Introduction
Congenital generalized lipodystrophy (CGL) is a
group of rare inherited lipodystrophies characterized by
lipoatrophy at birth or in early life (1,2). The
clinical presentation of CGL includes acromegaloid features,
hypertriglyceridemia and complications related to insulin
resistance (1). Four subtypes of
CGL have been reported, all of which are autosomal recessive
disorders (3). CGL1 (MIM #608594)
and CGL2 (MIM #269700) result from pathogenic variants in the
1-acylglycerol-3-phosphate O-Acyltransferase 2 gene and
Berardinelli-Seip congenital lipodystrophy 2 (BSCL2) gene,
respectively (3). These are the
major forms of CGL, accounting for 95% of reported CGL cases
(4). CGL4 (MIM #613327), which
accounts for ~5% of cases, is caused by pathogenic variants in the
polymerase 1 and transcript release factor (PTRF) gene
(3,4). CGL3 (MIM #612526), which is due to
pathogenic variants in the caveolin 1 (CAV1) gene, has been
rarely reported (5). Given the
genetic heterogeneity of this condition, exome sequencing has been
the method of choice for the molecular diagnosis of CGL. Although
whole exome sequencing (WES) can effectively detect
single-nucleotide variants (SNVs) and small insertions/deletions,
its use to detect copy number variations (CNVs) remains challenging
(6).
A founder deletion encompassing exon 3 of
BSCL2 had been reported in Peruvian patients (7). Other than that, exonic deletions in
BSCL2 are rare. In the present study a small heterozygous
insertion and a large heterozygous deletion similar to the Peruvian
deletion in BSCL2 were detected in the patient. It was
demonstrated that the deletions differ in size and breakpoints,
indicating that they arose independently.
Materials and methods
Case presentation
A 2-month-old male infant was referred to the
Pediatric Endocrine Department of Guangxi Maternal and Child Health
Hospital for skin hyperpigmentation and developmental delay on
April 20th, 2018. Written informed consent for the infant and his
parents was obtained from the infant's parents. The present study
was approved by the Medical Ethics Committee of Guangxi Maternal
and Child Health Hospital. Abdominal ultrasound was applied to
assess the presence of hepatomegaly, and cardiac function was
assessed by echocardiography and electrocardiography. Blood
glucose, blood lipid and C-peptide levels were determined as
previously reported (8). Serum
insulin and sex hormone levels were measured by electrochemical
luminescence immunoassay (Cobas E601; Roche Diagnostics).
WES and CNV analysis
Genomic DNA was extracted from 2 ml peripheral blood
of the infant and his parents in accordance with the standard
method of the Lab-Aid® DNA extraction kit (Xiamen Zeesan
Biotech Co, Ltd.). Human exome sequencing libraries were
constructed using Agilent SureSelect Human All Exon V5 kit (Agilent
Technologies, Inc.), and generated amplicons were sequenced with
Illumina HiSeq 2500 system (Illumina, Inc.). After sequencing,
reads were aligned to an indexed human reference genome
(GRCh37/hg19) with Burrows-Wheeler transformation 0.7.15-r1140
(9). Duplicate reads were removed
using Picard v.1.85 (http://picard.sourceforge.net) before further
processing. Base recalibration and variant calling were performed
using the Genome Analysis Toolkit v.2.3-4Lite (10). Finally, identified variants were
saved in variant call format. In addition, in the present study
there was an attempt to reveal CNVs with a read depth-based CNV
detection algorithm. Translational Genomics Expert (LifeMap
Sciences, Inc.) was used for variant prioritization. Integrative
Genomics Viewer 2.4.15 (IGV 2.4.15) was used to visualize WES data
and assess the coverage of the exons (http://software.broadinstitute.org/software/igv/).
Variants detected in the infant, but absent in the ClinVar
(www.ncbi.nlm.nih.gov/clinvar/), Human
Gene Mutation Database (www.hgmd.cf.ac.uk/ac/), Single Nucleotide Polymorphism
Database (dbSNP; www.ncbi.nlm.nih.gov/SNP), 1000 Genomes Project
database (http://www.internationalgenome.org/1000-genomes-browsers/),
Ensembl (http://grch37.ensembl.org/index.html) and Genome
Aggregation Database 2.1 (gnomAD 2.1; http://gnomad.broadinstitute.org) were considered
novel variants (11).
