Introduction
Cerebral ischemia/reperfusion (I/R) injury, which
coexists with ischemia over a certain time period, may result in
serious cerebral microcirculatory damage and is a major cause of
disability and mortality worldwide (1). Although considerable improvements in
diagnosis, treatment and rehabilitation have been achieved
(2), safe and effective
therapeutic agents for cerebral I/R are still urgently needed
(3).
Cerebral I/R injury involves multiple pathological
processes, among which neuroinflammation, apoptosis and oxidative
stress have been considered to play key roles in brain damage
during cerebral I/R (4–6). Accumulating evidence has demonstrated
the presence of excessive pro-inflammatory factors, pro-apoptotic
proteins and oxidative stress products in cerebral I/R injury
(6–8). Furthermore, previous studies
indicated that the phosphoinositide 3-kinase (PI3K)/protein kinase
B (Akt) pathway exerts neuroprotective effects via inhibiting
inflammation, oxidative stress and apoptosis (9–11).
Thus, targeting inflammation, oxidative stress and apoptosis may be
a viable therapy option for cerebral I/R injury.
C1q/tumor necrosis factor (TNF)-related protein-6
(CTRP6) is a member of the adiponectin paralogs family of proteins
designated as CTRPs. Although CTRPs share structural homology with
adiponectin, each CTRP has a unique tissue distribution and
exhibits diverse functions (12).
CTRP6, which is expressed in adipose tissue, placenta, heart and
brain, exerts modulating effects on energy metabolism and
inflammation (13,14). Considering that CTRP6 induces the
expression of interleukin (IL)-10 via extracellular
signal-regulated kinase 1/2 activation in macrophages (15), it may represent a novel target for
pharmacological therapy in inflammatory diseases. Of note, several
studies have confirmed the protective effects of CTRP6 on
inflammation-related diseases, such as myocardial fibrosis
(16), cardiac injury (17) and arthritis (18), but the role of CTRP6 in I/R injury
is rarely reported. Interestingly, as another member of the CTRP
family, CTRP9 has been reported to protect against myocardial I/R
injury via suppression of cell apoptosis (19,20),
but whether CTRP6 protects against cerebral I/R injury remains
elusive.
The small G protein Ras homolog family member A
(RhoA) and its downstream effector Rho-associated
coiled-coil-containing protein kinase (Rock) extensively
participate in the regulation of I/R injury in the myocardium,
brain, liver or kidney (21).
Previous evidence suggested that inhibition of RhoA/Rock signaling
may initiate phosphatase and tensin homologue deleted on chromosome
10 (PTEN) activity to activate the PI3K/Akt pathway, which, in
turn, prevents the inflammatory response in ischemic brain and
liver models (22,23). Moreover, CTRP6 was shown to improve
cardiac function or attenuate post-infarct cardiac fibrosis via
activation of Akt by targeting the RhoA pathway (16,17).
Therefore, the present study was designed to i)
determine the functions of CTRP6 in cerebral I/R injury; ii)
investigate whether CTRP6 regulates cerebral I/R injury induced by
oxygen-glucose deprivation and reperfusion (OGD/R) in PC12 cells
and iii) determine whether RhoA or Akt are responsible for the
actions of CTRP6 upon cerebral I/R injury, and elucidate the
detailed mechanisms.
Materials and methods
Cell culture
PC12 cells (American Type Culture Collection) were
cultured in standard growth medium (DMEM, Gibco; Thermo Fisher
Scientific, Inc.), supplemented with 10% FBS (Gibco; Thermo Fisher
Scientific, Inc.) and 1% penicillin-streptomycin at 37°C in a 5%
CO2 atmosphere.
OGD/R
For induction of an in vitro I/R injury
model, PC12 cells were incubated in a deoxygenated glucose-free
Hanks' Balanced Salt Solution within an anaerobic chamber (5%
CO2, 95% N2) at 37°C for 2 h. After OGD
treatment for 2 h, the cells were removed from the anoxic
atmosphere and transferred to a normal environment for 12 h.
Plasmid constructs and cell
transfection
Full-length cDNAs of rat CTRP6 or RhoA were cloned
into the pcDNA3.1 vector (Thermo Fisher Scientific, Inc.). A
pcDNA3.1 empty vector was used as a negative control. PC12 cells
were transfected with 12.5 mg/l pcDNA3.1-CTRP6, pcDNA3.1-RhoA or
pcDNA3.1 using Lipofectamine® 2000 reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. Following 48 h of transfection, the cells were collected
for subsequent experiments.
