Introduction
Diabetes mellitus is a complex metabolic syndrome,
which significantly affects systemic and cerebral vasculature
(1). Chronic and uncontrolled
diabetes mellitus is characterized by a persistent elevation in
blood glucose, which is in association with a number of long-term
complications, including ischemic stroke (1). Diabetes is an independent risk factor
for ischemic stroke (2). Diabetes
exacerbates cerebral ischemia injury in experimental and clinical
stroke subjects by accelerating neuronal damage and increasing
infarct volume (3–5). Patients with diabetes exhibit double
the risk of ischemic stroke compared with people without diabetes,
after correction for other risks, and these individuals are
predicted to exhibit increased morbidity and mortality (6,7).
However, the cellular and molecular mechanisms by which
hyperglycemia is associated with ischemic brain damage have not
been fully determined.
Oxidative stress serves a pivotal role in the
development of microvascular and macrovascular diabetes
complications (8). The
overproduction of reactive oxygen species (ROS), which is induced
by hyperglycemia, is a mediator of tissue damage that occurs during
diabetes, and can lead to cerebral dysfunction (8,9). The
brain is vulnerable to radical-mediated attack due to its limited
antioxidant defenses (10). When
the redox balance is impaired, free radicals and oxidative
stress-associated mechanisms can cause cell injury and necrosis
(11). During periods of oxidative
stress, Akt/mTOR pathways are closely integrated and have been
revealed to directly determine cell fate (12–14).
Research has demonstrated that the Akt/mTOR signaling cascade
serves an important role in the onset and progression of cerebral
ischemia injury (13). A number of
agents that increase the phosphorylation levels of Akt and mTOR
have been demonstrated to reduce brain injury in stroke models
(15,16). A previous study has also indicated
that lentiviral-mediated overexpression of cAkt can protect against
stroke-induced neuronal injury in vivo and in vitro,
and mTOR inhibition with rapamycin can block these protective
effects (17).
Excitotoxicity, which is induced by the
overactivation of glutamate, has been identified as a key factor in
the pathogenesis of cerebral ischemia (18). GLT1, which is predominantly located
on astrocytes, is responsible for up to 90% of glutamate clearance
to maintain glutamate homeostasis in adult brain tissue (19). However, the downregulation or
dysfunction of GLT1 following ischemia leads to the accumulation of
extracellular glutamate and neuronal death (20). It has been demonstrated that GLT1
knockdown exacerbates the neuronal death and neurological deficit
in rats with middle cerebral artery occlusion (MCAO) (21). Recent evidence has demonstrated
that mTOR is a downstream target of the PI3K/Akt pathway, which
regulates GLT1 expression (22).
mTOR complex1 (mTORC1) and mTOR complex2 (mTORC2) are associated
with GLT1 expression (23), and
oxidative stress and excitotoxic mechanisms have been suggested to
operate in tight conjunction to induce irreversible damage of brain
tissue (24). Chen et al
(25) proposed that
glutamate-mediated excitotoxicity with oxidative stress fulfill the
‘two-hit’ hypothesis that accelerates neurodegeneration. Therefore,
the current study hypothesized that oxidative stress causes the
downregulation of Akt/mTOR signaling, and mTOR participates in the
downregulation of GLT1, which can lead to further excitotoxicity,
and eventually exacerbate diabetic ischemic stroke.
Ginkgo biloba extract (GbE) is a
standardized mixture that is extracted from Ginkgo biloba
leaves, containing 22–27% Ginkgo flavone glycosides
(myricetin, quercetin, kaempferol and isorhamnetin) and 5–7%
terpene lactones (ginkgolide A, B, C and bilobalides) (26). GbE has been used as a
therapeutic agent for a number of cardiovascular and neurological
diseases (27,28). Although the exact mechanism is
unclear, an accumulation of evidence has demonstrated that
GbE exhibit a number of benefits, including improving
hemodynamics, inhibiting the platelet-activating factor, scavenging
ROS and relaxing vascular smooth muscles (29). These results demonstrate the
pharmacological use of GbE for the treatment of diabetic
ischemic stroke. Recent studies have demonstrated that GbE
protects against a number of diabetic complications, including
diabetic cataract (30), diabetic
nephropathy (31) and diabetic
cardiomyopathy (32). However, the
effect of GbE on diabetic ischemic stroke is yet to be
determined. Therefore, the present study was designed to evaluate
the protective effect and its possible mechanism of action in
diabetic rats with cerebral ischemia-reperfusion injury.
Materials and methods
Animals
Adult male Sprague-Dawley rats (8–10 weeks old;
180–220 g) were obtained from the Laboratory Animal Center of
Xuzhou Medical University, (license no. SCXK2007-2005; Xuzhou,
China), where an SPF level laboratory was founded, as authorized by
the Jiangsu province government. All animals were maintained at a
constant temperature of 25±2°C under a 12/12 h light/dark cycle.
Rats were allowed free access to food and water ad libitum.
Animal experiments were conducted in accordance to the principles
provided by National Institute of Health (NIH) Guideline for the
Care and use of Laboratory Animals. The approval to proceed with
this experiment was issued by the Animal Ethics Committee of Xuzhou
Medical University which also conforms to the Guidelines for
Ethical Conduct in the Care and Use of Animals. All efforts were
made to prevent unavoidable pain and distress when the approved
endpoint is reached, no animal death occurred during this study.
Euthanasia should result in rapid loss of consciousness, followed
by respiratory and cardiac arrest and ultimate loss of all brain
function. Death was confirmed after euthanasia and prior to
disposal of the animal. Each rat was weighed weekly, while the
measurement of blood glucose was performed using a glucometer via
the tail vein (Nanjing Jianqiao Medical Device Co. Ltd.).
Drugs
GbE was used in the current study and is an
extract of dried Ginkgo biloba leaves. GbE was
obtained from Shaanxi Huike Botanical Development Co., Ltd.
(Purity, >98%; cat. no. HK20121201). For administration in
vivo, GbE was dissolved in 1% CMC-Na at concentrations of 10,
20 and 40 mg/ml. Streptozotocin (STZ; cat. no. 0130) was purchased
from Sigma-Aldrich; Merck KGaA. Nimodipine (cat. no. 120554), which
was used as a positive control, was purchased from Yabao
Pharmaceutical Group Co., Ltd. and suspended in 1% CMC-Na
solution.
Diabetic model
The rats that were fasted overnight were subjected
to a single intraperitoneal injection of 60 mg/kg STZ that was
freshly dissolved in 0.1 mol/l cold citrate buffer at pH 4.3.
Age-matched normal rats were injected with an equal volume of
citrate buffer alone. Blood glucose was measured a period of one
week after STZ injection. Rats with fasting blood glucose of ≥13.88
mmol/l were considered diabetic and included in the present study
(30).
Focal cerebral ischemic model and
grouping
Normal and diabetic rats were placed in the supine
position on a heated pad and had a body temperature of 36.5–37.5°C,
which was monitored using a rectal thermometer. After being
anesthetized with an intraperitoneal injection of 10% chloral
hydrate (300 mg/kg) (33,34), rats were subjected to 30 min of
middle cerebral artery (MCA) occlusion followed by 24 h of
reperfusion. No signs of pain and peritonitis were observed
following administration of 10% chloral hydrate. Sham operation and
transient middle cerebral artery occlusion were performed as
previously described (35). The
right common carotid artery (CCA), external carotid artery, and
internal carotid artery (ICA) were isolated. A nylon filament,
which was purchased from Beijing Sunbio Biotech Co., Ltd., was
subsequently introduced into the CCA lumen and gently advanced to
the ICA until a slight resistance was felt.