Sanger sequencing and multiplex
ligation-dependent probe amplification (MLPA)
Sanger sequencing was performed to confirm the
variant identified by WES. Specific primers for amplification and
sequencing were designed in Primer3web version 4.1.0 (http://primer3.ut.ee/). The primer sequences of
BSCL2 exon 6 were as follows: forward,
5′-CTACTCAGGGGTGGTTGAGG-3′ and reverse, 5′-CCCCACTTCCAGTCTCTCAG-3′.
A pair of primers encompassing exon 3 was used to determine the
breakpoints (BSCL2_BP3-forward: 5′-TCCCTTAAGTCCCTGGTCCA-3′;
BSCL2_BP3-reverse: 5′-ACATTTCTAACACCTGCTGCAG-3′). The PCR
product was sequenced with the 3730×l Genetic Analyzer (Applied
Biosystems; Thermo Fisher Scientific, Inc.). Sequence data were
aligned with the consensus coding sequence [human genome assembly
37 in nucleotide Basic Local Alignment Search Tool (BLAST;
http://blast.ncbi.nlm.nih.gov/blast/)]. Mutation
Surveyor software (https://softgenetics.com/mutationSurveyor.php; version
4.0.4) was used in visualizing the sequences.
To confirm the suspected exon 3 deletion in
BSCL2, MLPA probes were designed and validated following a
previous method (12) and the
protocol from MRC-Holland BV (https://support.mlpa.com/downloads/files/designing-synthetic-mlpa-probes).
A total of 11 pairs of MLPA probes were designed for BSCL2
(Table SI). These probes were
dissolved and diluted in TE buffer, then added to the P200
reference probe mix, which was purchased from MRC-Holland BV and
included reference probes and control fragments. The MLPA
experiment was performed in accordance with the manufacturers'
protocols, with MLPA reagents provided by MRC-Holland BV. Capillary
electrophoresis was performed with the 3130 Genetic Analyzer
