Introduction
Progressive cardiac conduction defect (PCCD), also
known as Lenegre-Lev disease (Online Mendelian Inheritance in Man
entry no. 113900), is a rare bradycardic disorder (1,2).
PCCD is characterized by electrical deterioration of the conduction
system in the atrium and ventricle, which presents as temporal
prolongation of the PR interval, bradycardia, bundle branch block
and advanced atrioventricular block (AVB) (3). A genetic epidemiological study
reported that the PCCD frequency ranged from 0.21–2.28% in
>6,600 patients implanted with a pacemaker in western France
(4). In the past several decades,
the rapid development of molecular genetics has provided important
data on the genetic basis of PCCD (5).
Clinical and molecular studies have led to the
characterization of PCCD as an inherited disease (6). Several genes have been associated
with PCCD, including sodium voltage-gated channel α5, protein
kinase AMP-activated non-catalytic subunit γ2, NK2 homeobox 5 and
lamin A/C (LMNA) (5,7). LMNA is located on chromosome
1q21.2–21.3 and generates both lamin A and C by alternative
splicing of a single transcript (8). Lamin A and C are nuclear intermediate
filament proteins that form one of the major structural components
of the lamina network, which underlies and mechanically supports
the nuclear envelope (9). LMNA
mutations cause a spectrum of multisystem diseases, including PCCD,
dilated cardiomyopathy (DCM), muscular dystrophy and lipodystrophy
(6,9,10).
Although pleiotropic phenotypes are expressed in LMNA-associated
diseases, PCCD is one of the most malignant phenotypes, with an
untreated sudden cardiac death rate of ≤46% (11). Mortality occurred in 8% of LMNA
genotype-positive subjects at a mean age of 66±8 years, over a mean
follow-up period of 7.8±6.3 years (12). These findings illustrate the
importance of genetic screening and genetic counseling in patients
with PCCD and families with genetic mutations.
In the present study, whole-exome sequencing of
three patients with inherited PCCD and one healthy control subject
was performed, and a pathogenic mutation was identified. The
PCCD-associated mutation was identified as a nonsense LMNA
mutation, which truncates the LMNA protein. Family screening
identified LMNA genotype-positive carriers, potentially allowing
for improved management of mutation carriers in the future.