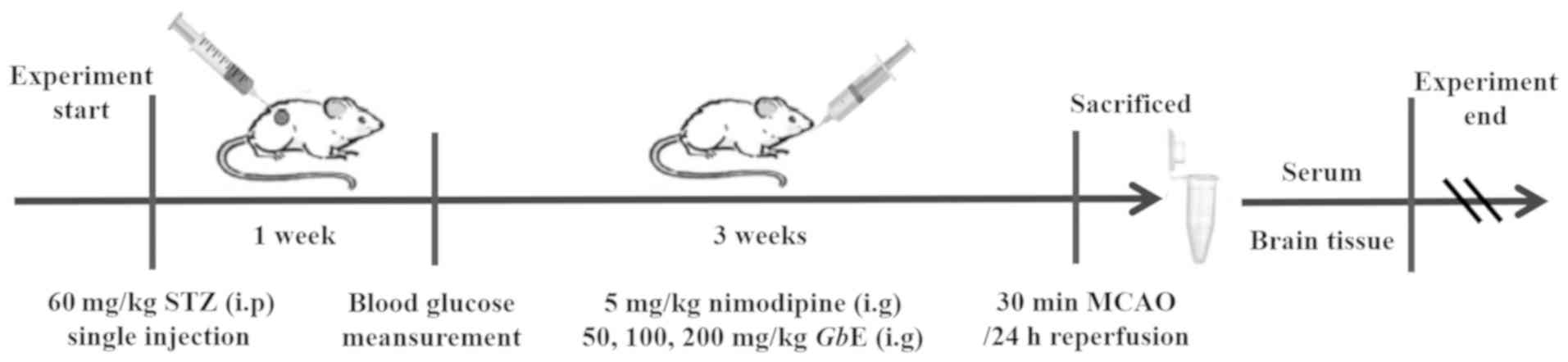
A total of eighty rats were randomly divided into
eight groups: The Con + sham group were nondiabetic and
sham-operated rats (Con + sham group; n=10); Con +
ischemia-reperfusion (I/R) rats were nondiabetic and received I/R
injury (Con + I/R group; n=10); diabetic and sham-operated rats
were in the STZ + sham group (STZ + sham group, n=10); diabetic
rats treated with ischemia-reperfusion injury were the STZ + I/R
group (STZ + I/R group; n=10). These aforementioned groups were
administered the same volume of 1% CMC solution for a period of
three weeks. Before ischemia-reperfusion injury, the diabetic rats
were administrated intragastrically with 50, 100 and 200 mg/kg
GbE for the GL group (low dose; n=10), the GM group
(moderate dose; n=10) and the GH group (high dose; n=10),
respectively. Diabetic rats that received nimodipine prior to
ischemia-reperfusion injury were the positive control group (Nimo
group; n=10; 5 mg/kg/day; intragastrically). All groups were
administered with the corresponding agents for three weeks before
induction of ischemia. All rats were anesthetized with
pentobarbital (45 mg/kg; intraperitoneal injection) and sacrificed
using cervical dislocation. Rat brains and blood samples were
collected. The experimental protocol is presented in Fig. 1.
Neurological deficit evaluation
Neurological function was evaluated 2 and 24 h after
reperfusion by an investigator blinded to the study groups: 0, no
deficit; 1, failure to extend right forelimb while the tail was
pulled; 2, spontaneous circling or walking to the contralateral
side; 3, stumble only when stimulated with a depressed level of
consciousness; 4, unresponsive to stimulation.
Infarct volume measurement
2,3,5-Triphenyltetrazolium chloride (TTC) staining
was performed according to previous descriptions (36) for the evaluation of the infarct
volume in experimental ischemic stroke. TTC stained the normal
cerebral areas deep red without any effect on the infarct tissue,
which enables identification of the healthy regions from the
infarcted areas. A total of 5 rats from each group were used for
infarct volume measurement (n=5 per group). Rats were euthanized
and brains were removed immediately. Brain samples were placed in a
brain matrix and sliced into 2 mm sections. The slices were
incubated in a 2% solution of TTC (cat. no. 129K1867V;
Sigma-Aldrich; Merck KGaA) at 37°C for 30 min, then fixed in a 4%
buffered paraformaldehyde solution and scanned using a scanner
(EPSON Perfection V33). The infarct area and the hemisphere area of
each section were traced and quantified using ImageJ software
(National Institutes of Health) and expressed as the percent of
infarct area in the whole brain.
S100B measurement
The amount of serum S100B protein was detected using
a commercially available ELISA kit (cat. no. 1302271; Shanghai
Bio-Tech Co., Ltd.), according to the manufacturer's protocol, and
expressed as ng/ml.
Measurement of malondialdehyde
level
The right striatum (50 mg) was homogenized with 450
µl 0.9% NaCl and centrifuged at 4°C at 12,000 × g for 15 min. A
total of 20 µl 6 mol/l NaOH was then added to 100 µl supernatant in
an Eppendorf tube and the sample was incubated in a water bath at
60°C for 30 min. The hydrolyzed sample was acidified with 50 µl 35%
(v/v) perchloric acid. The resulting suspension was
then mixed on a vortex for 30 sec and centrifuged at 12,000 × g for
10 min. A total of 200 µl top clear supernatant was transferred to
a 1.5 ml Eppendorf tube. The resultant supernatant was mixed with
20 µl 2,4-dinitrophenylhydrazine solution (5 mmol/l in 2 mol/l HCl;
pH=0.09) and incubated at room temperature for 30 min. After
derivatization, samples were filtered through a 0.22 µm filter.
Aliquots of 50 µl were injected into a HPLC system (37), in which an Agilent Zorbax SB-C18
column (250×4.6 mm; Agilent Technologies, Inc.) was used. The
mobile phase was acetonitrile-distilled water (38:62,
v/v) containing 0.2% (v/v) acetic acid
at a flow rate of 1.0 ml/min, and the wavelength of the UV detector
was set at 310 nm. The level of striatum malondialdehyde (MDA) was
expressed as µmol/g protein, and protein concentration was
determined using a bicinchoninic acid (BCA) assay.
Measurement of glutathione (GSH)
content
The level of GSH was measured as previously
described by Liu et al (38). The compound 3-carboxy-4-nitrophenyl
disulfide can react with sulfhydryl compounds (including GSH) and
form a yellow compound with a strong absorption at 420 nm. The
measurement of GSH was performed using a commercial kit (cat. no.
A006-1; Nanjing Jiancheng Bioengineering Institute), and the level
of striatum GSH was expressed as mg/g protein.
Superoxide dismutase (SOD) activity
assay
SOD is an important antioxidative enzyme, and, in
the current study, its activity was determined according to the
method of Sun et al (39).
This method uses the inhibition of nitroblue tetrazolium reduction
by the xanthine and xanthine oxidase system as a superoxide
generator. SOD activity was subsequently measured at 550 nm by the
degree of inhibition using a commercial kit (cat. no. A001-1;
Nanjing Jiancheng Bioengineering Institute). A total of one unit of
enzyme was defined as the amount of enzyme required at an
inhibitory rate of 50%. The activity of SOD was expressed as
units/mg protein.
Western blot analysis
A total of 5 rats from each group were used for
western blot analysis (n=5 per group). After weighing the rats, the
right hippocampus were dissected and homogenized using a sonicator
with six-fold volumes (w/v) of 50 mmol/l Tris buffer
(pH=7.4) containing 150 mmol/l NaCl, 1 mmol/l EDTA, 1 mmol/l PMSF,
1 mmol/l Na3VO4, 2 mmol/l DTT and 50 mmol/l
NaF, in an ice-cold bath. Samples were left at 4°C for at least 30
min, the homogenates were then centrifuged at 4°C at 10,000 × g for
15 min and the supernatant was collected and denatured in SDS. The
protein concentration in the supernatant was determined using a BCA
protein assay kit (Thermo Fisher Scientific, Inc.).
The same amount of protein (80 µg) was
electrophoresed on 8% SDS-PAGE and transferred to a PVDF membrane.
The membranes were incubated overnight at 4°C with primary
antibodies, including Akt Rabbit monoclonal antibody (1:1,000; cat.
no. 4691; Cell Signaling Technology, Inc.), p-Akt (Ser473) Rabbit
monoclonal antibody (1:1,000; cat. no. 4060; Cell Signaling
Technology, Inc.), mTOR Rabbit monoclonal antibody (1:1,000; cat.
no. 2983; Cell Signaling Technology, Inc.), p-mTOR (Ser2448) Rabbit
monoclonal antibody (1:1,000; cat. no. 5536; Cell Signaling
Technology, Inc.), GLT1 Rabbit polyclonal antibody (1:1,000; cat.
no. ab106289; Abcam), and β-actin Rabbit polyclonal antibody
(1:1,000; cat. no. AP0060; Bioworld Technology, Inc.),
respectively. The membranes were washed and incubated with alkaline
phosphatase-conjugated IgG (1:10,000; cat. no. E030220-02; EarthOx
Life Sciences) at room temperature for 2 h before being exposed to
BCIP/NBT alkaline phosphatase color developing reagent (cat. no.