(Applied Biosystems; Thermo Fisher Scientific, Inc.), and data were
analyzed with Coffalyser (https://www.mrcholland.com/).
Results
Clinical evaluation
The infant was the second child born to
nonconsanguineous healthy parents. He was a full-term baby with
normal delivery, and he had a healthy sister. His weight was 2,500
g (−1 SD) at birth and 3,900 g (−3 SD, which indicates that the
weight of the infant was lower than healthy infants) at 2 months.
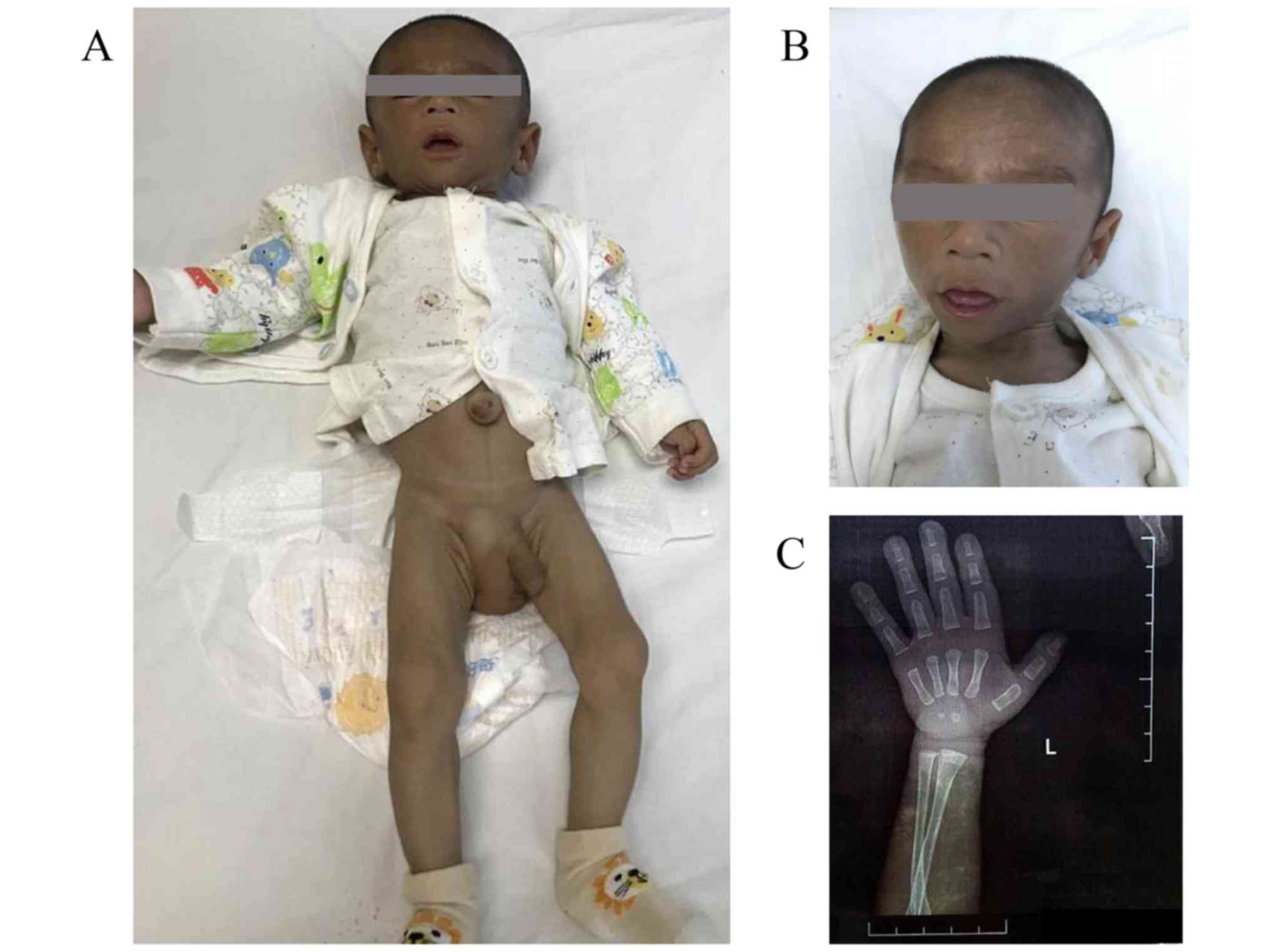
He had a thin appearance and advanced bone age (3 months).
Generalized lipoatrophy and skin hyperpigmentation were observed.
Moreover, he had a triangular face, marked umbilical prominence, a
long and large penis, and a swollen right scrotum (Fig. 1). Echocardiography and abdominal
ultrasound indicated atrial septal defect and hepatomegaly,
respectively, and no splenomegaly. Liver function tests were
normal. Other abnormal biochemical examinations were as follows:
Insulin and C-peptide levels were elevated [956.6 pmol/l, normal
range (NR): 17.8–173 pmol/l and >21.85 ng/ml, NR: 1.1–4.4 ng/ml,
respectively], which indicated insulin resistance. The triglyceride
level was increased (26.63 mmol/l, NR: 0.56–1.70 mmol/l), whereas
the levels of high-density lipoprotein cholesterol (0.52 mmol/l,
NR: 1.16–1.55 mmol/l), low-density lipoprotein cholesterol (2.36
mmol/l, NR: 2.70–3.13 mmol/l), apolipoprotein A1 (0.52 g/l, NR:
1.20–1.60 g/l), and apolipoprotein B (0.67 g/l, NR: 0.80–1.05 g/l)
were all decreased, indicating hypertriglyceridemia. Sex hormone
levels were decreased also (testosterone: 0.85 ng/ml, NR: 1.75–7.81
ng/ml; estradiol: 5.00 pg/ml, NR: 20.00–75.00 pg/ml).
