleus can be combined with target gene
promoters related to the hypoxia response element (22). HIF-1 can regulate numerous genes,
including VEGF, HO-1 and iNOS. These factors are involved in a
number of physiological responses, such as anaerobic metabolism,
angiogenesis, erythrocyte production, cell proliferation and
apoptosis (23,24).
HIF-1 is an important factor in the hypoxia response
and the HIF-1/HRE signaling pathway also plays an important role in
myocardial protection (25,26).
Zhao et al (27) found that
IPO could alleviate MIRI by upregulating HIF-1α in
normal/hyperlipidemic rats. In this experiment, it was also
demonstrated that IPO could reduce MIRI by activating the HIF-1/HRE
signal pathway. IPO increased the expression (genes and proteins)
of the HIF-1/HRE signaling pathway (HO-1, iNOS and VEGF), and it
also increased the protein level of HIF-1α. After the use of 2ME2,
the myocardial protection of IPO disappeared and the infarct area
in group IPO+2ME2 was increased compared with in group IPO. The
myocardial mitochondrial Flameng score in group IPO+2ME2 was
increased compared with in group IPO. The myocardial structure of
group IPO+2ME2 was largely the same as that in the I/R group and
the structure of the myocardium was damaged, in that vacuoles had
formed and mitochondria were swollen and ruptured. In group
IPO+2ME2, the downstream expression related proteins and genes
(HO-1, iNOS and VEGF) of the HIF-1/HRE pathway and HIF-1α were
lower than in group IPO. This experiment also showed that DPO was
similar to IPO.
Therefore, the present study showed that both IPO
and DPO can reduce the area of myocardial infarction caused by
MIRI, and reduce the damage to the myocardial cell structure by
activating the HIF-1/HRE pathway.
IPO has been the basis of the theoretical system of
‘trigger-regulating medium-terminal effectors’. IPO induces the
release of trigger factors, mediates the signaling pathway, acts on
a variety of effectors and exerts protective effects on cardiac
myocytes (28,29). A previous study showed that IPO
could activate the protein kinase C and reperfusion injury salvage
kinase pathways via the intracellular adenosine and NO
concentration, finally acting on mitochondrial permeability
transition pores and mitoKATP channels to play a role in
myocardial protection (30). DPO
can also open mitoKATP channels to reduce MIRI and the
blocking of the opening of mitoKATP channels can
eliminate myocardial protection (31). Therefore, as a result,
mitoKATP channels may play a key role in mitigating
MIRI.
As an eight-polymer channel located in the
mitochondrial inner membrane of the cell, the mitoKATP
channel allows inward access to potassium ions. The opening of the
mitoKATP channels leads to the influx of potassium and
the efflux of hydrogen, further alkalifying the mitochondrial
matrix. Mitochondrial matrix alkalinization can cause the
respiratory chain to produce ROS (32). ROS can regulate a variety of
signaling factors, which includes nuclear transcription factor
HIF-1 (33). In the present study,
it was also shown that both IPO and DPO could alleviate MIRI by
activating the HIF-1/HRE pathway. Therefore, it could be speculated
that the activation of the HIF-1/HRE pathway may be related to the
opening of mitoKATP channels in IPO/DPO.
Furthermore, the effects of 5HD (mitoKATP
channel specific inhibitor) on MIRI and the expression of HIF-1/HRE
pathway components were observed in the IPO/DPO groups. IPO/DPO
reduced the myocardial infarct area and mitigated myocardial
mitochondrial damage, increasing the expression of HIF-1/HRE
pathway components (HO-1, iNOS and VEGF) and HIF-1α protein. After
the use of 5HD, the above myocardial protective effect of IPO/DPO
disappeared and the expression (genes and proteins) of the
HIF-1/HRE pathway and HIF-1α protein decreased. It is suggested
that IPO/DPO can activate the HIF-1/HRE pathway to relieve MIRI by
opening the mitoKATP channels.
Based on the results of the present study, it is
possible to speculate that both IPO and DPO may open
mitoKATP channels and in turn activate the HIF-1/HRE
pathway to alleviate MIRI.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Science
Technology Fund Projects of Guizhou Province (grant nos. qian ke he
SY zi 2014 and 2188).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JL, WJZ, WC, YZ, HYW and TY participated in the
study design. JL, WC and WJZ performed the experiments. JL, WJZ,
WC, YZ, HYW and TY performed the data analysis. JL, WJZ and WC
wrote the manuscript. JL, WJZ, WC, YZ, HYW and TY read and approved
the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Ethics Committee on
Animal Laboratory of Zunyi Medical College. The use and processing
of animals was in accordance with the Guide for the Care and Use of
Laboratory Animals, published by the National Institute of Health
(NIH Publication 88.23, revised 1996).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jennings RB, Sommers HM, Smyth GA, Flack
HA and Linn H: Myocardial necrosis induced by temporary occlusion
of a coronary artery in the dog. Arch Pathol. 70:68–78.