C3206; Beyotime Institute of Biotechnology) for 15 min. Western
blot density was measured using Image J software (Rawak Software,
Inc.) and normalized using β-actin as an internal control.
Statistical analysis
All data in the different experimental groups were
expressed as the mean ± SD. Data were analyzed using GraphPad Prism
(version 5.0; GraphPad Software, Inc.). A comparison between groups
was conducted using one-way ANOVA, followed Tukey's multiple
comparisons tests. P<0.05 and P<0.01 were considered to
indicate a statistically significant difference.
Results
Effect of GbE on body weights and
fasting blood glucose
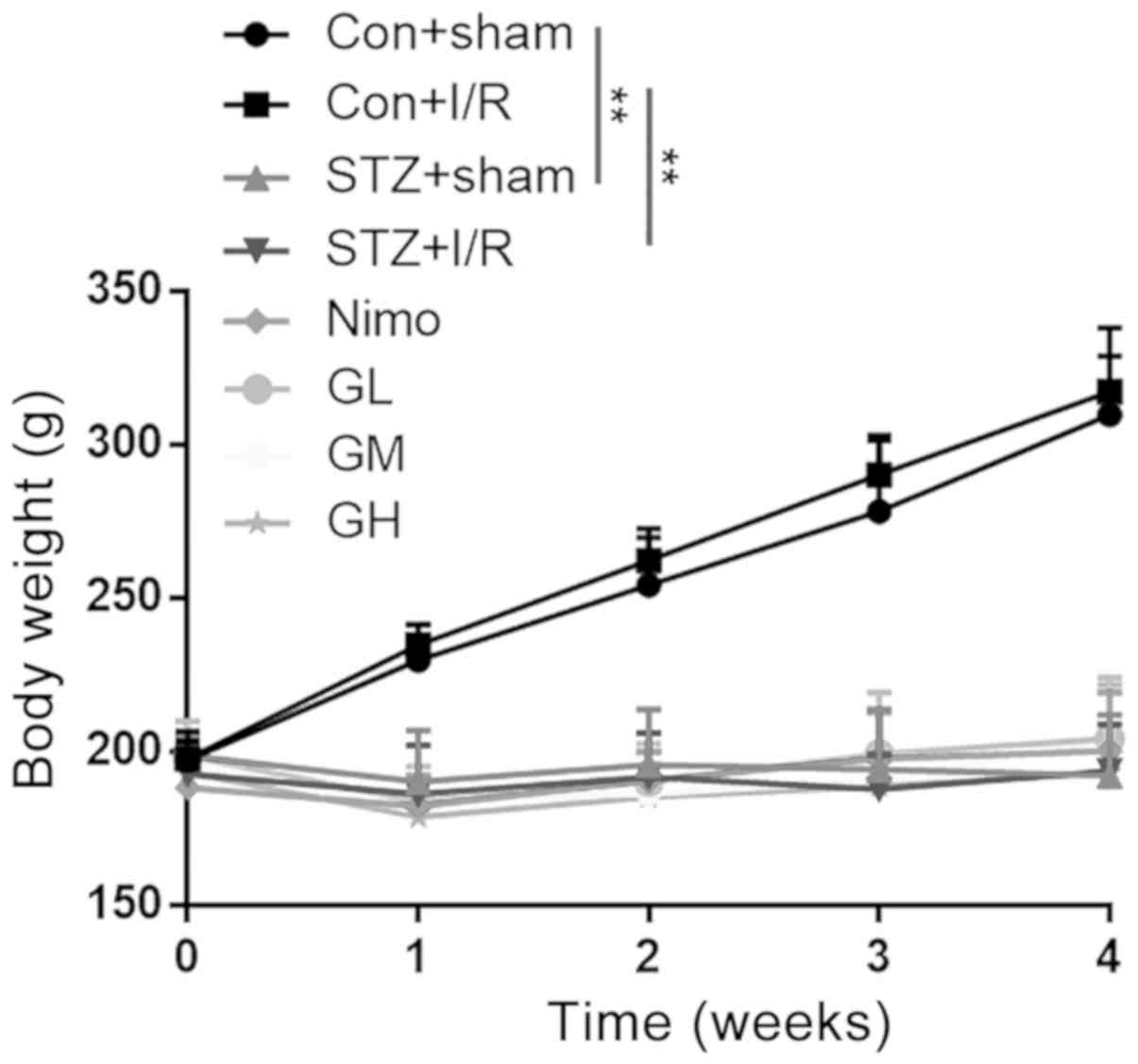
The body weights of rats were measured after STZ
injection (day 0) and on days 7, 14, 21 and 28. As presented in
Fig. 2, the original body weights
of eight groups were ~200 g. After STZ injection, the body weights
of diabetic rats (STZ + sham group and STZ + I/R group) remained
unchanged, whereas a marked increase was observed in nondiabetic
rats (Con + sham group and Con + I/R group) during the 4
consecutive weeks (P<0.01). GbE (50, 100 and 200
mg/kg/day) was administered intragastrically once per day from day
7 to day 28, and there were no significant differences among the
groups at each time point.
Fasting blood glucose levels were measured on days 7
(after a week of STZ injection) and 28 (after 3 weeks of
consecutive administration). On days 7 and 28, the fasting blood
glucose levels in diabetic rats was significantly increased
compared with nondiabetic rats (P<0.01; Table I). No significant differences in
fasting blood glucose levels were observed in the three GbE
groups and the Nimo group.
 | Table I.Effect of GbE on the fasting
blood glucose of STZ-induced diabetic rats subjected to 30 min
MCAO/24 h reperfusion. |
Table I.
Effect of GbE on the fasting
blood glucose of STZ-induced diabetic rats subjected to 30 min
MCAO/24 h reperfusion.
|
| Fasting blood
glucose (mmol/l) |
|---|
|
|
|
|---|
| Groups | Day 7 | Days 28 |
|---|
| Con + sham | 5.75±1.28 | 4.99±0.49 |
| Con + I/R | 5.89±1.48 | 5.16±0.59 |
| STZ + sham |
22.38±2.21a |
26.90±3.44a |
| STZ + I/R |
22.51±4.32b |
27.63±3.46b |
| Nimo | 19.42±1.87 | 24.44±6.81 |
| GL | 21.03±4.42 | 26.64±3.90 |
| GM | 21.22±4.74 | 27.85±5.39 |
| GH | 21.33±5.41 | 24.41±5.42 |
Effect of GbE on neurological
deficits
Neurological deficits were evaluated 2 and 24 h
after reperfusion. Compared with the 2 h time point after
reperfusion, I/R injury in nondiabetic rats (Con + I/R group) had
severe to mild neurological deficits (P<0.01), whereas I/R
injury in diabetic rats (STZ + I/R group) resulted in severe to
very severe neurological deficits 24 h after reperfusion
(P<0.01), and sham-operated animals did not exhibit any deficits
(Table II). The Nimo and three
GbE dose groups had significantly improved neurological
scores at 24 h of reperfusion (P<0.01; Table II).
| Table II.Effect of GbE on the
behavioral scores of neurological function of rats 2 and 24 h after
reperfusion. |
Table II.
Effect of GbE on the
behavioral scores of neurological function of rats 2 and 24 h after
reperfusion.
|
| Neurological scores
(mean ± SD) |
|
|
|---|
|
|
|
|
|
|---|
| Groups | 2 h after
reperfusion | 24 h after
reperfusion | n | Neurological
deficits |
|---|
| Con + sham | 0 | 0 | 10 | None |
| Con + I/R | 1.92±0.57 |
0.30±0.67a | 10 | Severe→mild |
| STZ + sham | 0 | 0 | 10 | None |
| STZ+I/R | 2.00±0.67 |
2.90±0.57a | 10 | Severe→very
severe |
| Nimo | 2.10±0.57 |
1.00±0.47a | 10 | Severe→mild |
| GL | 2.00±0.47 |
1.10±0.57a | 10 | Severe→mild |
| GM | 2.10±0.74 |
0.80±0.63a | 10 | Severe→mild |
| GH | 2.00±0.67 |
0.70±0.48a | 10 | Severe→mild |
Effect of GbE on cerebral infarct
volume
The effects of GbE on rat infarct volume were
investigated using TTC staining. No lesion was observed in
sham-operated groups. Con + I/R rats that were subjected to 30 min
MCAO followed by 24 h reperfusion presented smaller infarct volumes
of 9.80±1.48%. The STZ + I/R group exhibited markedly increased
infarct volume percentages (41.34±7.88%) compared with the Con +
I/R group (P<0.01; Fig. 3B).