Genetic findings
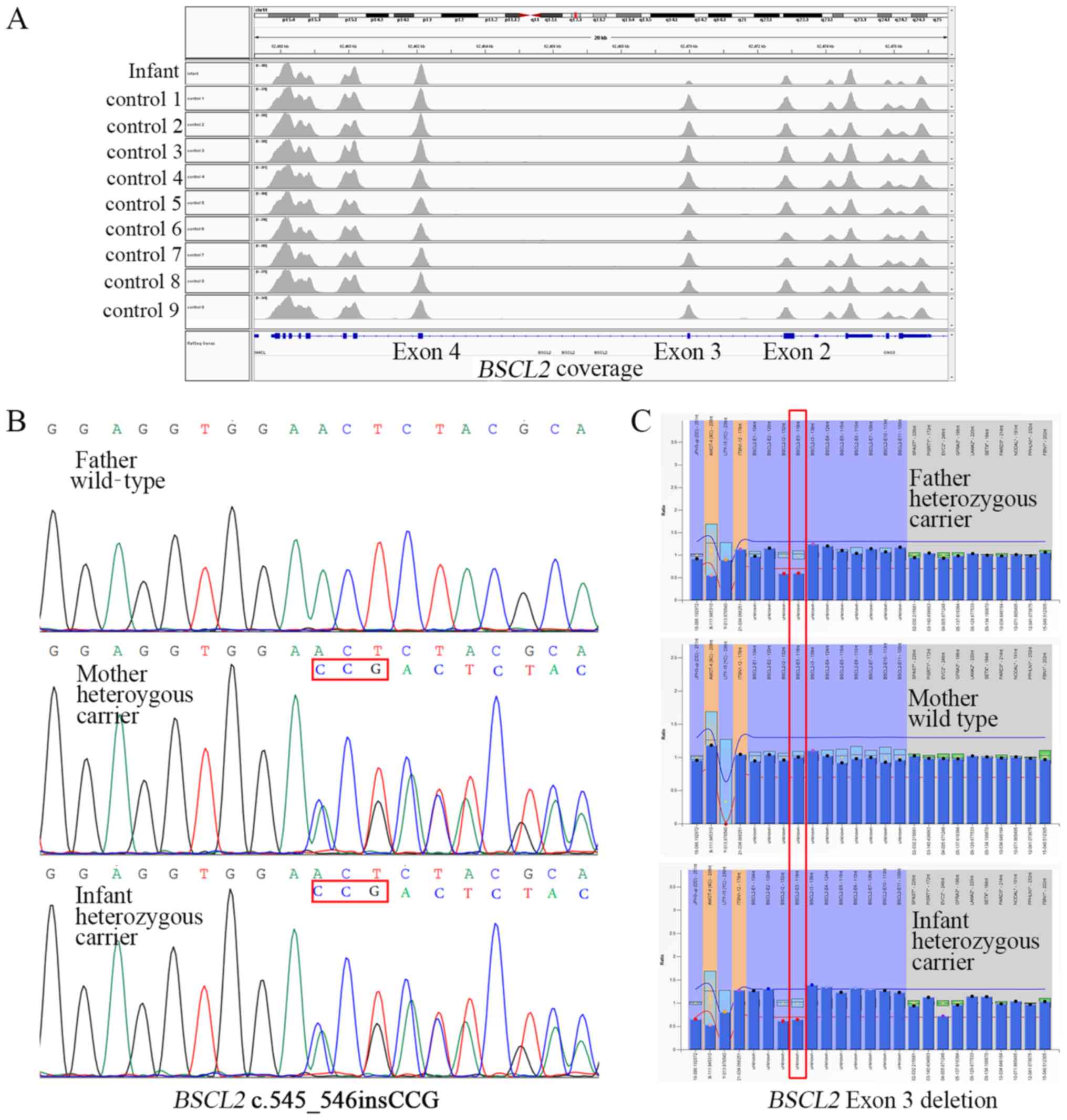
WES revealed a c.545_546insCCG heterozygous variant
in exon 6 of the BSCL2 gene (GenBank accession no.
NM_032667.6). This variant was confirmed by Sanger sequencing
(Fig. 2B). It led to a disruptive
inframe indel at position 182 (p.Glu182delinsAspArg). The variant
was inherited from the mother. This variant has been documented in
the dbSNP database (rs747297291) and at an extremely low frequency
in the gnomAD database. It has been detected in trans with a
pathogenic variant in a Chinese patient with CGL (13), indicating that it is a recurrent
pathogenic variant in the Chinese population.
There were no additional SNVs detected in
BSCL2 or any other rare SNVs that could explain the
phenotypes. CNV assessment by the visual review of BSCL2
coverage using the IGV browser suggested a heterozygous deletion at
11q12.3 (62,469,824-62,470,137) involving exon 3 of BSCL2
(Fig. 2A). To confirm this
heterozygous deletion, MLPA probes covering exon 3 of BSCL2
and other control regions were designed. The copy number ratios of
the BSCL2 exon 3 of the infant, his father and his mother
were 0.65, 0.62 and 1.01, respectively (Fig. 2C), indicating the presence of an
Ex3del in the infant and his father. The Ex3del was a null variant
and absent in the dbSNP, the 1000 Genomes Project database,
Ensembl, gnomAD and local normal controls. Notably, a similar exon
3 deletion has been reported as a founder mutation in Peruvian
patients with CGL (7).