Western blot analysis
To determine protein expression levels, total
protein was extracted from PC12 cells using RIPA lysis buffer
(Thermo Fisher Scientific, Inc.). All protein samples were
quantified by a bicinchoninic acid kit (Thermo Fisher Scientific,
Inc) and equal amounts (50 µg) of each sample were separated by
8–12% SDS-PAGE. Separated proteins were transferred onto PVDF
membranes (Bio-Rad Laboratories, Inc.) and then blocked with 5%
non-fat milk at 37°C for 1 h, after which time the membranes were
incubated with the following primary antibodies (Abcam) overnight
at 4°C: Anti-CTRP6 (cat. no. ab36900; 1:1,000), anti-Bcl2 (cat. no.
ab32124; 1:1,000), anti-Bax (cat. no. ab32503; 1:1,000),
anti-cleaved caspase-3 (cat. no. ab2302; 1:500), anti-caspase-3
(cat. no. ab13847; 1:500), anti-cleaved caspase-9 (cat. no.
ab32539; 1:1,000), anti-caspase-9 (cat. no. ab2324; 1:400),
anti-RhoA (cat. no. ab187027; 1:5,000), anti-Rock1 (cat. no.
ab45171; 1:2,000), anti-Rock2 (cat. no. ab71598; 1:1,000),
anti-PTEN (cat. no. ab32199; 1:10,000), anti-Akt (cat. no. ab8805;
1:500), anti-PI3K (cat. no. ab109006; 1:1,000),
anti-phosphorylated-PI3K (cat. no. ab133595; 1:1,000),
anti-phosphorylated-Akt (cat. no. ab81283; 1:5,000) and anti-GAPDH
(Santa Cruz Biotechnology, Inc.; cat. no. Sc-47724; 1:1,000).
Finally, the membranes were incubated with a horseradish-conjugated
secondary antibody (goat anti-rabbit IgG; cat. no. ab205718;
1:10,000; Abcam) at room temperature for 2 h. Protein bands were
visualized using an electrochemiluminescence system (Amersham
Imager 600; GE Healthcare) and an Immun-Star HRP chemiluminescent
substrate kit (cat. no. 1705041; Bio-Rad Laboratories, Inc.) at
room temperature for 2 min. ImageJ version 1.8.0_112 software
(National Institutes of Health) was used to quantify the expression
levels of each protein.
RNA isolation and reverse
transcription-quantitative PCR (RT-qPCR) analysis
Total RNA was extracted from PC12 cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol, and 1 µg total RNA
was converted to cDNA using the PrimeScript™ RT reagent kit with
gDNA Eraser (Takara Biotechnology Co., Ltd.), according to the
manufacturer's protocol. The following RT temperature protocol was
used: 37°C for 15 min and 85°C for 5 sec. qPCR was performed using
TB Green® Fast qPCR Mix (Takara Biotechnology Co.,
Ltd.), according to the manufacturer's protocol. Differential
expression of mRNA was calculated using the 2−ΔΔCq
method (24). The primers used
were as follows: CTRP6, forward 5′-CCATCCTGAAAGGTGACAAAGG-3′ and
reverse 5′-AGTAATGCGTCTGGCACGAG-3′; RhoA, forward
5′-CCAAAATGAAGCAGGAGCCG-3′ and reverse 5′-ATGAGGCACCCCGACTTTTT-3′;
and GAPDH, forward 5′-AGTGCCAGCCTCGTCTCATA-3′ and reverse
5′-TGAACTTGCCGTGGGTAGAG-3′. The following thermocycling conditions
were used for the qPCR: Initial denaturation at 95°C for 30 sec;
and 40 cycles of 95°C for 5 sec and 60°C for 15 sec; followed by a
default melting curve.
MTT assay
The cell viability of PC12 cells under different
conditions was assessed by MTT assay (Beyotime Institute of
Biotechnology; cat. no. C0009), according to the manufacturer's
protocol. In brief, cells that transfected with or without
indicated plasmids were seeded in 96-well plates for 24 h and then
incubated with 20 µl MTT solution for 4 h. After discarding MTT
medium dissolving the remaining formazan crystals with DMSO, the
cell viability was tested at 570 nm by a microplate reader (Thermo
Fisher Scientific, Inc).
Detection of inflammatory factors and
oxidative stress levels
The levels of the pro-inflammatory cytokines TNF-α
(cat. no. ab100785), IL-1β (cat. no. ab100768), IL-6 (cat. no.
ab100772) and IL-10 (cat. no. ab100765) were detected in PC12 cells
using their respective ELISA kits (Abcam), according to the
manufacturer's protocol. A cellular reactive oxygen species (ROS;
cat. no. ab113851) assay kit, superoxide dismutase (SOD; cat. no.
ab65354) activity assay kit and lipid peroxidation product
malondialdehyde (MDA; cat. no. ab118970) assay kit (Abcam) were
used to determine ROS, SOD and MDA activity, respectively,
according to the manufacturers' protocols.
Flow cytometry
Cell apoptosis was assessed by flow cytometry using
propidium iodide (PI) staining. Briefly, cells were gently washed
twice with PBS, digested with 0.25% trypsin and centrifuged at 200
× g at 4°C for 5 min. After the resuspension of the cell pellet
with 1 ml NaCl/Pi supplemented with 100 µg/ml RNase, the cells were
incubated with PI at 4°C for 15 min in the dark and immediately
analyzed using a BD FACSCanto™ II flow cytometer (BD Biosciences).
Data were analyzed using FlowJo 7.6 software (FlowJo LLC).
Statistical analysis
All data are expressed as the mean ± standard
deviation and the statistical analyses were conducted using
GraphPad Prism 6 (GraphPad Software, Inc.). Differences were
calculated by Student's t-test or one-way ANOVA followed by
Turkey's post hoc test for ≥3 samples. P<0.05 was considered to
indicate statistically significant differences.