Intermediate and high doses of GbE, and nimodipine markedly
reduced the infarct volume (P<0.01; Fig. 3B). However, the low dose of
GbE had little effect on it.
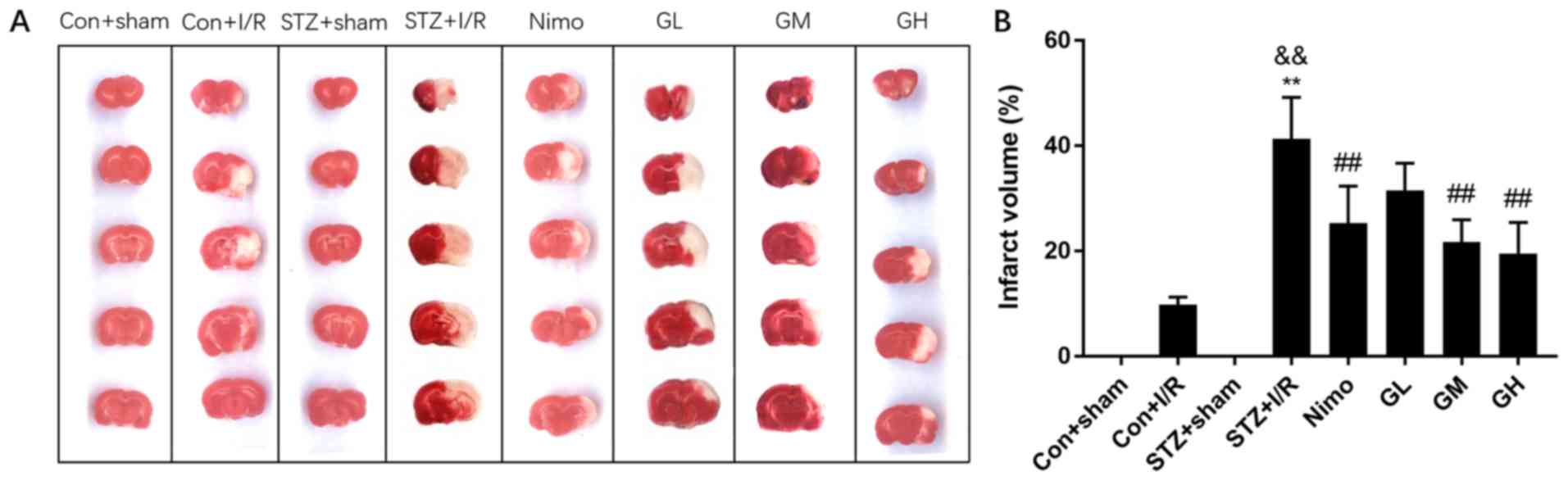 | Figure 3.Effect of GbE on the infarct
volume of STZ-induced diabetic rats subjected to 30 min MCAO/24 h
reperfusion. TTC stained brain slices of rats in different groups.
(A) The normal brain areas are red and the infarct areas are white.
(B) Infarct volume of rats in different groups. Data are expressed
as mean ± standard deviation (n=5). **P<0.01 vs. STZ + sham
group; &&P<0.01 vs. Con + I/R group;
##P<0.01 vs. STZ + I/R group. GbE, Ginkgo
biloba extract; STZ, streptozotocin; MCAO, middle cerebral
artery occlusion; TTC, 2,3,5-Triphenyltetrazolium chloride; Con,
control; I/R, ischemia-reperfusion; Nimo, nimodipine group; GL,
GbE low dose group; GM, GbE moderate dose group; GH,
GbE high dose group. |
Effect of GbE on S100B in serum of
rats
S100 calcium-binding protein B (S100B), which is a
biomarker of traumatic brain injury, is primarily expressed in the
central nervous system by astrocytes (40). Ischemia is associated with the
increased expression of S100B, which may be released from damaged
astrocytes (41). The level of
serum S100B is an indicator of brain injury following stroke
(42). Therefore, the
concentration of S100B was examined using an ELISA to investigate
the neuroprotective effect of GbE. As presented in Fig. 4, in diabetic rats, I/R injury
significantly increased the level of serum S100B (P<0.01).
STZ-induced diabetic increased S100B level (P<0.01) in STZ + I/R
group compared with the nondiabetic rats with I/R injury (Con + I/R
group). However, pretreatment with nimodipine and GbE
significantly decreased S100B compared with the STZ + I/R group
(P<0.01).
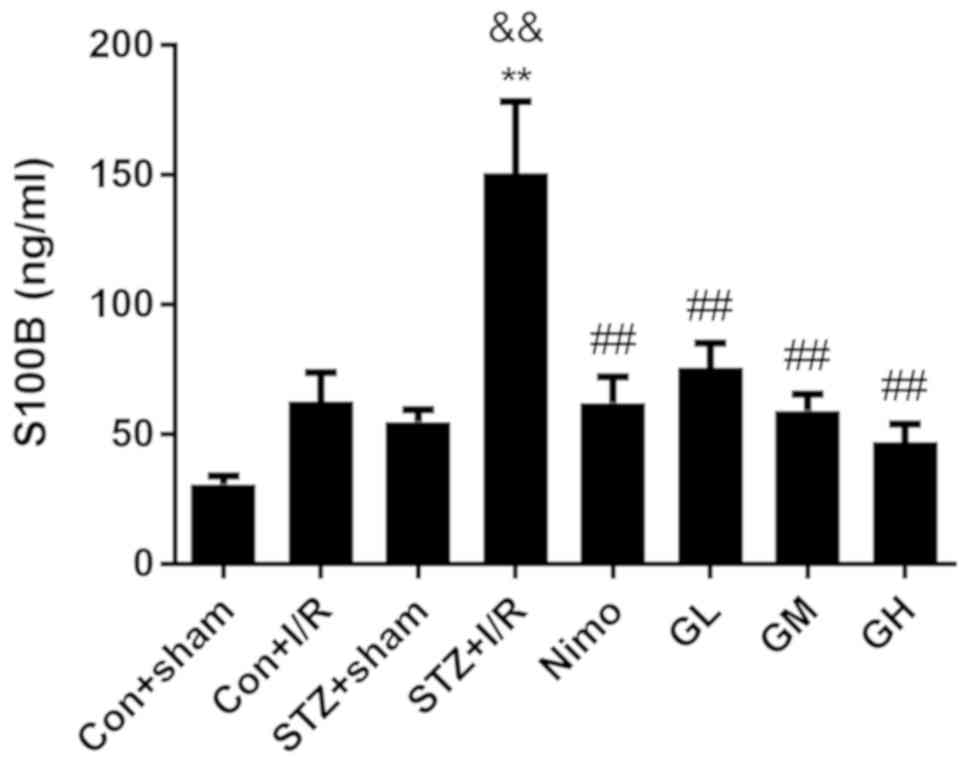 | Figure 4.Effect of GbE on the content
of serum S100B in diabetic rats subjected to 30 min MCAO/24 h
reperfusion. Data are expressed as mean ± standard deviation
(n=10). **P<0.01 vs. STZ + sham group,
&&P<0.01 vs. Con + I/R group,
##P<0.01 vs. STZ + I/R group. GbE, Ginkgo
biloba extract; STZ, streptozotocin; MCAO, middle cerebral
artery occlusion; Con, control; I/R, ischemia-reperfusion; Nimo,
nimodipine group; GL, GbE low dose group; GM, GbE
moderate dose group; GH, GbE high dose group. |
Increasing doses of GbE resulted in a stepped
decrease of infarct volume and S100B level. The moderate dose of
GbE (100 mg/kg, GM) was subsequently used for the following
experiments.
Effect of GbE on oxidative stress in
the rat striatum
One potential mechanism for diabetes and its
complications is oxidative stress (43). To investigate whether the
neuroprotective effect of GbE was associated with the
decrease in oxidative stress levels, three associated molecules
were examined, including MDA, GSH and SOD. As presented in Fig. 5, in nondiabetic rats, compared with
the Con + sham group, I/R injury significantly decreased GSH level
(P<0.05). In diabetic rats, I/R injury significantly caused
oxidative stress damage, and this was indicated by increased MDA
level (P<0.01), and decreased GSH (P<0.05) and decreased SOD
activity (P<0.05). Compared with nondiabetic rats with I/R
injury (Con + I/R group), STZ-induced diabetic rats exhibited
increased MDA level (P<0.01). However, GbE pretreatment
significantly suppressed MDA level (P<0.01), and inhibited the
decrease of GSH (P<0.05) and SOD (P<0.01).