Gap-PCR was performed to further confirm and map the
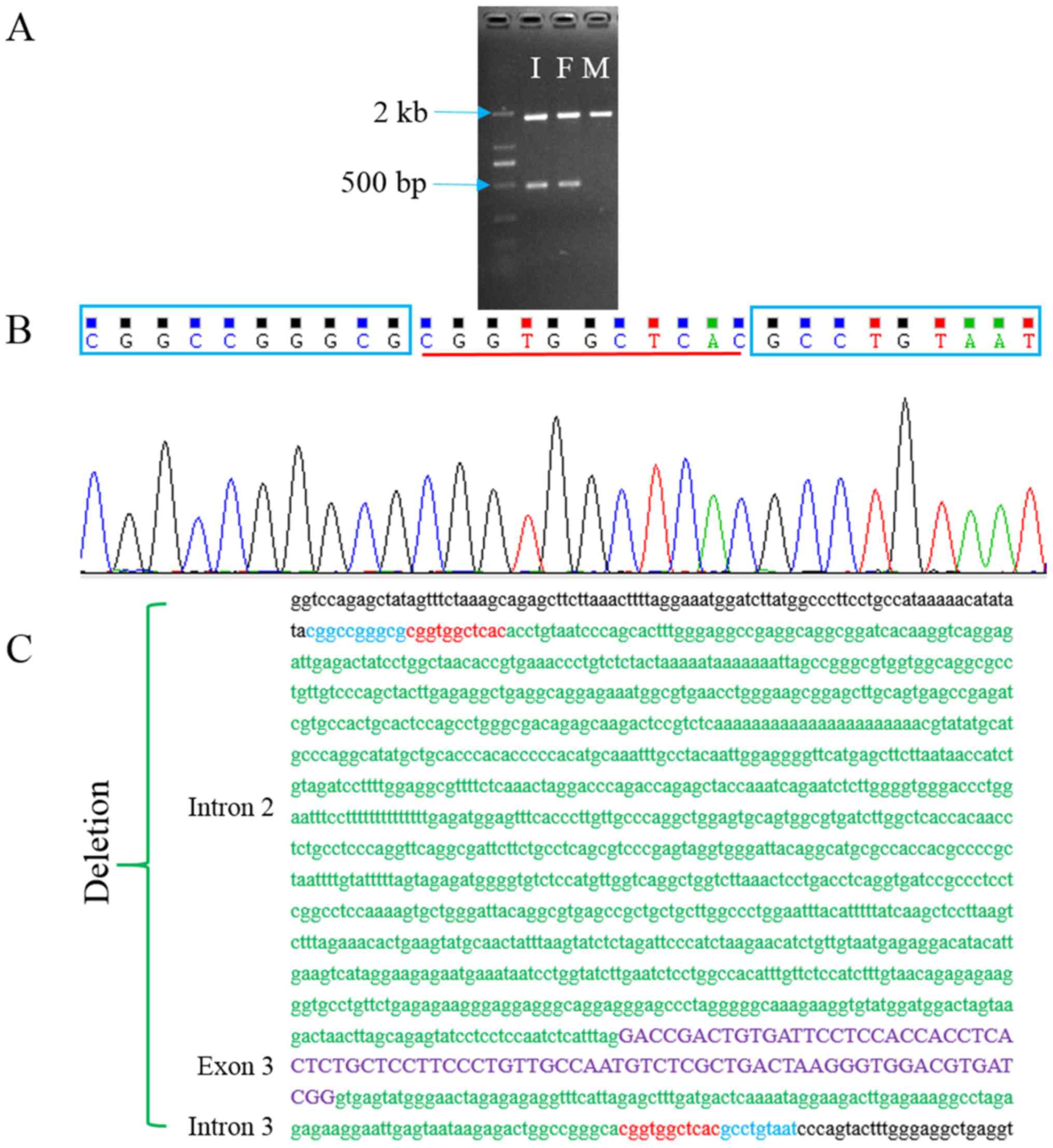
exact breakpoints of the exon 3 deletion. As revealed in Fig. 3A, a 483-bp amplicon that
represented the deleted allele was present in the proband and in
the father but not in the mother. Sequencing this amplicon
confirmed the deletion and revealed the breakpoints of a 1,274 bp
deletion (chr11:62,469,829-62,471,102; c.213-1081_c.294+111),
including complete exon 3 and part of introns 2 and 3 (Fig. 3B and C). An 11-bp microhomology was
identified at the breakpoints. Thus, this deletion is different
from the founder deletion variant detected in Peruvian patients
with CGL (7). The deletion would
lead to a frameshift and premature termination (p.Thr72Cysfs*2)
that is predicted to result in nonsense-mediated mRNA decay.