Results
CTRP6 expression is markedly
downregulated after OGD/R
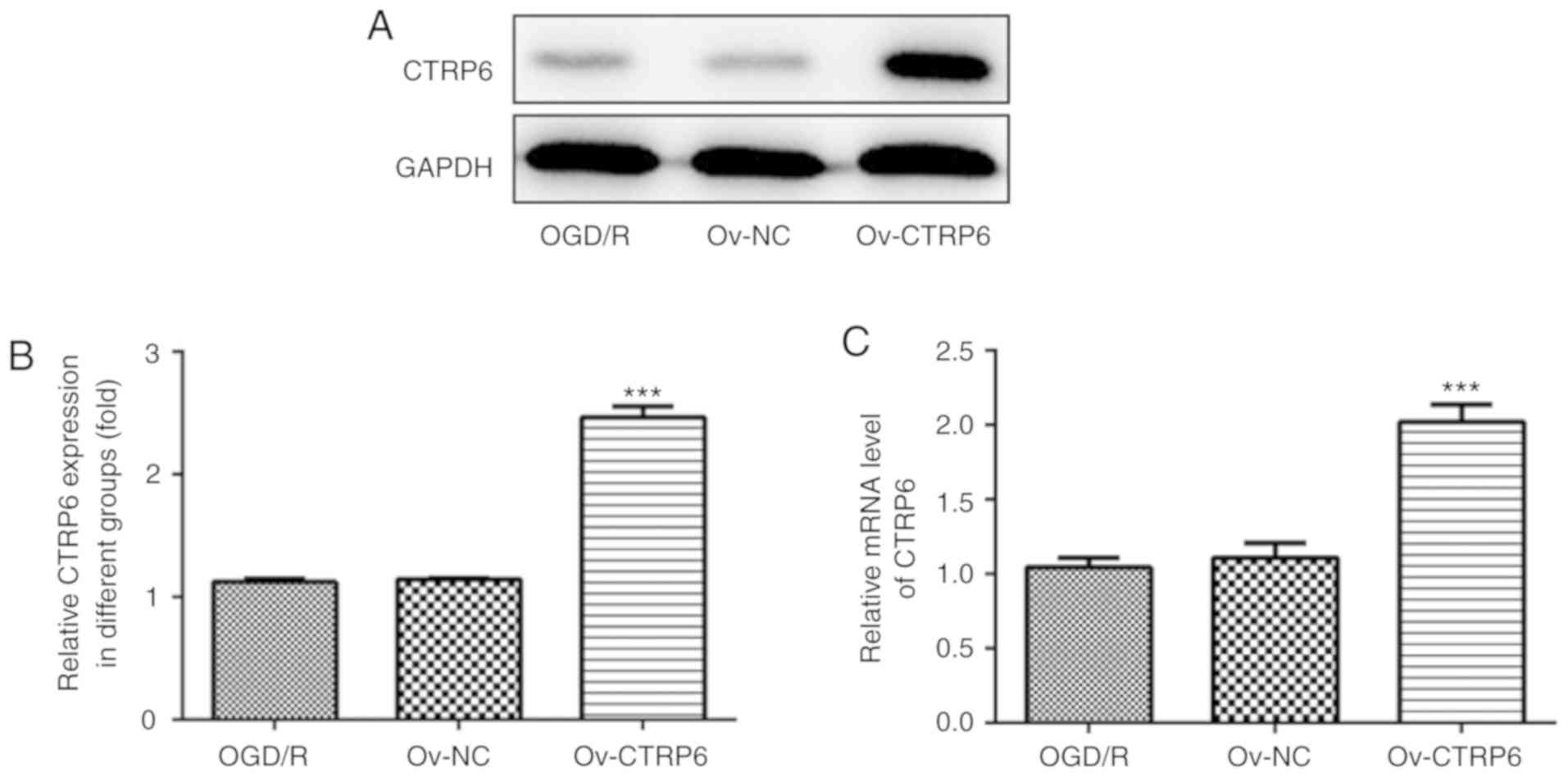
CTRP6 expression was first detected in PC12 cells
subjected to OGD/R treatment and it was observed that CTRP6 protein
expression was significantly reduced in OGD/R-treated cells
(Fig. 1A and B). Further detection
revealed that the CTRP6 mRNA level also decreased following OGD/R
treatment (Fig. 1C). The
alteration of CTRP6 expression suggested a role for CTRP6 in
OGD/R-induced cerebral I/R injury in PC12 cells.
CTRP6 overexpression protects against
OGD/R-induced inflammation, oxidative stress and cell
apoptosis
To explore the specific role of CTRP6 in cerebral
I/R injury, recombinant plasmids expressing CTRP6 were transfected
into PC12 cells to overexpress CTRP6. As shown in Fig. 2A and B, CTRP6 protein and mRNA
expression was significantly increased in cells treated with
pcDNA3.1-CTRP6, suggesting the efficiency of CTRP6
overexpression.
To determine whether CTRP6 could alleviate
OGD/R-induced inflammation, the activities of inflammatory
molecules, including TNF-α, IL-1β, IL-6 and IL-10, were detected. A
significant increase in TNF-α, IL-1β and IL-6 and a decrease in the
anti-inflammatory cytokine IL-10 were observed in OGD/R-treated
cells (Fig. 3B-E), indicating the
occurrence of inflammation induced by OGD/R. In addition, CTRP6
overexpression successfully rescued the activities of these
inflammatory factors (Fig. 3B-E).