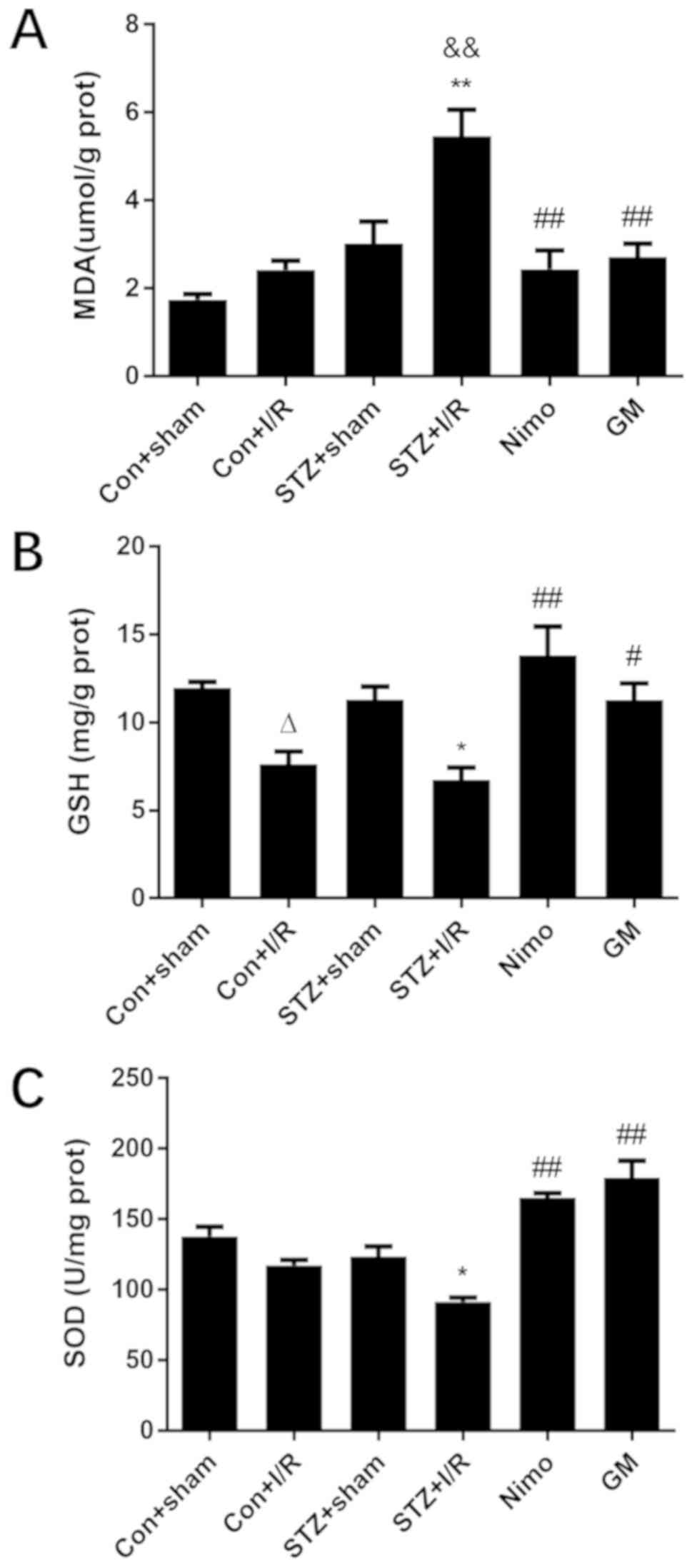 | Figure 5.Effect of GbE on oxidative
stress in the striatums of rats. (A) MDA, (B) GSH and (C) SOD were
determined. Data are expressed as mean ± standard deviation (n=5).
∆P<0.05 vs. Con + sham group; *P<0.05 or
**P<0.01 vs. STZ + sham group; &&P<0.01
vs. Con + I/R group; #P<0.05 or
##P<0.01 vs. STZ + I/R group. GbE, Ginkgo
biloba extract; MDA, malondialdehyde; GSH, glutathione; SOD,
superoxide dismutase; STZ, streptozotocin; Con, control; I/R,
ischemia-reperfusion; Nimo, nimodipine group; GM, GbE
moderate dose group. |
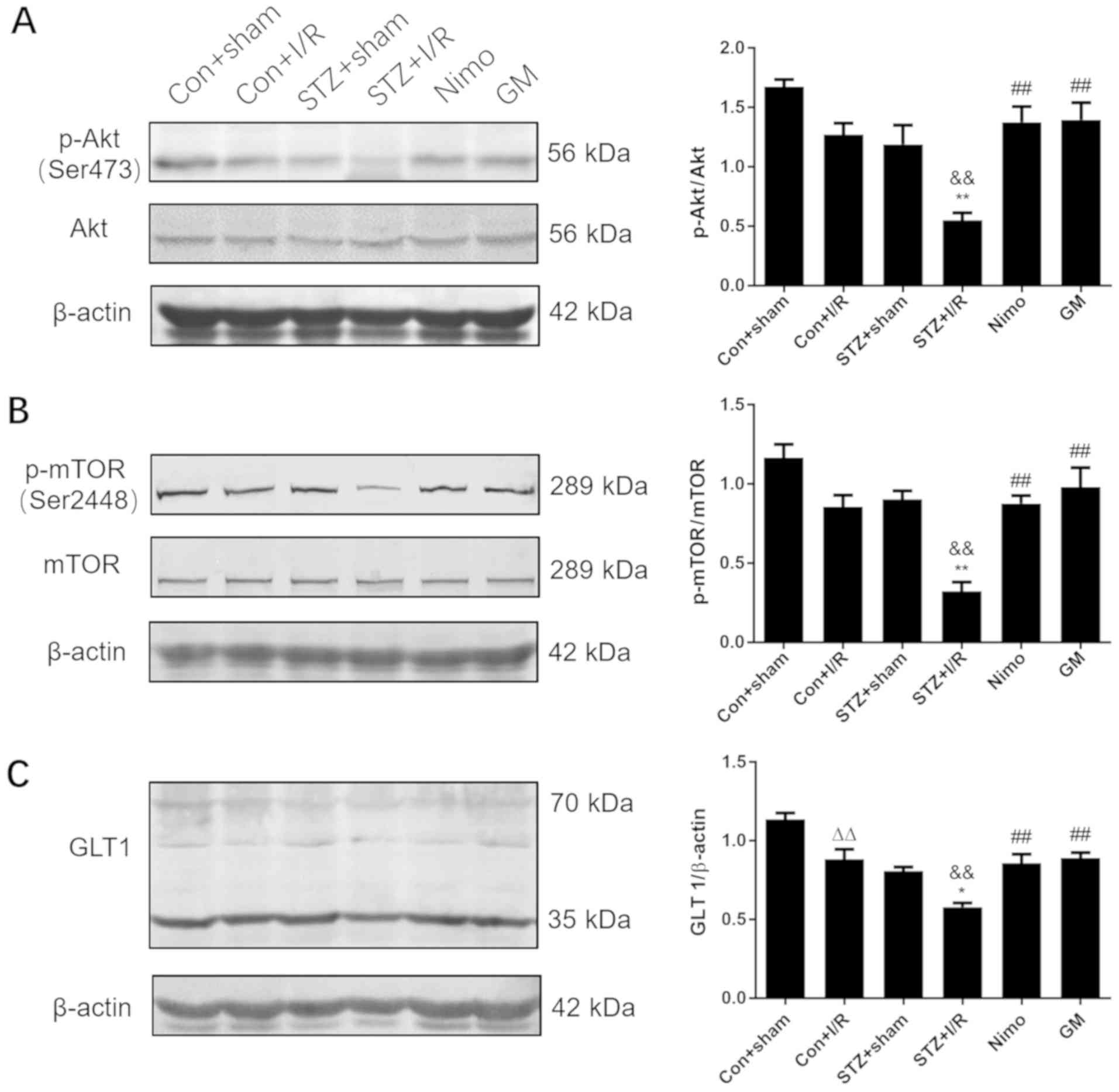
Effect of GbE on the quantities of
p-Akt and p-mTOR in the hippocampus of rats
It was previously revealed that GbE
pretreatment improved neurological deficits, reduced the infarct
volume and relieved oxidative stress following cerebral I/R injury
in diabetic rats. To elucidate the mechanism by which GbE
ameliorated neuronal damage, western blot analysis was used to
identify associated protein expression. The results indicated that
in diabetic rats, I/R injury decreased the expression of p-Akt
(P<0.01; Fig. 6A). Compared
with nondiabetic rats with I/R injury (Con + I/R group),
STZ-induced diabetic decreased p-Akt/Akt ratio (P<0.01) in the
STZ + I/R group. However, in the nimodipine and
GbE-pretreated groups, the ratio of p-Akt/Akt was
significantly increased compared with the STZ + I/R group
(P<0.01; Fig. 6A). Variation in
the p-mTOR/mTOR ratio was consistent with the change in the
p-Akt/Akt ratio (Fig. 6B).