The maternally inherited variant c.545_546insCCG is
rare in the general population (PM2) (13,14).
It has been reported in trans with a pathogenic variant at least
twice (PM3_Strong) and shortens the protein product (PM4) (13). The patient's phenotype is
consistent and specific with CGL (PP4). This variant is classified
as likely pathogenic on the basis of ACMG/AMP guidelines for
variant classification (15). In
the present study, it was speculated that Ex3del was a
loss-of-function variant (PVS1) that was absent from the general
population (PM2), and PP4 was also applicable; thus, it can be
classified as pathogenic.
Discussion
The main clinical feature of CGL is adipose tissue
loss resulting in abnormal carbohydrate and lipid metabolism. The
accumulation of ectopic fat can lead to hepatomegaly, dyslipidemia,
insulin resistance and diabetes (16). The patient reported in this study
presented with facial abnormalities, marasmus, a macropenis,
hepatomegaly and hypertriglyceridemia, which were in accordance
with the diagnosis of CGL.
Given that CGL is a genetically heterogeneous
disorder, identifying the causative mutation for patients by Sanger
sequencing is challenging. In the present study, WES was
demonstrated to be useful in detecting small insertions, as well as
CNVs that cannot be identified by Sanger sequencing.
CNV detection may increase the diagnostic yield of
WES (17), an important issue in
genetic diagnosis. There are several algorithms to call CNV from
WES data, but no one can detect all CNVs in different size ranges
(18–20). A read-depth based algorithm was
developed to predict CNVs, which are confirmed in the IGV browser.
Although it cannot identify the precise breakpoint of the deletion,
the CNV-calling program was successful at predicting heterozygous
deletions, including BSCL2 exon 3.
CNV analysis based on exome data is useful but must
be confirmed with orthogonal methods. CNV validation methods
include chromosomal microarray analysis, reverse
transcription-quantitative PCR and MLPA (21). In the present study, MLPA probes
were designed to confirm the Ex3del by designing specific MLPA
probes for the region. Further breakpoint analysis by Sanger
sequencing revealed a 1,274 bp deletion
(chr11:62,469,829-62,471,102), including the complete exon 3 and
part of introns 2 and 3. A BLAST search of the flanking junction
sequences of the breakpoints was performed, and it was detected
that the breakpoint in intron 2 is part of the 303-bp repeat
AluY (chr11:62,470,820-62,471,122), and the breakpoint in
intron 3 is part of the 293-bp repeat AluSp
(chr11:62,469,545-62,469,837). The overall similarity of
AluSp and AluY is 80%, and both of the Alu elements
are at the same orientation at the two sites. In particular, an
overlapping sequence CGGTGGCTCAC is present at
chr11:62,471,103-62,471,114 and chr11:62,469,817-62,469,828. The
overlapping 11-bp sequences may mediate a crossover event that lead
to deletion. To date, nearly one-quarter of the mutations in
BSCL2 reported in patients with type 2 CGL are missense
mutations; the others are null mutations, including nonsense,
splicing, small indels, small insertions and deletions (3,22).
Large deletions have rarely been reported, except for the exon 3
deletion. A 3.3-kb homozygous deletion of exon 3 was reported as a
founder mutation in Peruvian population, which appeared to be
mediated by recombination between AluSq2 and AluSx3
in introns 2 and 3 (7). The size
of the deletions and the breakpoints of Ex3del are different in the
Chinese and Peruvian CGL patients, indicating they are independent
events (7). However, as
aforementioned, both events may be mediated by a similar mechanism
and result in the same consequence. Thus, this independent deletion
is suggestive of a recurrent event involving exon 3. Therefore, the
exon 3 deletion should be considered for patients with CGL2 when
only one BSCL2 variant was detected by exome sequencing.