These results suggested that CTRP6 was able to inhibit
OGD/R-induced inflammation in PC12 cells.
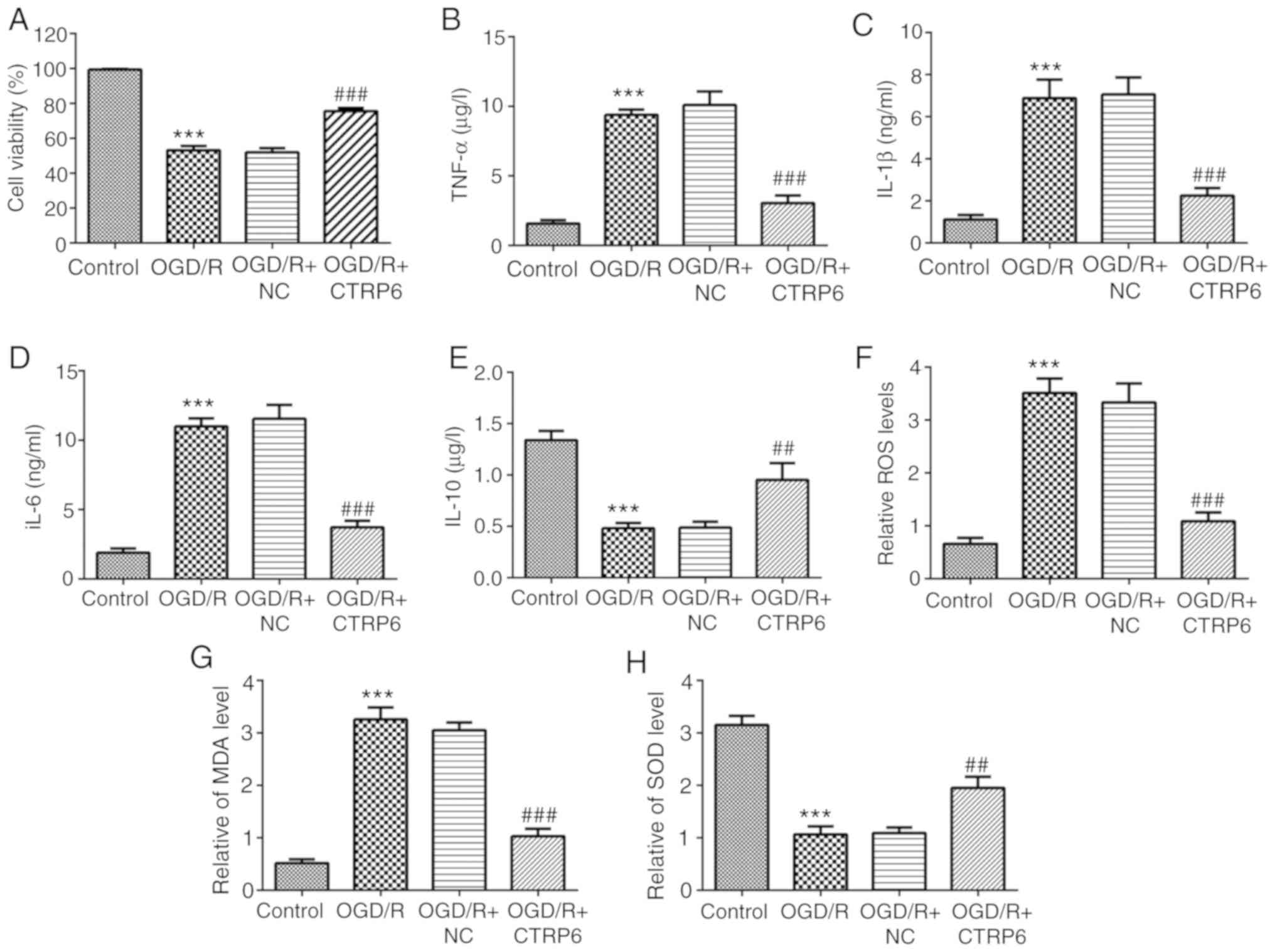 | Figure 3.CTRP6 overexpression inhibits
OGD/R-induced inflammation and oxidative stress in PC12 cells.
Alteration of (A) cell viability and inflammatory cytokines (B)
TNF-α, (C) IL-1β, (D) IL-6 and (E) IL-10 levels (n=3). Alteration
of oxidative products (F) ROS, (G) MDA and (H) antioxidant SOD
levels (n=3). ***P<0.001 vs. control, ##P<0.01 and
###P<0.001 vs. OGD/R + NC. CTRP6, C1q/tumor necrosis
factor-related protein-6; OGD/R, oxygen-glucose deprivation and
reperfusion; NC, negative control; TNF, tumor necrosis factor; IL,
interleukin; ROS, reactive oxygen species; MDA, malondialdehyde;
SOD, superoxide dismutase. |
Oxidative stress also plays a key role in cerebral
I/R injury (4) and the present
results demonstrated that the levels of the oxidative products ROS
and MDA were upregulated following OGD/R treatment (Fig. 3F and 3G), while the level of the
antioxidant enzyme SOD was significantly reduced (Fig. 3H). Of note, the levels of ROS, MDA
and SOD were also partially restored by CTRP6 overexpression
(Fig. 3F-H).