Case report
Patients and DNA samples
The proband (III-4), a 57 year-old Chinese woman,
initially presented with chest discomfort, repeated attacks of
dizziness and amaurosis. The patient presented with the
aforementioned symptoms 12 years ago (in 2000) and received no
treatment prior to their first hospitalization in 2012. Due to
persistent symptoms, the patient was admitted to Department of
Cardiology in Fuwai Hospital (July 2012) for comprehensive
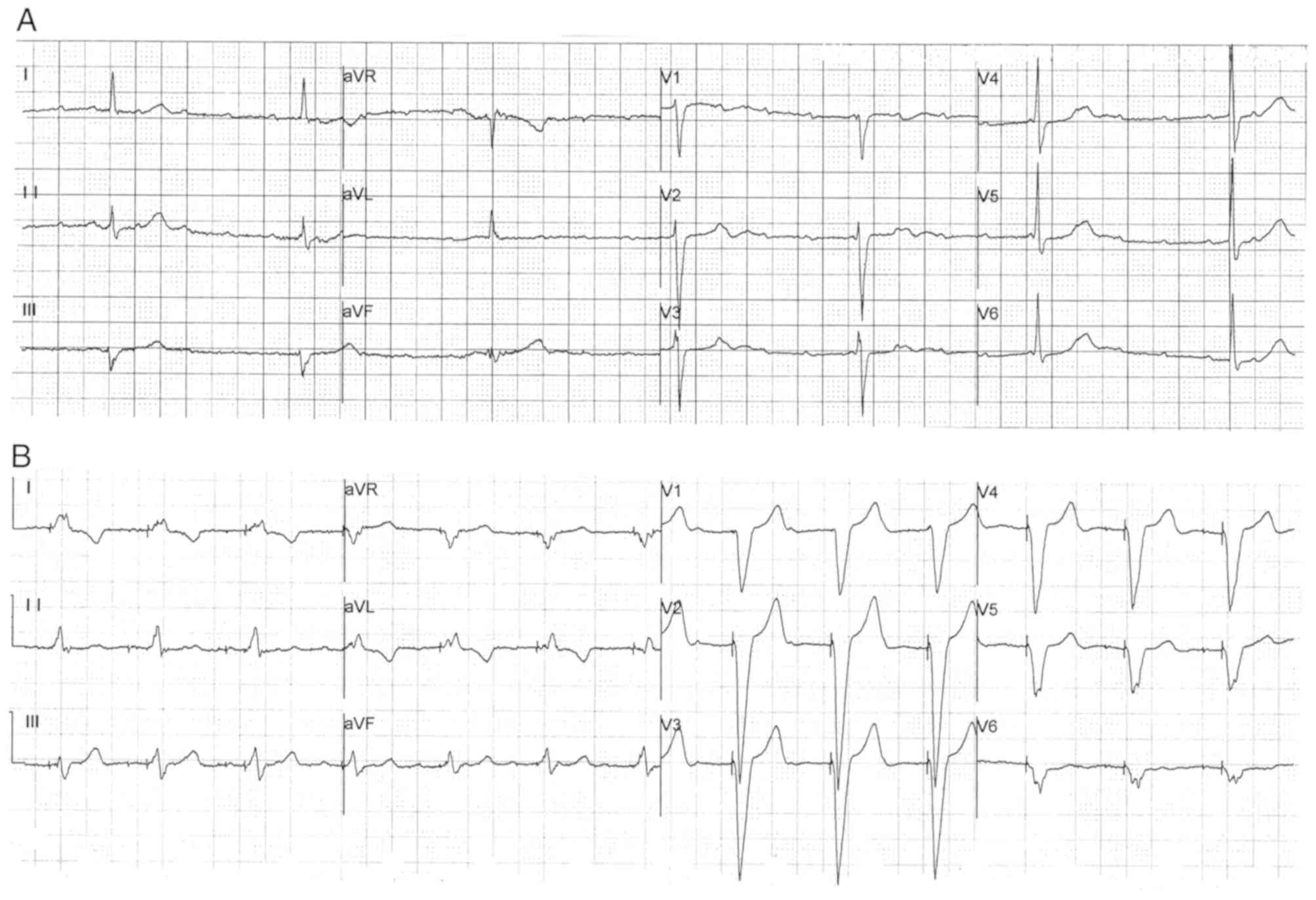
diagnosis and treatment. The 24 h ambulatory electrocardiogram
(ECG) results identified sinus bradycardia [average heart rate, 48
beats per minute (bpm); minimum heart rate, 23 bpm] and advanced
AVB in the patient (Fig. 1A). An
echocardiogram identified an enlargement of the left atrium (left
atrial diameter, 43 mm). Therefore, the patient received a
permanent pacemaker with a normal pacing ECG (Fig. 1B), which alleviated the
aforementioned symptoms. By investigation of the familial cardiac
history of the patient, a further ten relatives also experiencing
dizziness, syncope and bradycardia were identified. Among these
symptomatic family members, six participants had an implanted
pacemaker (II-10 also had an implantable cardioverter
defibrillator), and four participants experienced sudden death. In
addition, other common cardiovascular diseases were excluded in the
family, including coronary heart disease, DCM and myocarditis. The
direct family history, including cases of bradycardia, pacemaker
implantation and sudden death, raised suspicion of an inherited
arrhythmia disease.
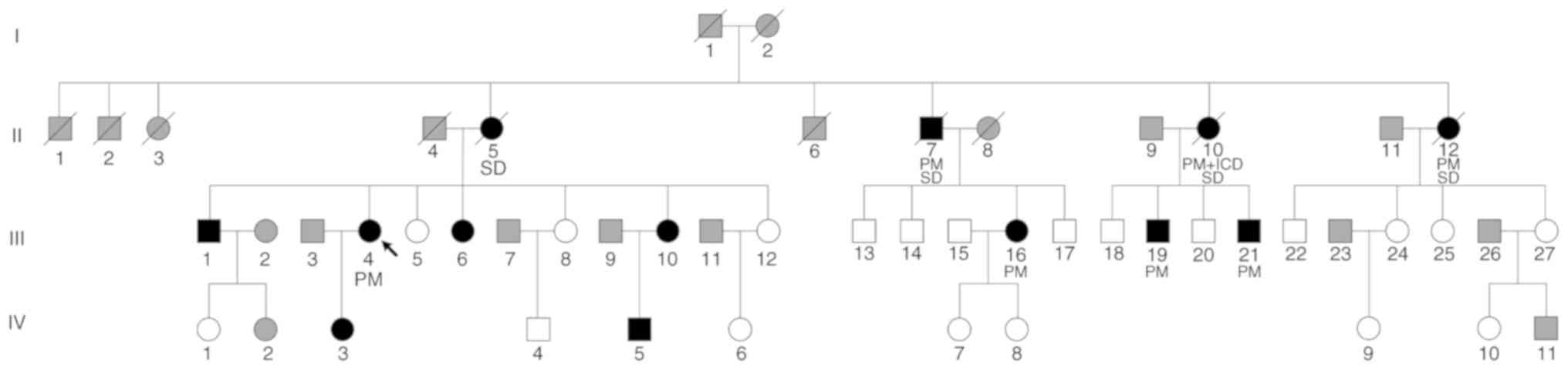
A total of 39 participants (male patients, 10;
female patients, 29; age range, 13–68 years; mean age, 42.59 years)
from a Chinese family were included in Department of Cardiology in
Fuwai Hospital (September 2017). The pedigree chart is presented in
Fig. 2. Each participant provided
peripheral venous blood for biochemical and genetic analyses and
was examined by an ECG and echocardiography. A clinical survey of
the cardiac disease history of each participant was also performed.
Then, 2 ml anticoagulated venous blood samples from each
participant were contained in EDTA vacuum tubes. Genomic DNA was
extracted from each blood sample using the QIAamp DNA Blood Mini
kit (Qiagen GmbH), according to the manufacturer's
instructions.