Glu182, a site that is highly conserved among
numerous species, is localized in the conserved loop of the ER
lumen (13,23). The variant of c.545_546insCCG
resulted in a disruptive inframe indel at position 182
(p.Glu182delinsAspArg), which may cause the misfolding of the
secondary structure of the seipin protein due to the insertion of
the basic amino acid arginine and could destabilize the protein
structural domain. Given that this variant has only been reported
in Chinese patients with CGL (13,14),
it is a recurrent pathogenic variant in the Chinese population.
In conclusion, a novel compound heterozygous variant
(p.Glu182delinsAspArg/Ex3del) in the BSCL2 gene was
identified in a Chinese infant with CGL by WES. The present study
revealed an independent recombination event in introns 2 and 3 that
resulted in exon 3 deletion (c.213-1081_c.294+111). This finding
expanded the mutational spectrum of the BSCL2 gene while
providing further evidence of exonic deletion in the BSCL2
gene in patients with CGL.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Major
Research Plan of the Provincial Science and Technology Foundation
of Guangxi (grant no. AB16380214); the National key research and
development program (grant no. 2018YFC1002501); the ‘Eastern
Scholar’ Fund; the ‘Guangxi Bagui Scholar’ fund; the Natural
Science Foundation of China (grant no. 81873633) and GeneScience
Pharmaceuticals Co., Ltd. (grant no. xixueyanjiubu-20150808-1). The
funding body played no direct role in the design of the study, or
in the collection, analysis and interpretation of data, or in the
writing of the manuscript.
Availability of data and materials
The data used and analyzed during the present study
are available from the corresponding author or first author on
reasonable request.
Authors' contributions
YS and BX designed the study and experiments. JL and
XF analyzed the clinical phenotype and followed up the infant. ZQ
and FS exacted DNA and performed the pathogenicity evaluation of
gene variation. SY and JW worked on the experiments and
bioinformatics analysis of WES. QY performed the Sanger sequencing
experiments. YL performed the MLPA experiments and collected the
data. BX designed the probes for MLPA and wrote the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by Guangxi Maternal
and Child Health Hospital Medical Ethics Committee [approval no.
(2018) Lun Han Shen (5–5)] and written informed consent for the
infant and his parents was obtained from the infant's parents.
Patient consent for publication
Written informed consent for the infant was obtained
from his parents for the publication of this report, including
images and data.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
BSCL2
|
Berardinelli-Seip congenital
lipodystrophy 2
|
|
WES
|
Whole exome sequencing
|
|
MLPA
|
multiplex ligation-dependent probe
amplification
|
|
CNV
|
copy number variation
|
References
|
1
|
Dantas de Medeiros JL, Carneiro Bezerra B,
Brito de Araújo TA, Craveiro Sarmento AS, de Azevedo Medeiros LB,
Peroni Gualdi L, Luna Cruz MDS, Xavier Nobre TT, Gomes Lima J and
Araújo de Melo Campos JT: Impairment of respiratory muscle strength
in Berardinelli-Seip congenital lipodystrophy subjects. Respir Res.
19:1732018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cheema HA, Malik HS, Waheed N, Mushtaq I,
Fayyaz Z and Anjum MN: Berardinelli-Seip Congenital Generalised
Lipodystrophy. J Coll Physicians Surg Pak. 28:406–408. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Patni N and Garg A: Congenital generalized
lipodystrophies - new insights into metabolic dysfunction. Nat Rev
Endocrinol. 11:522–534. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Boutet E, El Mourabit H, Prot M, Nemani M,
Khallouf E, Colard O, Maurice M, Durand-Schneider AM, Chrétien Y,
Grès S, et al: Seipin deficiency alters fatty acid Delta9
desaturation and lipid droplet formation in Berardinelli-Seip
congenital lipodystrophy. Biochimie. 91:796–803. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim CA, Delépine M, Boutet E, El Mourabit
H, Le Lay S, Meier M, Nemani M, Bridel E, Leite CC, Bertola DR, et
al: Association of a homozygous nonsense caveolin-1 mutation with
Berardinelli-Seip congenital lipodystrophy. J Clin Endocrinol
Metab. 93:1129–1134. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yao R, Zhang C, Yu T, Li N, Hu X, Wang X,
Wang J and Shen Y: Evaluation of three read-depth based CNV
detection tools using whole-exome sequencing data. Mol Cytogenet.