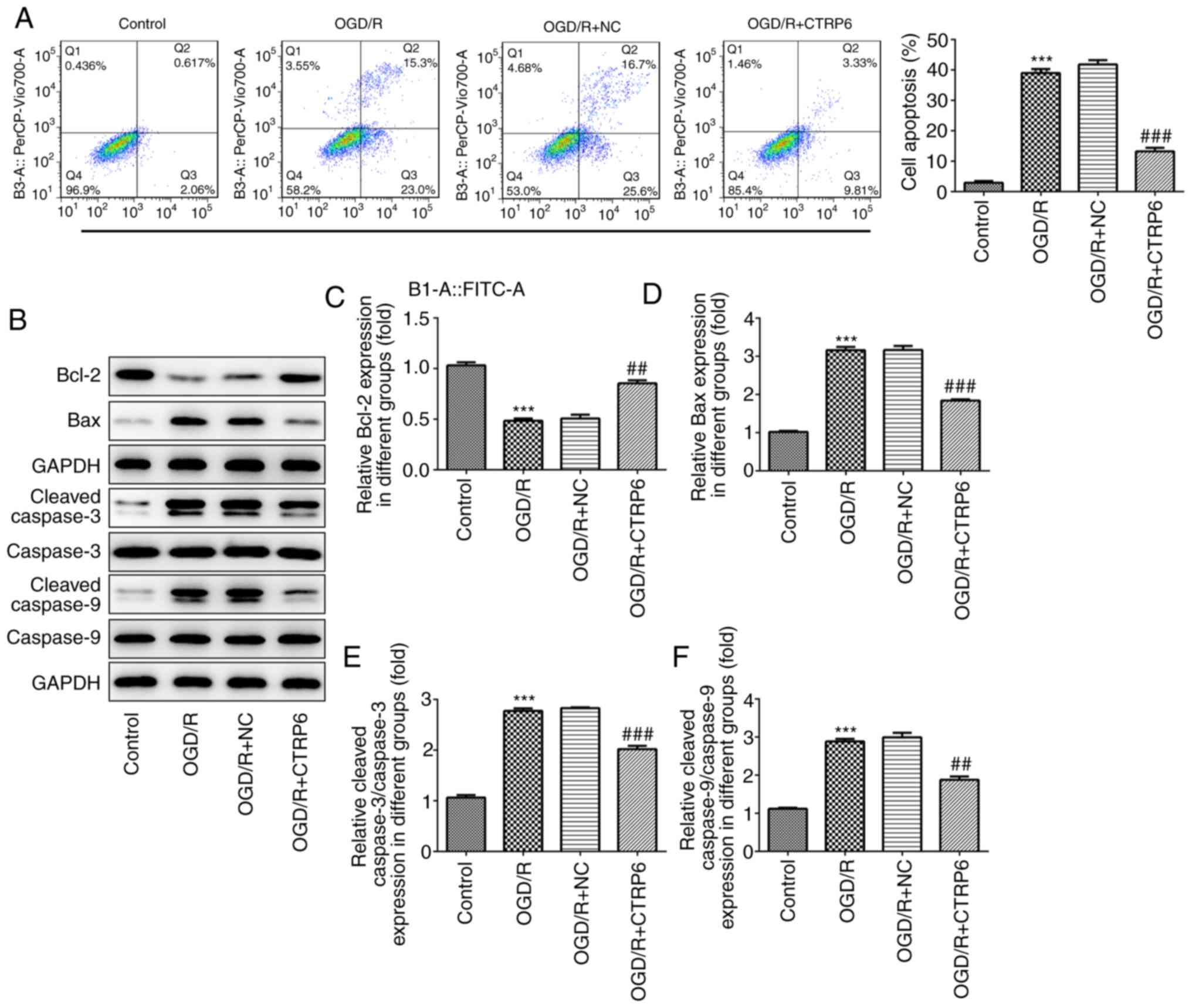
The OGD/R-induced apoptosis was significantly
rescued by CTRP6 overexpression. As shown in Fig. 4A, CTRP6 reduced the ratio of
apoptotic cells. However, the expression of apoptosis-related
proteins, including Bcl-2, Bax, cleaved caspase-3 and caspase-9,
was partially recovered when OGD/R treatment was given in
combination with CTRP6 overexpression (Fig. 4B-F). Taken together, these findings
indicate that CTRP6 may alleviate cerebral I/R injury via
inhibiting inflammation, oxidative stress and apoptosis.