The present study was approved by the Ethics
Committee of Fuwai Hospital. Written informed consent was obtained
from each participant.
Whole-exome sequencing (WES)
WES was performed on blood samples from three
participants with PCCD (III-4, III-16 and III-21) and a healthy
family member (III-5) from the same generation of the family using
an Illumina HiSeq 2000 sequencing system (Illumina, Inc.). The
average coverage depth of the WES was >200-fold. The SureSelect
Human All Exon V5 (Agilent Technologies, Inc.) was used for exome
capture and Burrows-Wheeler Aligner (version 0.7.12) (13) was used to align the reads to the
National Center for Biotechnology Information human reference
genome (GRCh37/hg19) (https://www.ncbi.nlm.nih.gov/projects/genome/guide/human/index.shtml),
after removing sequence adaptors and low-quality reads (the
proportion of reads with undetermined base information in
single-end sequencing >10%; single read >50% low quality
(<5) nucleotides). Picard (version 1.119; sourceforge.net/projects/picard) and Genome Analysis
Toolkit (version 3.7) (14) tools
were used for duplicate removal, local realignment and base quality
recalibration. Single nucleotide polymorphism (SNP) and Indel
calling were performed using Sequence Alignment Map tools (version
1.10; samtools.sourceforge.net/) and were annotated using
Annotate Variation (version 20170825) (15), according to the SNP database
(www.ncbi.nlm.nih.gov/SNP) and the 1000
Genomes Project database (www.1000genomes.org/index.html).
Genotype identification
Firstly, all potentially significant variants were
identified by in silico analysis, using the Protein
Variation Effect Analyzer (version 1.1.5; provean.jcvi.org/index.php), Polymorphism Phenotyping
(version 2.0; genetics.bwh.harvard.edu/pph2), Sorting Intolerant
From Tolerant (version 4.0.3; sift.jcvi.org)
and Mutation Taster (version 2.0; www.mutationtaster.org) bioinformatics tools. The
conservation status of the candidate variants was estimated by the
Genome Evolutionary Rate Processing (version 2.0; mendel.stanford.edu/sidowlab/downloads/gerp/index.html),
Phylogenetic Analysis with Space/Time Models (PHAST) Cons (version
1.3; compgen.bscb.cornell.edu/phast) and PhyloP (version
1.3; compgen.bscb.cornell.edu/phast/help-pages/phyloP.txt)
software.
In addition, Sanger sequencing was used to identify
the validity of the variants. PCR of every potential variant was
amplified DNA from the proband. A primer pair was used to amplify
the LMNA variant c.1443C>A (forward, 5′-AGATGGAAGGAGAGGCCTCA-3′
and reverse, 5′-AGAAAGGGGCCCTGAATTGG-3′). PCR was performed with a
TaKaRa LA Taq kit with GC buffer (cat. no. RR02AG; Takara
Biomedical Technology Co., Ltd.) according to manufacturer's
protocol. The amplification process was: Initial denaturation for 1
min at 94°C, followed by 30 cycles of 30 sec at 94°C, 30 sec at
60°C, 2 min at 72°C; and a final extension step at 72°C for 5 min.
Subsequently, the candidate LMNA variants were sequenced in other
family members and 100 healthy controls of matched ancestry
(without any cardiovascular disease). The control group (male
patients, 53; female patients, 47; age range, 18–60 years; mean
age, 40.35 years) was recruited from September 2016 to August 2017.
The PCR products were sequenced using an ABI Prism® 377
DNA sequencer (Applied Biosystems; Thermo Fisher Scientific, Inc.).
The identification of the variants was performed using Chromas
software (version 2.22; Technelysium Pty, Ltd.). The co-segregation
of genotype and phenotype was analyzed.
Results
Clinical examinations
The ECG examinations identified similar symptoms
among the participants in terms of bradycardia, extended PR
interval and advanced AVB (Table
I). The most common characteristic was bradycardia, which was
observed in 12 patients from the family. The minimum heart rate
recorded for each participant was ≤50 bpm. An extended PR interval
>200 msec was recorded in seven participants (III-1, III-4,
III-6, III-10, III-16, III-19 and III-21). A cardiac conduction
block manifested in different forms, including a right bundle
branch block, an intraventricular block and various degrees of AVB.
Advanced AVB was identified in nine patients, of which eight
(except III-1) required the implantation of a pacemaker. Moreover,
II-10 had also received an implantable cardioverter defibrillator
to prevent sudden death. However, one identified mutation carrier
(IV-3) displayed no abnormal ECG findings, and although individual
IV-5 presented with bradycardia, the PR interval in the ECG was
within the normal range.
 | Table I.Clinical features of patients with
PCCD and asymptomatic lamin A/C mutation carriers. |
Table I.