1960.PubMed/NCBI
|
|
2
|
Hausenloy DJ, Boston-Griffiths E and
Yellon DM: Cardioprotection during cardiac surgery. Cardiovasc Res.
94:253–265. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Frankenreiter S, Groneberg D, Kuret A,
Krieg T, Ruth P, Friebe A and Lukowski R: Cardioprotection by
ischemic postconditioning and cyclic guanosine
monophosphate-elevating agents involves cardiomyocyte nitric
oxide-sensitive guanylyl cyclase. Cardiovasc Res. 114:822–829.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pachauri P, Garabadu D, Goyal A and
Upadhyay PK: Angiotensin (1–7) facilitates cardioprotection of
ischemic preconditioning on ischemia-reperfusion-challenged rat
heart. Mol Cell Biochem. 430:99–113. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hao YL, Fang HC, Zhao HL, Li XL, Luo Y, Wu
BQ, Fu MJ, Liu W, Liang JJ and Chen XH: The role of microRNA-1
targeting of MAPK3 in myocardial ischemia-reperfusion injury in
rats undergoing sevoflurane preconditioning via the PI3K/Akt
pathway. Am J Physiol Cell Physiol. 315:C380–C388. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang FW, Tong J, Yan YS, Chen QQ and Zhao
XP: ω-3 polyunsaturated fatty acid postconditioning protects the
isolated perfused rat heart from ischemia-reperfusion injury.
Cardiorenal Med. 8:173–182. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang YH, Zhang Y, Chen W, Wang Y, Cao S,
Yu T and Wang H: Pinacidil-postconditioning is equivalent to
ischemic postconditioning in defeating cardiac ischemia-reperfusion
injury in rat. Eur J Pharmacol. 780:26–32. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Testai L, Marino A, Piano I, Brancaleone
V, Tomita K, Di Cesare Mannelli L, Martelli A, Citi V, Breschi MC,
Levi R, et al: The novel H2S-donor 4-carboxyphenyl
isothiocyanate promotes cardioprotective effects against
ischemia/reperfusion injury through activation of
mitoKATP channels and reduction of oxidative stress.
Pharmacol Res. 113:290–299. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hnatiuk AP, Ong SG, Olea FD, Locatelli P,
Riegler J, Lee WH, Jen CH, De Lorenzi A, Giménez CS, Laguens R, et
al: Allogeneic mesenchymal stromal cells overexpressing mutant
human hypoxia-inducible factor 1-α (HIF1-α) in an ovine model of
acute myocardial infarction. J Am Heart Assoc. 5:e0037142016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao HX, Wang XL, Wang YH, Wu Y, Li XY, Lv
XP, Zhao ZQ, Zhao RR and Liu HR: Attenuation of myocardial injury
by postconditioning: Role of hypoxia inducible factor-1alpha. Basic
Res Cardiol. 105:109–118. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Si J, Wang N, Wang H, Xie J, Yang J, Yi H,
Shi Z, Ma J, Wang W, Yang L, et al: HIF-1α signaling activation by
post-ischemia treatment with astragaloside IV attenuates myocardial
ischemia-reperfusion injury. PLoS One. 9:e1078322014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jin C, Wu J, Watanabe M, Okada T and
Iesaki T: Mitochondrial K+ channels are involved in
ischemic postconditioning in rat hearts. J Physiol Sci. 62:325–332.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Penna C, Perrelli MG, Tullio F, Angotti C,
Camporeale A, Poli V and Pagliaro P: Diazoxide postconditioning
induces mitochondrial protein S-nitrosylation and a redox-sensitive
mitochondrial phosphorylation/translocation of RISK elements: No
role for SAFE. Basic Res Cardiol. 108:3712013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Flagg TP, Enkvetchakul D, Koster JC and
Nichols CG: Muscle KATP channels: Recent insights to energy sensing
and myoprotection. Physiol Rev. 90:799–829. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Olejnickova V, Novakova M and Provaznik I:
Isolated heart models: Cardiovascular system studies and
technological advances. Med Biol Eng Comput. 53:669–678. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Flameng W, Borgers M, Daenen W and
Stalpaert G: Ultrastructural and cytochemical correlates of
myocardial protection by cardiac hypothermia in man. J Thorac
Cardiovasc Surg. 79:413–424. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang L, Xie P, Wu J, Yu J, Yu T, Wang H,
Wang J, Xia Z and Zheng H: Sevoflurane postconditioning improves
myocardial mitochondrial respiratory function and reduces
myocardial ischemia-reperfusion injury by up-regulating HIF-1. Am J
Transl Res. 8:4415–4424. 2016.PubMed/NCBI
|
|
19
|
Eckle T, Kohler D, Lehmann R, El KK and
Eltzschig HK: Hypoxia-inducible factor-1 is central to
cardioprotection: A new paradigm for ischemic preconditioning.