GbE inhibits ischemia-induced
downregulation of GLT1 in diabetic rats subjected to 30 min MCAO/24
h reperfusion
A recent study has revealed that glutamate uptake
exhibits a protective function in hippocampal astrocytes (44). Therefore, in the current study, the
hippocampus was collected to elucidate the possible mechanism
underlying the neuroprotective effect of GbE against injury.
The results indicated that whether in nondiabetic or diabetic rats,
I/R injury decreased the expression of GLT1 (P<0.01 or
P<0.05; Fig. 6C). Compared with
nondiabetic rats with I/R injury (Con + I/R group), STZ-induced
diabetic rats exhibited decreased GLT1 expression (P<0.01) in
the STZ + I/R group. However, in the nimodipine and
GbE-pretreated groups, the expression of GLT1 was
significantly increased compared with the STZ + I/R group
(P<0.01; Fig. 6C).
Discussion
Ischemic stroke generally occurs in diabetic
patients with poor glycemic control (1). It has been well established that
patients with hyperglycemia are four to five times more likely to
suffer from a stroke compared with patients with normoglycemia,
with worse neurologic outcomes (5,45).
The present work demonstrated in vivo evidence of the
protective effect of GbE against cerebral ischemic injury in
diabetic rats. These results indicated that cerebral injury of 30
min MCAO/24 h reperfusion in STZ induced diabetic rats causes more
damage compared with nondiabetic rats. However, pretreatment with
GbE protected against cerebral I/R injury in diabetic rats,
which may be associated with the inhibition of oxidative stress,
the activation of Akt/mTOR signaling cascade and the upregulation
of GLT1 expression.
The current study indicated that a single injection
of STZ significantly prevented weight gain and increased fasting
blood glucose level compared with normal control rats. However,
GbE exhibited no effect on body weight or fasting blood
glucose level. Furthermore, it was demonstrated that GbE
exhibited a protective effect on cerebral ischemic injury in
diabetic rats, which was assessed by measuring the neurological
scores, infarct volume and serum S100B level. The current study
revealed that whether in nondiabetic or diabetic rats, deficits in
performance due to I/R injury, significantly reduced the
neurological deficit following 2 h of reperfusion. However, long
term reperfusion ameliorated injury of nondiabetic rats and
aggravated diabetic rats, which may be due to high glucose levels
preventing the repair of ischemic penumbra and resultant brain
tissue damage (46). GbE
pretreatment exhibited a markedly decreased neurological deficit.
The cerebral infarct volume of 24 h following reperfusion was
consistent with the results of neurological deficits. Furthermore,
it was revealed that the dose-dependent administration of
GbE significantly decreased serum S100B level. The mechanism
by which GbE protects against cerebral ischemic injury in
diabetic rats was subsequently assessed.
A large number of ROS are produced during cerebral
ischemia and excessively consume the endogenous antioxidative
proteins, leading to the changes in the expression and activity of
SOD and GSH (47). The decreased
activity of antioxidant enzymes reduces the ability of brain tissue
to scavenge ROS (48). MDA is the
final product of lipid peroxidation and indirectly reflects changes
in ROS content (49). The results
of the current study indicated that GbE enhanced antioxidant
enzyme activities and reduced lipid peroxidation.
The hippocampus is an essential brain region which
plays roles in memory forming, organizing, and storing. Brian
ischemia leads to movement, visual, sensory, and behavioral
disorders, especially aphasia and impaired spatial learning
(50,51). Neurons in the hippocampal CA1
regions are very sensitive, and they quickly react to brain I/R
(52,53). Studies have demonstrated that
hippocampal areas of the mouse brain are more vulnerable to neuron
death following an ischemic insult (54). Several researchers chose the
hippocampus to study Akt pathway in rat models of ischemic brain
damage (54–56). The phosphatidylinositol-3 (PI3)
kinase/Akt signal pathway enhances cell survival, proliferation and
differentiation (57). It has been
demonstrated that ROS regulates the PI3K/Akt pathway, which leads
to changes in a number of downstream signaling proteins and induces
a variety of pathophysiological responses (58,59).
A study performed by Wang et al (60) demonstrated that ROS overproduction,
which was induced by high glucose in astrocytes, can lead to
decreased cell viability and apoptosis. A number of studies have
indicated that oxidative stress-induced neuronal damage and death
following cerebral ischemia can be inhibited by activation of the
PI3K/Akt/mTOR signaling pathway (16). In the present study, the results
demonstrated that I/R injury significantly decreased the ratio of
p-Akt/Akt and p-mTOR/mTOR, and this was exacerbated by diabetes.
However, GbE pretreatment markedly reversed this decreased
expression by upregulating the ratio of p-Akt/Akt and p-mTOR/mTOR.
This result suggested that GbE, which is a free radical
scavenger, could attenuate I/R injury in diabetic rats by
activating the Akt/mTOR signal cascade.
Glutamate is the most abundant neurotransmitter in
the cerebral neural system (61).
GLT1, which is also known as EAAT2, is the predominant subtype that
performs the majority of glutamate reuptake (19). Extracellular glutamate
concentration is mediated primarily by the astrocytic glutamate
transporter GLT1. However, disruption of glutamate transporter
activity and expression can lead to excitotoxicity and is
implicated in ischemic events (20,21).
It has been demonstrated that Akt is a key regulator of GLT1
expression (23). Additionally,
mTOR, which is a downstream target of the PI3K/Akt pathway, is
associated with the regulation of GLT1 (22). Previous reports have demonstrated
that GLT1 knock-down exacerbates neuronal damage in ischemia rats
(21), whereas GLT1 upregulation
reduces cerebral ischemic injury in hyperglycemic rats (34). In the current study, I/R injury was
demonstrated to downregulate GLT1 expression in diabetic rats, and
GbE resulted in the upregulation of GLT1 expression,
suggesting that GbE might serve an important role in
protecting against I/R injury in diabetic rats via resistance to
glutamate excitotoxicity. The results of the present study are
supported by a study performed by Mdzinarishvili et al
(62) that demonstrated that
EGb761 reduces the release of glutamate in the brain of ischemic
mice by monitoring extracellular glutamate concentration, however
the underlying mechanism for this was not determined.
Previous studies have identified that the
neuron-dependent regulation of GLT 1 transcription requires NF-κB
and κB motif-binding phosphoprotein (23,63).
Therefore, the downstream transcription factor(s) require
identification in future studies. Additionally, nimodipine, which
is a positive control drug used in the present study, is a
Ca2+ channel blocker. The protective effect of
nimodipine is positive, which indicated that the experimental
design of the current study is feasible. Nimodipine worked as well
as GbE in the present study, however nimodipine can lead to
hypotension (64), while the
incidence of adverse reactions by GbE is low. Additionally,
GbE may also serve a role in lowering blood glucose
(65). As an effective adjuvant
drug, GbE exhibits a glycemic control value in the clinical
treatment of patients with T2DM (66).
In conclusion, the results of the current study
demonstrated that GbE pretreatment ameliorated neurological
deficit, reduced infarct volume, decreased S100B level, inhibited
oxidative stress, and upregulated the expression of Akt/mTOR and
GLT1 in diabetic rats with cerebral ischemia-reperfusion injury.
However, further studies using cell models are required to
determine the in vitro protective effect and to clarify the
accurate mechanism of the effect of GbE in cerebral
ischemia-reperfusion injury.
Acknowledgements
Not applicable.