Clinical features of patients with
PCCD and asymptomatic lamin A/C mutation carriers.
| Subject | Onset age
(years) | Minimum heart rate
(bpm) | ECG | PR (msec) | Prognosis |
|---|
| II-5a | 43 | 40 | Bradycardia,
advanced AVB | NA | SD |
| II-7a | 45 | 44 | Bradycardia, III
AVB | NA | SD |
| II-10a | 42 | 35 | Bradycardia,
advanced AVB | NA | PM, ICD + SD |
| II-12a | 40 | 37 | Bradycardia,
advanced AVB | NA | PM + SD |
| III-1 | 38 | 36 | AF, bradycardia,
intraventricular block, III AVB | 305 | – |
| III-4 | 45 | 23 | Bradycardia,
advanced AVB | 384 | PM |
| III-6 | 24 | 50 | AF, RBBB,
bradycardia, II AVB | 238 | – |
| III-10 | 40 | 50 | Bradycardia, atrial
premature | 200 | – |
| III-16 | 37 | 47 | Bradycardia,
advanced AVB | 314 | PM |
| III-19 | 40 | 30 | Bradycardia, III
AVB | 296 | PM |
| III-21 | 42 | 39 | Bradycardia,
advanced AVB | 280 | PM |
| IV-3 | – | 61 | – | 182 | – |
| IV-5 | 8 | 50 | Bradycardia | 187 | – |
None of the participants displayed hypertension or
muscle weakness. Investigation of serum creatinine phosphokinase
displayed no abnormalities in each participant. There was no
evidence supporting the diagnosis of DCM, although several patients
displayed abnormalities in the echocardiograph. Participants III-1
and III-19 had enlarged hearts, mitral and tricuspid insufficiency
and a normal ejection fraction. Participant III-6 had an atrial
septal defect and right atrial and ventricular enlargement (right
ventricular anteroposterior diameter, 39 mm). After repair of the
atrial septal defect, the right ventricular anteroposterior
diameter decreased to 21 mm. Participant III-16 had a mild
enlargement of the left heart due to mitral and tricuspid
insufficiency. Participant III-21 suffered from patent foramen
ovale. Other mutation carriers (III-10, IV-3 and IV-5) displayed a
normal echocardiograph.
Identification of pathogenic
mutations
Whole-exome sequencing was performed in three
participants (III-4, III-16 and III-21) and a healthy family member
(III-5). A total of 5,401 non-synonymous variants were detected in
the exonic and untranslated regions after filtering by frequency
<1% from the 1000 Genomes Project database. Co-association
analysis identified six candidate genes (LMNA, ARFGEF3, FNDC1,
MAN1A2, ODF2L and YY1AP1) associated with cardiac disease.
Furthermore, Sanger sequencing identified all possible pathogenic
variants among all participants. A nonsense LMNA mutation p.Tyr481*
(Fig. 3A) was identified as a
pathogenic mutation in the patients with PCCD. The genotype and
phenotype were only completely co-associated for this variant in
the family included in the present study. In 100 healthy controls,
the same mutation was not detected (Fig. 3B). To investigate whether the LMNA
mutation caused significant alterations to gene and protein
expression, several bioinformatics tools were employed (Table II). The nucleotide at position
1,443 and the amino acid at position 481 were fully conserved in
nine vertebrates (Fig. 3C). To
predict the functional effects of the LMNA mutation, in
silico analyses were conducted and suggested that the nonsense
mutation was a risk factor for PCCD. It was further suggested that
the nonsense LMNA mutation generated a truncated LMNA protein,