10:302017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Purizaca-Rosillo N, Mori T, Benites-Cóndor
Y, Hisama FM, Martin GM and Oshima J: High incidence of BSCL2
intragenic recombinational mutation in Peruvian type 2
Berardinelli-Seip syndrome. Am J Med Genet A. 173:471–478. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lima JG, Nobrega LH, de Lima NN, do
Nascimento Santos MG, Baracho MF and Jeronimo SM: Clinical and
laboratory data of a large series of patients with congenital
generalized lipodystrophy. Diabetol Metab Syndr. 8:232016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li H and Durbin R: Fast and accurate
long-read alignment with Burrows-Wheeler transform. Bioinformatics.
26:589–595. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Van der Auwera GA, Carneiro MO, Hartl C,
Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen
D, Thibault J, et al: From FastQ Data to High Confidence Variant
Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr
Protoc Bioinformatics. 43:11.10.1–11.10.33. 2013.
|
|
11
|
Karczewski KJ, Francioli LC, Tiao G,
Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A,
Birnbaum DP, et al: Variation across 141,456 human exomes and
genomes reveals the spectrum of loss-of-function intolerance across
human protein-coding genes. bioRxiv. 5312102019.
|
|
12
|
Shen Y and Wu BL: Designing a simple
multiplex ligation-dependent probe amplification (MLPA) assay for
rapid detection of copy number variants in the genome. J Genet
Genomics. 36:257–265. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu Y, Li D, Ding Y, Kang L, Jin Y, Song
J, Li H and Yang Y: Further delineation of AGPAT2 and BSCL2 related
congenital generalized lipodystrophy in young infants. Eur J Med
Genet. 62:1035422019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Su X, Lin R, Huang Y, Sheng H and Li X,
Ting TH, Liu L and Li X: Clinical and Mutational Features of Three
Chinese Children with Congenital Generalized Lipodystrophy. J Clin
Res Pediatr Endocrinol. 9:52–57. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al ACMG
Laboratory Quality Assurance Committee, : Standards and guidelines
for the interpretation of sequence variants: A joint consensus
recommendation of the American College of Medical Genetics and
Genomics and the Association for Molecular Pathology. Genet Med.
17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Garg A and Misra A: Lipodystrophies: Rare
disorders causing metabolic syndrome. Endocrinol Metab Clin North
Am. 33:305–331. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Marchuk DS, Crooks K, Strande N,
Kaiser-Rogers K, Milko LV, Brandt A, Arreola A, Tilley CR, Bizon C,
Vora NL, et al: Increasing the diagnostic yield of exome sequencing
by copy number variant analysis. PLoS One. 13:e02091852018.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Samarakoon PS, Sorte HS, Kristiansen BE,
Skodje T, Sheng Y, Tjønnfjord GE, Stadheim B, Stray-Pedersen A,
Rødningen OK and Lyle R: Identification of copy number variants
from exome sequence data. BMC Genomics. 15:6612014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tan R, Wang Y, Kleinstein SE, Liu Y, Zhu
X, Guo H, Jiang Q, Allen AS and Zhu M: An evaluation of copy number
variation detection tools from whole-exome sequencing data. Hum
Mutat. 35:899–907. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Miyatake S, Koshimizu E, Fujita A, Fukai
R, Imagawa E, Ohba C, Kuki I, Nukui M, Araki A, Makita Y, et al:
Detecting copy-number variations in whole-exome sequencing data
using the eXome Hidden Markov Model: An ‘exome-first’ approach. J
Hum Genet. 60:175–182. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yao R, Yu T, Qing Y, Wang J and Shen Y:
Evaluation of copy number variant detection from panel-based
next-generation sequencing data. Mol Genet Genomic Med.
7:e005132019. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Magré J, Delépine M, Khallouf E,
Gedde-Dahl T Jr, Van Maldergem L, Sobel E, Papp J, Meier M,
Mégarbané A, Bachy A, et al BSCL Working Group, : Identification of
the gene altered in Berardinelli-Seip congenital lipodystrophy on
chromosome 11q13. Nat Genet. 28:365–370. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wee K, Yang W, Sugii S and Han W: Towards
a mechanistic understanding of lipodystrophy and seipin functions.
Biosci Rep. 34:342014. View Article : Google Scholar
|