Funding
The current study was supported by the Open
Foundation (grant no. KJS1107) from the Key Laboratory of
Anesthesiology and Discipline Construction Projects of Jiangsu
Province, the College Graduate Research and Innovation Projects in
Jiangsu Province (grant no. CXZZ_1000).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ML and SG designed the study. MY, ML, ZS, TM and XM
performed the experiments. MY and ML collected and analyzed the
experimental data. MY drafted the manuscript. All authors read and
approved the final manuscript.
Ethics approval and patient consent to
participate
All experimental and surgical procedures for animals
were strictly performed in accordance with the Guiding Principles
for Care and Use of Laboratory Animals of Xuzhou Medical
University. The present study was approved by the Ethics Committee
of Xuzhou Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Luitse MJ, Biessels GJ, Rutten GE and
Kappelle LJ: Diabetes, hyperglycaemia, and acute ischaemic stroke.
Lancet Neurol. 11:261–271. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Barrett-Connor E and Khaw KT: Diabetes
mellitus: An independent risk factor for stroke? Am J Epidemiol.
128:116–123. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Muranyi M, Fujioka M, He Q, Han A, Yong G,
Csiszar K and Li PA: Diabetes activates cell death pathway after
transient focal cerebral ischemia. Diabetes. 52:481–486. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rizk NN, Rafols J and Dunbar JC: Cerebral
ischemia induced apoptosis and necrosis in normal and diabetic
rats. Brain Res. 1053:1–9. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Els T, Klisch J, Orszagh M, Hetzel A,
Schulte-Mönting J, Schumacher M and Lucking CH: Hyperglycemia in
patients with focal cerebral ischemia after intravenous
thrombolysis: Influence on clinical outcome and infarct size.
Cerebrovasc Dis. 13:89–94. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Emerging Risk Factors Collaboration, ;
Sarwar N, Gao P, Seshasai SR, Gobin R, Kaptoge S, Di Angelantonio
E, Ingelsson E, Lawlor DA, Selvin E, et al: Diabetes mellitus,
fasting blood glucose concentration, and risk of vascular disease:
A collaborative meta-analysis of 102 prospective studies. Lancet.
9733:2215–2222. 2010.
|
|
7
|
Almdal T, Scharling H, Jensen JS and
Vestergaard H: The independent effect of type 2 diabetes mellitus
on ischemic heart disease, stroke and death: A population-based
study of 13,000 men and women with 20 years of follow-up. Arch
Intern Med. 164:1422–1426. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Giacco F and Brownlee M: Oxidative stress
and diabetic complications. Circ Res. 107:1058–1070. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brownlee M: Biochemistry and molecular
cell biology of diabetic complications. Nature. 6865:813–820. 2001.
View Article : Google Scholar
|
|
10
|
Adibhatla RM and Hatcher JF: Lipid
oxidation and peroxidation in CNS health and disease: from
molecular mechanisms to therapeutic opportunities. Antioxid Redox
Signal. 12:125–169. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ran Z, Zhang Y, Wen X and Ma J: Curcumin
inhibits high glucose-induced inflammatory injury in human retinal
pigment epithelial cells through the ROSPI3K/AKT/mTOR signaling
pathway. Mol Med Rep. 19:1024–1031. 2019.PubMed/NCBI
|
|
12
|
Pan Y, Wang N, Xia P, Wang E, Guo Q and Ye
Z: Inhibition of Rac1 ameliorates neuronal oxidative stress damage
via reducing Bcl-2/Rac1 complex formation in mitochondria through
PI3K/Akt/mTOR pathway. Exp Neurol. 300:149–166. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Maiese K, Chong ZZ, Wang S and Shang YC:
Oxidant stress and signal transduction in the nervous system with
the PI3-K, Akt and mTOR cascade. Int J Mol Sci. 13:13830–13866.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dong L, Zhou S, Yang X, Chen Q, He Y and
Huang W: Magnolol protects against oxidative stress-mediated neural
cell damage by modulating mitochondrial dysfunction and PI3K/Akt
signaling. J Mol Neurosci. 50:469–481. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Koh PO: Melatonin prevents ischemic brain
injury through activation of the mTOR/p70S6 kinase signaling
pathway. Neurosci Lett. 444:74–78. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yan BC, Wang J, Rui Y, Cao J, Xu P, Jiang
D, Zhu X, Won MH, Bo P and Su P: Neuroprotective effects of
gabapentin against cerebral ischemia reperfusion-induced neuronal
autophagic injury via regulation of the PI3K/Akt/mTOR signaling
pathways. J Neuropathol Exp Neurol. 78:157–171. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xie R, Cheng M, Li M, Xiong X, Daadi M,
Sapolsky RM and Zhao H: Akt isoforms differentially protect against
stroke-induced neuronal injury by regulating mTOR activities. J
Cereb Blood Flow Metab. 33:1875–1885. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tymianski M: Emerging mechanisms of
disrupted cellular signaling in brain ischemia. Nat Neurosci.
14:1369–1373. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Danbolt NC: Glutamate uptake. Prog
Neurobiol. 65:1–105. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Camacho A and Massieu L: Role of glutamate
transporters in the clearance and release of glutamate during
ischemia and its relation to neuronal death. Arch Med Res.
37:11–18. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rao VL, Dogan A, Todd KG, Bowen KK, Kim
BT, Rothstein JD and Dempsey RJ: Antisense knockdown of the glial
glutamate transporter GLT-1, but not the neuronal glutamate
transporter EAAC1, exacerbates transient focal cerebral
ischemia-induced neuronal damage in rat brain. J Neurosci.
21:1876–1883. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu X, Kihara T, Akaike A, Niidome T and
Sugimoto H: PI3K/Akt/mTOR signaling regulates glutamate transporter
1 in astrocytes. Biochem Biophys Res Commun. 393:514–518. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ji YF, Zhou L, Xie YJ, Xu SM, Zhu J, Teng
P, Shao CY, Wang Y, Luo JH and Shen Y: Upregulation of glutamate
transporter GLT-1 by mTOR-Akt-NF-κB cascade in astrocytic
oxygen-glucose deprivation. Glia. 61:1959–1975. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Trotti D, Danbolt NC and Volterra A:
Glutamate transporters are oxidant-vulnerable: A molecular link
between oxidative and excitotoxic neurodegeneration? Trends
Pharmacol Sci. 19:328–334. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen MJ, Ng JM, Peng ZF, Manikandan J, Yap
YW, Llanos RM, Beart PM and Cheung NS: Gene profiling identifies
commonalities in neuronal pathways in excitotoxicity: Evidence
favouring cell cycle re-activation in concert with oxidative
stress. Neurochem Int. 62:719–730. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chan PC, Xia Q and Fu PP: Ginkgo
biloba leave extract: Biological, medicinal, and toxicological
effects. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev.
25:211–244. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ross R: Atherosclerosis-an inflammatory
disease. N Engl J Med. 340:115–126. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Springer TA: Traffic signals for
lymphocyte recirculation and leukocyte emigration: The multistep
paradigm. Cell. 76:301–314. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Akisu M, Kultursay N, Coker I and
Huseyinov A: Platelet-activating factor is an important mediator in
hypoxic ischemic brain injury in the newborn rat. Flunarizine and
Ginkgo biloba extract reduce PAF concentration in the brain.
Biol Neonate. 74:439–444. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lu Q, Hao M, Wu W, Zhang N, Isaac AT, Yin
J, Zhu X, Du L and Yin X: Antidiabetic cataract effects of GbE,
rutin and quercetin are mediated by the inhibition of oxidative
stress and polyol pathway. Acta Biochim Pol. 65:35–41. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lu Q, Zuo WZ, Ji XJ, Zhou YX, Liu YQ, Yao
XQ, Zhou XY, Liu YW, Zhang F and Yin XX: Ethanolic Ginkgo
biloba leaf extract prevents renal fibrosis through Akt/mTOR
signaling in diabetic nephropathy. Phytomedicine. 22:1071–1078.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Saini AS, Taliyan R and Sharma PL:
Protective effect and mechanism of Ginkgo biloba extract-EGb
761 on STZ-induced diabetic cardiomyopathy in rats. Pharmacogn Mag.
10:172–178. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang J, Wang A, He H, She X, He Y, Li S,
Liu L, Luo T, Huang N, Luo H and Zou K: Trametenolic acid B
protects against cerebral ischemia and reperfusion injury through
modulation of microRNA-10a and PI3K/Akt/mTOR signaling pathways.