lacking the final 184 amino acids (Fig. 3D).
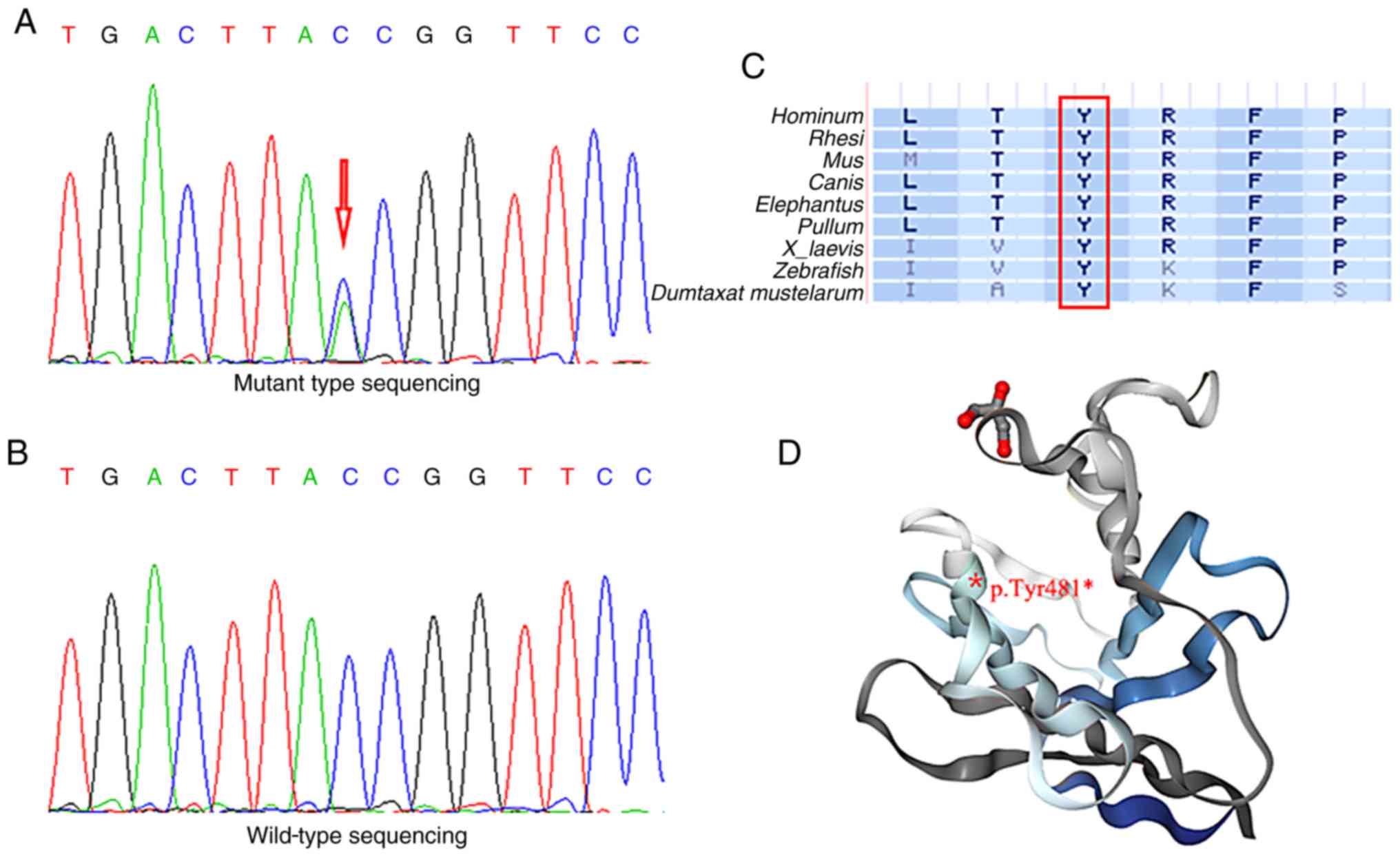 | Figure 3.Genotype identification of the LMNA
mutation. Electropherograms of sequencing displaying (A) mutant
site of LMNA gene (c.1443C>A), indicated by a red arrow, and (B)
wild-type sequencing of LMNA. (C) Amino acid at position 481 were
fully conserved in nine vertebrates. Red box indicates the location
of codon 481, bold letters indicate different amino acids and
normal letters indicate nine types of vertebrates. (D) Position of
the mutant amino acid is located in the structure of the LMNA
globular domain (protein data bank deposition no. 1IFR), and the
truncated protein is denoted by the grey fragment. LMNA, lamin
A/C.; L, leucine; M, methionine; I, isoleucine; T, threonine; V,
valine; A, alanine; Y, tyrosine; R, arginine; K, lysine; F,
phenylalanine; P, proline; S, serine. |
| Table II.In silico prediction of the
consequences of a nonsense mutation in lamin A/C. |
Table II.
In silico prediction of the
consequences of a nonsense mutation in lamin A/C.
| cDNA change | Amino acid
change | Prediction
tools | Score | Consequences of
mutation |
|---|
| c.1443C>A | Tyr481* | PROVEAN | −265.622 | Deleterious |
|
|
| MutationTaster | 1.000 | Disease
causing |
Discussion
In the present study, a large Chinese family with
inherited PCCD were investigated and an LMNA mutation, p.Tyr481*,
was identified by whole-exome sequencing. The present study
suggested that the LMNA mutation played a crucial role in the
genetic pathogenesis of PCCD in the aforementioned family. Genetic
testing of LMNA offers the possibility for hereditary diagnosis of
patients with PCCD and the opportunity to take precautionary
actions to prevent sudden death as early as possible (16).
As a hereditary syndrome, PCCD is related to genetic
variants in multiple genes, including LMNA (5,17).
At present, a large number of disease-causing LMNA mutations have
been described (16,18). The LMNA mutations are predominantly
missense mutations, but nonsense, frameshift and intragenic
deletions and duplications have also been reported (16). Non-missense mutations in LMNA have
an influence on the pathogenesis of PCCD (19). Nishiuchi et al (20) reported that truncation mutation
carriers presented a worse prognosis than those with a missense
mutation, based on gene risk stratification of cardiac disorders in
77 LMNA mutation carriers from 45 Japanese families. van Rijsingen
et al (21) evaluated risk
factors for malignant ventricular arrhythmias in a multicenter
cohort of 269 LMNA mutation carriers, and reported that
non-missense mutations were an independent risk factor for
malignant ventricular arrhythmias. In the present study, the
participants had a high risk of advanced AVB and six subjects
(II-10, II-12, III-4, III-16, III-19 and III-21) were implanted
with a pacemaker. Therefore, regarding the family investigated in
the present study, it could be suggested that other mutation
carriers should be kept under careful observation.
LMNA mutations are associated with several cardiac
phenotypes, including cardiac conduction disorders, DCM, malignant
ventricular arrhythmia and left ventricular dysfunction (17,22).