CTRP6 inhibits RhoA/Rock/PTEN
signaling and activates the PI3K/Akt pathway
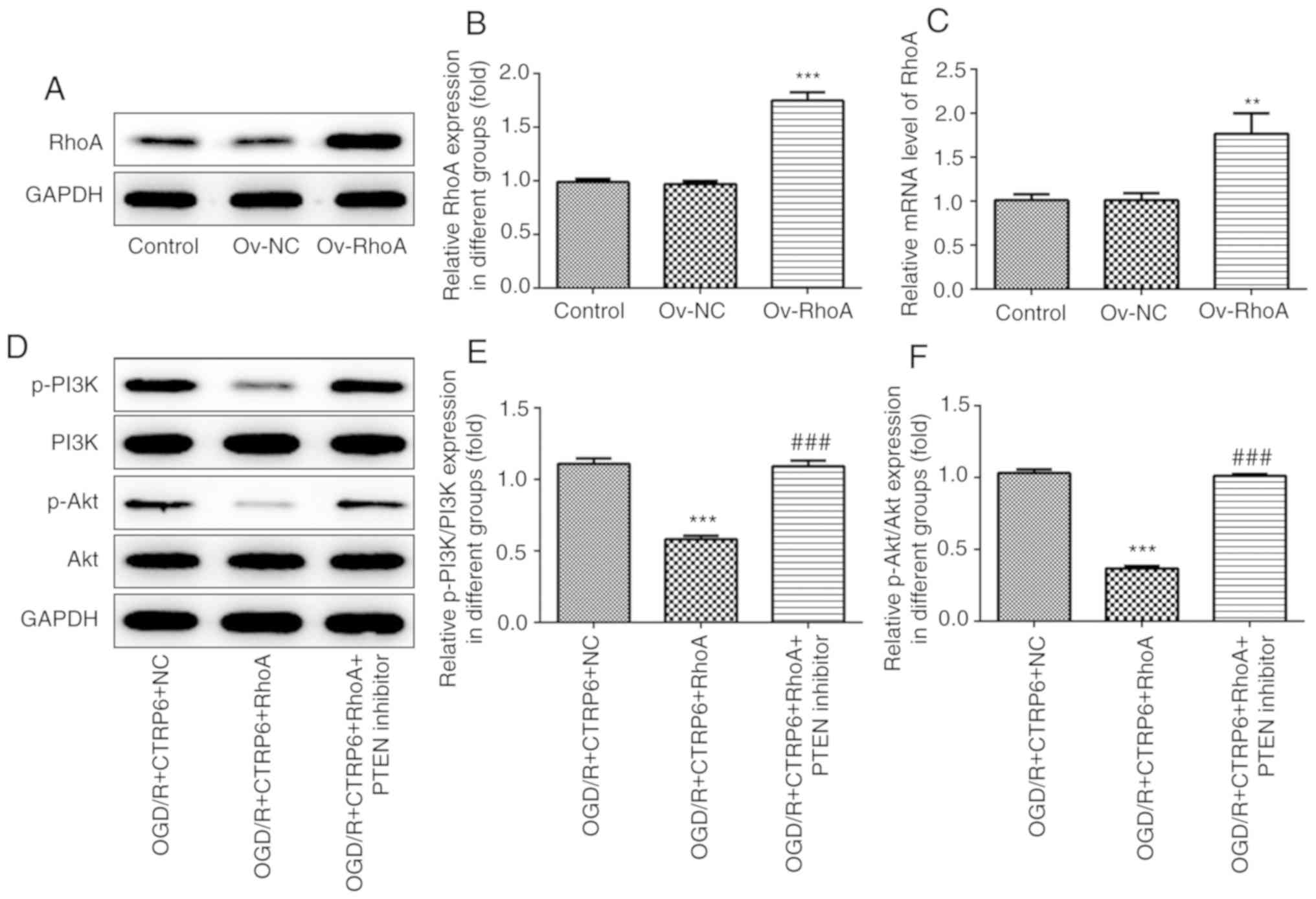
To explore whether RhoA and Akt-associated signaling
are the targets of CTRP6, western blotting was used to detect the
protein expression of GTP-RhoA, Rock1, PTEN, phosphorylated
(p)-PI3K and p-Akt. The data revealed enhanced expression of
GTP-RhoA, Rock1 and PTEN, and reduced p-PI3K and p-Akt levels upon
OGD/R treatment (Fig. 5),
indicating that the RhoA/Rock/PTEN and PI3K/Akt pathways are
involved in cerebral I/R injury. Next, CTRP6 overexpression was
used to determine its effect on RhoA/Rock/PTEN and PI3K/Akt
signaling, and it was observed that CTRP6 overexpression inhibited
RhoA, Rock and PTEN expression, but reactivated PI3K and Akt
expression in a cerebral I/R injury cell model (Fig. 5).