Circulation. 118:166–175. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ong SG and Hausenloy DJ: Hypoxia-inducible
factor as a therapeutic target for cardioprotection. Pharmacol
Ther. 136:69–81. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xie J, Liao Y, Yang L, Wu J, Liu C, Xuan
W, Li M, Zhang L, Liu Y, Wu P and Bin J: Ultrasound molecular
imaging of angiogenesis induced by mutant forms of
hypoxia-inducible factor-1α. Cardiovasc Res. 92:256–266. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tian YM, Mole DR, Ratcliffe PJ and Gleadle
JM: Characterization of different isoforms of the HIF prolyl
hydroxylase PHD1 generated by alternative initiation. Biochem J.
397:179–186. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tekin D, Dursun AD and Xi L: Hypoxia
inducible factor 1 (HIF-1) and cardioprotection. Acta Pharmacol
Sin. 31:1085–1094. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zimna A and Kurpisz M: Hypoxia-inducible
factor-1 in physiological and pathophysiological angiogenesis:
Applications and therapies. Biomed Res Int. 2015:5494122015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Poynter JA, Manukyan MC, Wang Y, Brewster
BD, Herrmann JL, Weil BR, Abarbanell AM and Meldrum DR: Systemic
pretreatment with dimethyloxalylglycine increases myocardial HIF-1α
and VEGF production and improves functional recovery after acute
ischemia/reperfusion. Surgery. 150:278–283. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li X, Zhao H, Wu Y, Zhang S, Zhao X, Zhang
Y, Wang J, Wang J and Liu H: Up-regulation of hypoxia-inducible
factor-1alpha enhanced the cardioprotective effects of ischemic
postconditioning in hyperlipidemic rats. Acta Biochim Biophys Sin
(Shanghai). 46:112–118. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao H, Wang Y, Wu Y, Li X, Yang G, Ma X,
Zhao R and Liu H: Hyperlipidemia does not prevent the
cardioprotection by postconditioning against myocardial
ischemia/reperfusion injury and the involvement of hypoxia
inducible factor-1alpha upregulation. Acta Biochim Biophys Sin
(Shanghai). 41:745–753. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Granfeldt A, Jiang R, Wang NP, Mykytenko
J, Eldaif S, Deneve J, Zhao ZQ, Guyton RA, Tønnesen E and
Vinten-Johansen J: Neutrophil inhibition contributes to
cardioprotection by postconditioning. Acta Anaesthesiol Scand.
56:48–56. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang ZH, Liu JL, Wu L, Yu Z and Yang HT:
Concentration-dependent wrestling between detrimental and
protective effects of H2O2 during myocardial
ischemia/reperfusion. Cell Death Dis. 5:e12972014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ferdinandy P, Schulz R and Baxter GF:
Interaction of cardiovascular risk factors with myocardial
ischemia/reperfusion injury, preconditioning, and postconditioning.
Pharmacol Rev. 59:418–458. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ichinomiya T, Cho S, Higashijima U,
Matsumoto S, Maekawa T and Sumikawa K: High-dose fasudil preserves
postconditioning against myocardial infarction under hyperglycemia
in rats: Role of mitochondrial KATP channels. Cardiovasc Diabetol.
11:282012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Andrukhiv A, Costa AD, West IC and Garlid
KD: Opening mitoKATP increases superoxide generation from complex I
of the electron transport chain. Am J Physiol Heart Circ Physiol.
291:H2067–H2074. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Koshikawa N, Hayashi J, Nakagawara A and
Takenaga K: Reactive oxygen species-generating mitochondrial DNA
mutation up-regulates hypoxia-inducible factor-1alpha gene
transcription via phosphatidylinositol 3-kinase-Akt/protein kinase
C/histone deacetylase pathway. J Biol Chem. 284:33185–33194. 2009.
View Article : Google Scholar : PubMed/NCBI
|