The most prominent cardiac feature of LMNA mutations is PCCD
(23). The majority of the
participants in the present study presented with dizziness and
chest discomfort and suffered from bradycardia on initial ECG
examination. As the disease progressed with age, certain
participants developed advanced AVB and required the implantation
of a pacemaker. Nevertheless, clinical variability has been
observed among mutation carriers with the same LMNA mutation in
present study. The age of onset for the majority of the
participants in the present study was ~40 years. Participants IV-3
(age, 13 years) and IV-5 (age, 18 years) did not present with
typical symptoms, potentially due to their young age. Therefore, it
may be important for these individuals to attend future follow-up
appointments.
Variable phenotypes in LMNA mutations may be caused
by factors, including gender, onset age and mutation location
(16). A number of studies have
reported significant gender differences in cardiac phenotypes
(17,21,24),
and it has been further reported that estrogen may play a role in
protecting against cardiac dysfunction (25,26).
Ollila et al (22) reported
that male LMNA mutation carriers have an earlier onset of disease
than females. However, no gender differences in the cardiac
phenotypes were identified in the family included in the present
study. In addition, patients with LMNA mutations display
age-dependent differences in the manifestation of cardiac
phenotypes and prognosis (27).
Consistent with the literature, in the present study, the greater
the age of the patient with PCCD, the more severe the cardiac
phenotype and prognosis (16,27).
Furthermore, the LMNA mutation location can predict the severity of
the cardiac phenotype (16). LMNA
mutations upstream of the nuclear localization signal or the
C-terminal tail domain are associated with a more severe cardiac
phenotype overall than those occurring downstream (16). The mutation identified in the
present study, p.Tyr481*, was located in the C-terminal tail domain
of LMNA, which is consistent with other severe cardiac phenotypes
(16,17).
In the same codon at position 481, other nucleotide
mutant forms and phenotypes have been reported (24,28–32).
A missense mutation (c.1442A>G, p.Tyr481Cys) was detected in
patients with DCM by clinical DNA sequencing (28–30).
Kitaguchi et al (31) first
reported a case of limb-girdle muscular dystrophy with
atrioventricular conduction block resulting from an LMNA missense
mutation (c.1441T>C, p.Tyr481His). In terms of nonsense
mutations, Sylvius et al (32) identified a proband with DCM
carrying a heterozygous mutation (c.1443C>G, p.Tyr481*).
Furthermore, in a multicenter cohort study of 269 LMNA mutation
carriers, the same mutation (c.1443C>A, p.Tyr481*) was
identified without detailed data of phenotypes or family history
(24). The present study, which
provided clinical and genetic data for a large family with PCCD,
has enriched the association of genotype and phenotype for LMNA
mutations.
Early-onset PCCD in a structurally normal heart
should prompt consideration of PCCD genetic testing, particularly
if there is a family history of PCCD, pacemaker implantation or
sudden death (33). Once a proband
with a known pathogenic LMNA mutation has been screened, it is
necessary to perform a family screening to identify other carriers
of the mutation (34). For
relatives of a sudden death victim with an unknown genotype, family
screening for an LMNA mutation is a class IIa level of
recommendation, based on the 2015 European Society of Cardiology
Guidelines for the Management of Patients With Ventricular
Arrhythmias and the Prevention of Sudden Cardiac Death (34). The number of asymptomatic relatives
identified with LMNA mutations and subclinical phenotypes is
increasing (35). Family screening
distinguished genotype positives and negatives in the family
included in the present study. Individuals IV-3 and IV-5 were
genotype positive but did not present with typical symptoms or
advanced AVB, therefore it could be suggested that these
individuals should be offered life-long follow-up care (36).
In conclusion, the present study suggested that the
c.1443C>A, p.Tyr481* mutation in LMNA may cause PCCD. However,
further investigation is required to fully understand the
functional changes of this mutation. Family genetic counseling and
sequencing are vital to enable early identification of LMNA
genotype-positive family members, risk stratification and
determination of therapeutic timing. Further functional
identification would be beneficial for improving the existing
knowledge of the pathogenesis of PCCD.
Acknowledgements
Not applicable.
Funding
The present study was supported by the CAMS
Innovation Fund for Medical Sciences (grant no. 2016-I2M-1-002),
the National Key Research and Development Program of China (grant
no. 2016YFC1300100), the National Natural Science Foundation of
China (grant no. 81600305), the Beijing Municipal Science and
Technology Commission (grant no. Z151100003915078), the PUMC Youth
Fund and the Fundamental Research Funds for the Central
Universities (grant nos. 3332015108, 3332015205, 3332018058 and
3332019134), the PUMC Graduate Innovation Fund (grant no.
2018-1002-01-14).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
LPW and XLZ designed and supervised the research.
PF, DZ and KQY collected the samples and the clinical information.