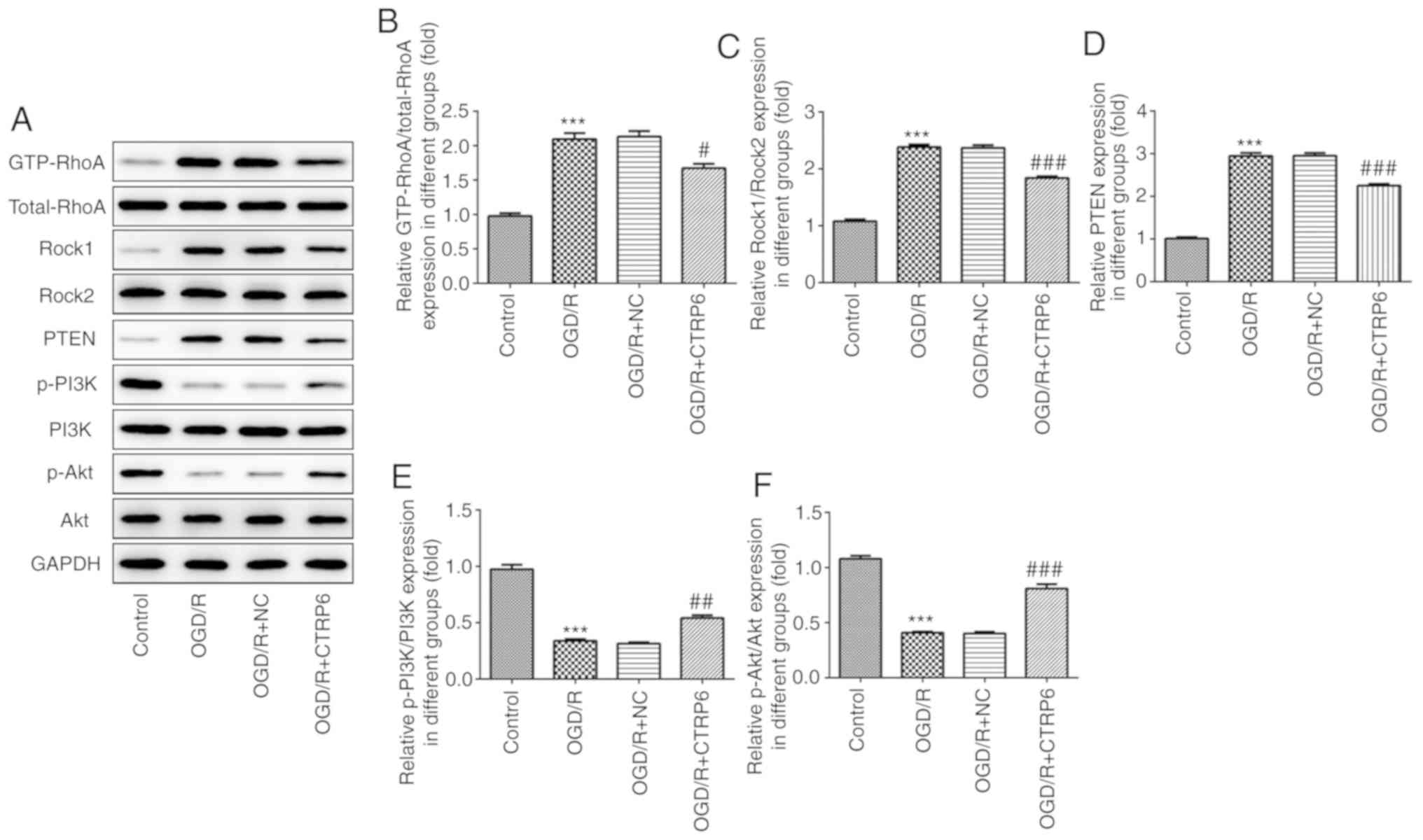 | Figure 5.CTRP6 overexpression inhibits
RhoA/Rock/PTEN and activates the PI3K/Akt signaling pathway. (A)
Representative immunoblot analysis for RhoA, Rock1, PTEN, p-PI3K
and p-Akt. Relative protein expression of (B) RhoA, (C) Rock1, (D)
PTEN, (E) p-PI3K and (F) p-Akt after normalization to GAPDH (n=3).
***P<0.001 vs. control, #P<0.05,
##P<0.01 and ###P<0.001 vs. OGD/R + NC.
CTRP6, C1q/tumor necrosis factor-related protein-6; OGD/R,
oxygen-glucose deprivation and reperfusion; NC, negative control;
RhoA, Ras homolog family member A; Rock, Rho-associated
coiled-coil-containing protein kinase; PTEN, phosphatase and tensin
homologue deleted on chromosome 10; PI3K, phosphoinositide
3-kinase; Akt, protein kinase B; p, phosphorylated. |
Overexpression of RhoA abolishes,
while the PTEN inhibitor recovers, the protective effects of CTRP6
against OGD/R-induced inflammation, oxidative stress and cell
apoptosis
To further confirm the role of the RhoA/Rock/PTEN
and PI3K/Akt pathways in the protective effects of CTRP6 against
cerebral I/R injury, recombinant plasmids expressing RhoA were
transfected into PC12 cells to overexpress RhoA (Fig. 6A-C). The results of western
blotting demonstrated that the CTRP6-stimulated activation of the
PI3K/Akt pathway was blunted by RhoA overexpression (Fig. 6D-F). However, the expression of
PI3K and the level of p-Akt were rescued when the cells were
treated with a PTEN inhibitor (Fig.
6D-F). Consequently, it was confirmed that CTRP6 activates the
PI3K/Akt pathway via inhibiting RhoA/Rock/PTEN signaling in
OGD/R-induced PC12 cells.