Biomed Pharmacother. 112:1086922019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Guan T, Qian Y, Tang X, Huang M, Huang L,
Li Y and Sun H: Maslinic acid, a natural inhibitor of glycogen
phosphorylase, reduces cerebral ischemic injury in hyperglycemic
rats by GLT-1 up-regulation. J Neurosci Res. 89:1829–1839. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniectomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang Y, Shuaib A and Li Q: Quantification
of infarct size on focal cerebral ischemia model of rats using a
simple and economical method. J Neurosci Methods. 84:9–16. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pilz J, Meineke I and Gleiter CH:
Measurement of free and bound malondialdehyde in plasma by
high-performance liquid chromatography as the
2,4-dinitrophenylhydrazine derivative. J Chromatogr B Biomed Sci
Appl. 742:315–325. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu YW, Zhu X, Li W, Lu Q, Wang JY, Wei YQ
and Yin XX: Ginsenoside Re attenuates diabetes-associated cognitive
deficits in rats. Pharmacol Biochem Behav. 101:93–98. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sun Y, Oberley LW and Li Y: A simple
method for clinical assay of superoxide dismutase. Clin Chem.
34:497–500. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Metting Z, Wilczak N, Rodiger LA, Schaaf
JM and van der Naalt J: GFAP and S100B in the acute phase of mild
traumatic brain injury. Neurology. 78:1428–1433. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rothermundt M, Peters M, Prehn JH and
Arolt V: S100B in brain damage and neurodegeneration. Microsc Res
Tech. 60:614–632. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Foerch C, Wunderlich MT, Dvorak F, Humpich
M, Kahles T, Goertler M, Alvarez-Sabin J, Wallesch CW, Molina CA,
Steinmetz H, et al: Elevated serum S100B levels indicate a higher
risk of hemorrhagic transformation after thrombolytic therapy in
acute stroke. Stroke. 38:2491–2495. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Stadler K: Oxidative stress in diabetes.
Adv Exp Med Biol. 2012:272–287. 2012.
|
|
44
|
Ouyang YB, Voloboueva LA, Xu LJ and
Giffard RG: Selective dysfunction of hippocampal CA1 astrocytes
contributes to delayed neuronal damage after transient forebrain
ischemia. J Neurosci. 27:4253–4260. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kaarisalo MM, Räihä I, Sivenius J,
Immonen-Räihä P, Lehtonen A, Sarti C, Mähönen M, Torppa J,
Tuomilehto J and Salomaa V: Diabetes worsens the outcome of acute
ischemic stroke. Diabetes Res Clin Pract. 69:293–298. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kidwell CS, Alger JR and Saver JL:
Evolving paradigms in neuroimaging of the ischemic penumbra.
Stroke. 35 (11 Suppl 1):S2662–S2665. 2004. View Article : Google Scholar
|
|
47
|
Tanaka N, Ikeda Y, Ohta Y, Deguchi K, Tian
F, Shang J, Matsuura T and Abe K: Expression of Keap1-Nrf2 system
and antioxidative proteins in mouse brain after transient middle
cerebral artery occlusion. Brain Res. 1370:246–253. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Manickam DS, Brynskikh AM, Kopanic JL,
Sorgen PL, Klyachko NL, Batrakova EV, Bronich TK and Kabanov AV:
Well-defined cross-linked antioxidant nanozymes for treatment of
ischemic brain injury. J Control Release. 162:636–645. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Weismann D, Hartvigsen K, Lauer N, Bennett
KL, Scholl HP, Charbel Issa P, Cano M, Brandstatter H, Tsimikas S,
Skerka C, et al: Complement factor H binds malondialdehyde epitopes
and protects from oxidative stress. Nature. 478:76–81. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Martinic-Popovic I, Lovrencic-Huzjan A and
Demarin V: Assessment of subtle cognitive impairment in stroke-free
patients with carotid disease. Acta Clin Croat. 48:231–240.
2009.PubMed/NCBI
|
|
51
|
Lee K, Kim EH, Song D, Kim YD, Nam HS, Lee
HS and Heo JH: Lenticulostriate artery involvement is predictive of
poor outcomes in superficial middle cerebral artery territory
infarction. Yonsei Med J. 58:123–130. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Cahill SP, Yu RQ, Green D, Todorova EV and
Snyder JS: Early survival and delayed death of developmentally-born
dentate gyrus neurons. Hippocampus. 27:1155–1167. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wegener S, Weber R, Ramos-Cabrer P,
Uhlenkueken U, Sprenger C, Wiedermann D, Villringer A and Hoehn M:
Temporal profile of T2-weighted MRI distinguishes between
pannecrosis and selective neuronal death after transient focal
cerebral ischemia in the rat. J Cereb Blood Flow Metab. 26:38–47.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Echeverry R, Wu J, Haile WB, Guzman J and
Yepes M: Tissue-type plasminogen activator is a neuroprotectant in
the mouse hippocampus. J Clin Invest. 120:2194–2205. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Miyawaki T, Ofengeim D, Noh KM,
Latuszek-Barrantes A, Hemmings BA, Follenzi A and Zukin RS: The
endogenous inhibitor of Akt, CTMP, is critical to ischemia-induced
neuronal death. Nat Neurosci. 12:618–626. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Liu Y, Wang H, Liu N, Du J, Lan X, Qi X,
Zhuang C, Sun T, Li Y and Yu J: Oxymatrine protects neonatal rat
against hypoxic-ischemic brain damage via PI3K/Akt/GSK3β pathway.
Life Sci. May 16–2019, https://doi.org/10.1016/j.lfs.2019.04.070
|
|
57
|
Cheng SM, Ho TJ, Yang AL, Chen IJ, Kao CL,
Wu FN, Lin JA, Kuo CH, Ou HC, Huang CY and Lee SD: Exercise
training enhances cardiac IGFI-R/PI3K/Akt and Bcl-2 family
associated pro-survival pathways in streptozotocin-induced diabetic
rats. Int J Cardiol. 167:478–485. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kim GD, Oh J, Park HJ, Bae K and Lee SK:
Magnolol inhibits angiogenesis by regulating ROS-mediated apoptosis
and the PI3K/AKT/mTOR signaling pathway in mES/EB-derived
endothelial-like cells. Int J Oncol. 43:600–610. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Qi S, Xin Y, Guo Y, Diao Y, Kou X, Luo L
and Yin Z: Ampelopsin reduces endotoxic inflammation via repressing
ROS-mediated activation of PI3K/Akt/NF-κB signaling pathways. Int
Immunopharmacol. 12:278–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Wang J, Li G, Wang Z, Zhang X, Yao L, Wang
F, Liu S, Yin J, Ling EA, Wang L and Hao A: High glucose-induced
expression of inflammatory cytokines and reactive oxygen species in
cultured astrocytes. Neuroscience. 202:58–68. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yang Y, Gozen O, Watkins A, Lorenzini I,
Lepore A, Gao Y, Vidensky S, Brennan J, Poulsen D, Won Park J, et
al: Presynaptic regulation of astroglial excitatory
neurotransmitter transporter GLT1. Neuron. 61:880–894. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Mdzinarishvili A, Sumbria R, Lang D and
Klein J: Ginkgo extract EGb761 confers neuroprotection by reduction
of glutamate release in ischemic brain. J Pharm Pharm Sci.
15:94–102. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ghosh M, Yang Y, Rothstein JD and Robinson
MB: Nuclear factor-κB contributes to neuron-dependent induction of
glutamate transporter-1 expression in astrocytes. J Neurosci.
31:9159–9169. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Mitra R, Dube SK and Jain V: Isolated
diastolic hypotension: A unique complication of intra-arterial
nimodipine infusion!! J Clin Anesth. 59(1)2020.PubMed/NCBI
|
|
65
|
Rhee KJ, Lee CG, Kim SW, Gim DH, Kim HC
and Jung BD: Extract of Ginkgo biloba ameliorates
Streptozotocin-induced type 1 diabetes mellitus and High-fat
diet-induced type 2 diabetes mellitus in mice. Int J Med Sci.
12:987–994. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Aziz TA, Hussain SA, Mahwi TO, Ahmed ZA,
Rahman HS and Rasedee A: The efficacy and safety of Ginkgo
biloba extract as an adjuvant in type 2 diabetes mellitus
patients ineffectively managed with metformin: A double-blind,
randomized, placebo-controlled trial. Drug Des Devel Ther.
12:735–742. 2018. View Article : Google Scholar : PubMed/NCBI
|