PF, DZ and TT performed the experiments. PF, KQY, FL and YXL
performed the data analysis. PF wrote the manuscript. YXL, LPW and
XLZ edited the manuscript. All authors reviewed the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Fuwai Hospital. Each participant provided written
informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lenegre J and Moreau P: Chronic
auriculo-ventricular block. Anatomical, clinical and histological
study. Arch Mal Coeur Vaiss. 56:867–888. 1963.(In French).
PubMed/NCBI
|
|
2
|
Lev M, Kinare SG and Pick A: The
pathogenesis of atrioventricular block in coronary disease.
Circulation. 42:409–425. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kawaguchi T, Hayashi H, Miyamoto A,
Yoshino T, Taniguchi A, Naiki N, Sugimoto Y, Ito M, Xue Q, Murakami
Y and Horie M: Prognostic implications of progressive cardiac
conduction disease. Circ J. 77:60–67. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gourraud JB, Kyndt F, Fouchard S, Rendu E,
Jaafar P, Gully C, Gacem K, Dupuis JM, Longueville A, Baron E, et
al: Identification of a strong genetic background for progressive
cardiac conduction defect by epidemiological approach. Heart.
98:1305–1310. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Baruteau AE, Probst V and Abriel H:
Inherited progressive cardiac conduction disorders. Curr Opin
Cardiol. 30:33–39. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rankin JS and Ellard S: The laminopathies:
A clinical review. Clin Genet. 70:261–274. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rogozhina Y, Mironovich S, Shestak A,
Adyan T, Polyakov A, Podolyak D, Bakulina A, Dzemeshkevich S and
Zaklyazminskaya E: New intronic splicing mutation in the LMNA gene
causing progressive cardiac conduction defects and variable
myopathy. Gene. 595:202–206. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lin F and Worman HJ: Structural
organization of the human gene encoding nuclear lamin A and nuclear
lamin C. J Biol Chem. 268:16321–16326. 1993.PubMed/NCBI
|
|
9
|
Mattout A, Dechat T, Adam SA, Goldman RD
and Gruenbaum Y: Nuclear lamins, diseases and aging. Curr Opin Cell
Biol. 18:335–341. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mestroni L and Taylor MR: Lamin A/C gene
and the heart: How genetics may impact clinical care. J Am Coll
Cardiol. 52:1261–1262. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
van Berlo JH, de Voogt WG, van der Kooi
AJ, van Tintelen JP, Bonne G, Yaou RB, Duboc D, Rossenbacker T,
Heidbuchel H, de Visser M, et al: Meta-analysis of clinical
characteristics of 299 carriers of LMNA gene mutations: Do lamin
A/C mutations portend a high risk of sudden death? J Mol Med
(Berl). 83:79–83. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hasselberg NE, Haland TF, Saberniak J,
Brekke PH, Berge KE, Leren TP, Edvardsen T and Haugaa KH: Lamin A/C
cardiomyopathy: Young onset, high penetrance, and frequent need for
heart transplantation. Eur Heart J. 39:853–860. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li H and Durbin R: Fast and accurate short
read alignment with burrows-wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
DePristo MA, Banks E, Poplin R, Garimella
KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA,
Hanna M, et al: A framework for variation discovery and genotyping
using next-generation DNA sequencing data. Nat Genet. 43:491–498.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Captur G, Arbustini E, Syrris P,
Radenkovic D, O'Brien B, McKenna WJ and Moon JC: Lamin mutation
location predicts cardiac phenotype severity: Combined analysis of
the published literature. Open Heart. 5:e0009152018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kawakami H, Ogimoto A, Tokunaga N,
Nishimura K, Kawakami H, Higashi H, Iio C, Kono T, Aono J, Uetani
T, et al: A novel truncating LMNA mutation in patients with cardiac
conduction disorders and dilated cardiomyopathy. Int Heart J.
59:531–541. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Captur G, Arbustini E, Bonne G, Syrris P,
Mills K, Wahbi K, Mohiddin SA, McKenna WJ, Pettit S, Ho CY, et al:
Lamin and the heart. Heart. 104:468–479. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Priori SG, Blomstrom-Lundqvist C, Mazzanti
A, Blom N, Borggrefe M, Camm J, Elliott PM, Fitzsimons D, Hatala R,
Hindricks G, et al: 2015 ESC guidelines for the management of
patients with ventricular arrhythmias and the prevention of sudden
cardiac death: The task force for the management of patients with
ventricular arrhythmias and the prevention of sudden cardiac death
of the european society of cardiology (ESC). Endorsed by:
Association for European paediatric and congenital cardiology
(AEPC). Eur Heart J. 36:2793–2867. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nishiuchi S, Makiyama T, Aiba T, Nakajima
K, Hirose S, Kohjitani H, Yamamoto Y, Harita T, Hayano M,
Wuriyanghai Y, et al: Gene-based risk stratification for cardiac
disorders in LMNA mutation carriers. Circ Cardiovasc Genet.
10:e0016032017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
van Rijsingen IA, Arbustini E, Elliott PM,
Mogensen J, Hermans-van Ast JF, van der Kooi AJ, van Tintelen JP,
van den Berg WP, Pilotto A, Pasotti M, et al: Risk factors for
malignant ventricular arrhythmias in lamin a/c mutation carriers a
European cohort study. J Am Coll Cardiol. 59:493–500. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ollila L, Nikus K, Holmstrom M, Jalanko M,
Jurkko R, Kaartinen M, Koskenvuo J, Kuusisto J, Karkkainen S,
Palojoki E, et al: Clinical disease presentation and ECG
characteristics of LMNA mutation carriers. Open Heart.
4:e0004742017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Brodt C, Siegfried JD, Hofmeyer M, Martel
J, Rampersaud E, Li D, Morales A and Hershberger RE: Temporal
relationship of conduction system disease and ventricular
dysfunction in LMNA cardiomyopathy. J Card Fail. 19:233–239. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
van Rijsingen IA, Nannenberg EA, Arbustini
E, Elliott PM, Mogensen J, Hermans-van Ast AJ, van der Kooi AJ, van
Tintelen JP, van den Berg MP, Grasso M, et al: Gender-specific
differences in major cardiac events and mortality in lamin A/C
mutation carriers. Eur J Heart Fail. 15:376–384. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Du XJ: Gender modulates cardiac phenotype
development in genetically modified mice. Cardiovasc Res.
63:510–519. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Babiker FA, De Windt LJ, van Eickel M,
Grohe C, Meyer R and Doevendans PA: Estrogenic hormone action in
the heart: Regulatory network and function. Cardiovasc Res.
53:709–719. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nakajima K, Aiba T, Makiyama T, Nishiuchi
S, Ohno S, Kato K, Yamamoto Y, Doi T, Shizuta S, Onoue K, et al:
Clinical manifestations and long-term mortality in lamin A/C
mutation carriers from a Japanese multicenter registry. Circ J.
82:2707–2714. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Walsh R, Thomson KL, Ware JS, Funke BH,
Woodley J, McGuire KJ, Mazzarotto F, Blair E, Seller A, Taylor JC,
et al: Reassessment of mendelian gene pathogenicity using 7,855
cardiomyopathy cases and 60,706 reference samples. Genet Med.
19:192–203. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ito K, Patel PN, Gorham JM, McDonough B,
De Palma SR, Adler EE, Lam L, MacRae CA, Mohiuddin SM, Fatkin D, et
al: Identification of pathogenic gene mutations in LMNA and MYBPC3
that alter RNA splicing. Proc Natl Acad Sci U S A. 114:7689–7694.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pugh TJ, Kelly MA, Gowrisankar S, Hynes E,
Seidman MA, Baxter SM, Bowser M, Harrison B, Aaron D, Mahanta LM,
et al: The landscape of genetic variation in dilated cardiomyopathy
as surveyed by clinical DNA sequencing. Genet Med. 16:601–608.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kitaguchi T, Matsubara S, Sato M, Miyamoto
K, Hirai S, Schwartz K and Bonne G: A missense mutation in the exon
8 of lamin A/C gene in a Japanese case of autosomal dominant
limb-girdle muscular dystrophy and cardiac conduction block.
Neuromuscul Disord. 11:542–546. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sylvius N, Bilinska ZT, Veinot JP,
Fidzianska A, Bolongo PM, Poon S, McKeown P, Davies RA, Chan KL,
Tang AS, et al: In vivo and in vitro examination of the functional
significances of novel lamin gene mutations in heart failure
patients. J Med Genet. 42:639–647. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ackerman MJ, Priori SG, Willems S, Berul
C, Brugada R, Calkins H, Camm AJ, Ellinor PT, Gollob M, Hamilton R,
et al: HRS/EHRA expert consensus statement on the state of genetic
testing for the channelopathies and cardiomyopathies: This document
was developed as a partnership between the heart rhythm society
(HRS) and the European heart rhythm association (EHRA). Heart
Rhythm. 8:1308–1339. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Priori SG, Blomstrom-Lundqvist C, Mazzanti
A, Blom N, Borggrefe M, Camm J, Elliott PM, Fitzsimons D, Hatala R,
Hindricks G, et al: 2015 ESC guidelines for the management of
patients with ventricular arrhythmias and the prevention of sudden
cardiac death. Rev Esp Cardiol (Engl Ed). 69:1762016.PubMed/NCBI
|
|
35
|
Pinto YM, Elliott PM, Arbustini E, Adler
Y, Anastasakis A, Bohm M, Duboc D, Gimeno J, de Groote P, Imazio M,
et al: Proposal for a revised definition of dilated cardiomyopathy,
hypokinetic non-dilated cardiomyopathy, and its implications for
clinical practice: A position statement of the ESC working group on
myocardial and pericardial diseases. Eur Heart J. 37:1850–1858.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mogensen J, van Tintelen JP, Fokstuen S,
Elliott P, van Langen IM, Meder B, Richard P, Syrris P, Caforio AL,
Adler Y, et al: The current role of next-generation DNA sequencing
in routine care of patients with hereditary cardiovascular
conditions: A viewpoint paper of the European society of cardiology
working group on myocardial and pericardial diseases and members of
the European society of human genetics. Eur Heart J. 36:1367–1370.
2015. View Article : Google Scholar : PubMed/NCBI
|