To elucidate the direct effect of RhoA and PTEN on
CTRP6 against OGD/R-induced cerebral I/R injury, the levels of
cytokines and proteins associated with inflammation, oxidative
stress and apoptosis were detected when cells overexpressed RhoA or
were treated with a PTEN inhibitor. First, an ELISA was applied to
evaluate the activities of inflammation-related factors. As seen
from the data, the CTRP6-stimulated decrease in the levels of
TNF-α, IL-1β, IL-6 and increase in the levels of IL-10 was
diminished by RhoA overexpression, whereas it was rescued by the
PTEN inhibitor (Figs. 3B-E and
7B-E). In the same manner, RhoA
overexpression cancelled, while PTEN inhibitor rescued the
alleviation of oxidative stress caused by CTRP6 (Figs. 3F-H and 7F-H). Finally, the ratio of apoptotic
cells and apoptosis-related protein expression were also evaluated
by flow cytometry and western blotting. RhoA overexpression and
PTEN inhibitor exerted the same effect on the protective action of
CTRP6 against apoptosis (Figs. 4
and 8).
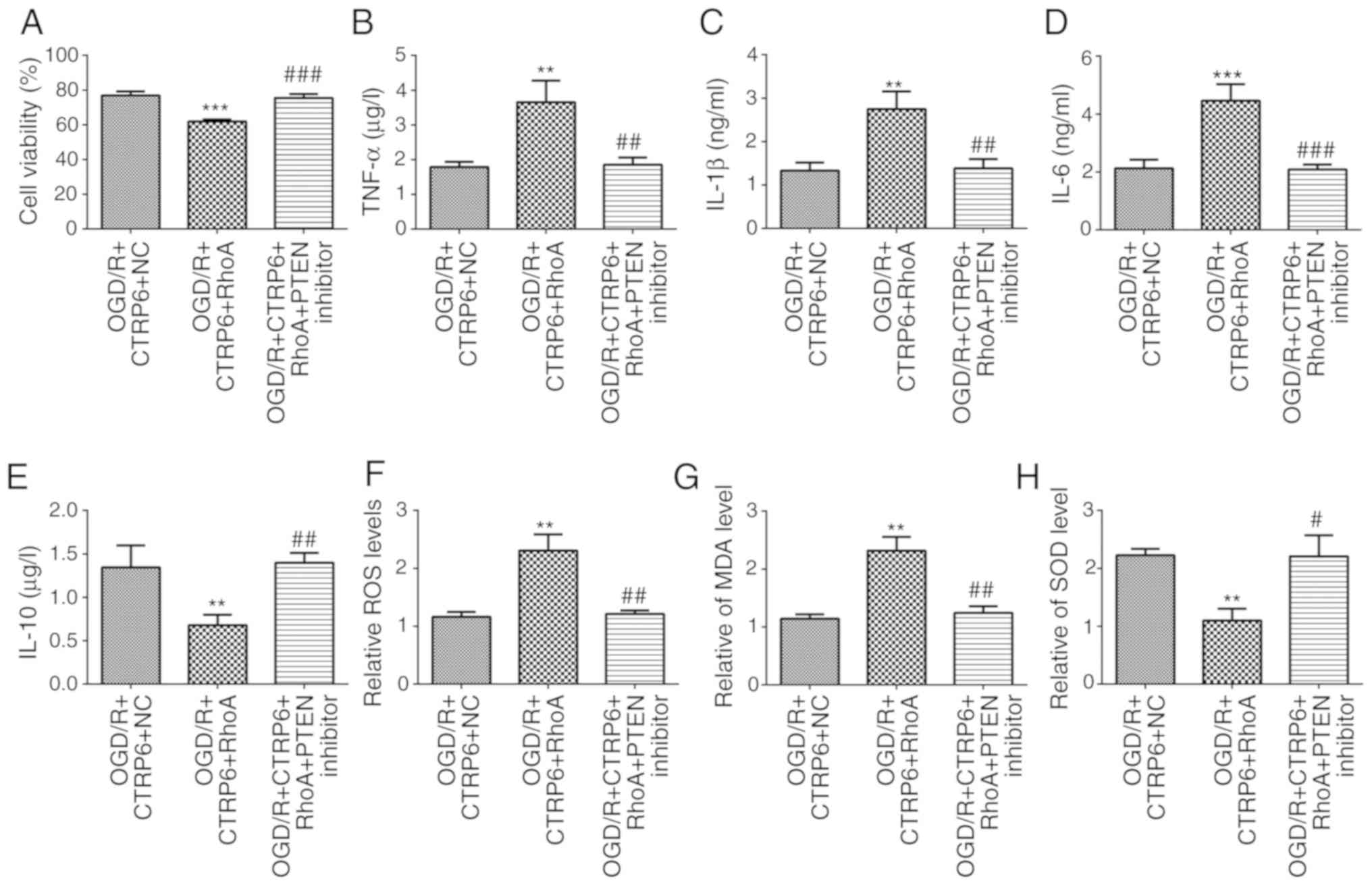 | Figure 7.RhoA overexpression abolishes, while
the PTEN inhibitor recovers, the protective effects of CTRP6
against OGD/R-induced inflammation and oxidative stress. Alteration
of (A) cell viability and inflammatory cytokines (B) TNF-α, (C)
IL-1β, (D) IL-6 and (E) IL-10 levels in different groups (n=3).
Alteration of oxidative products (F) ROS, (G) MDA and (H)
antioxidant SOD levels in different groups (n=3). **P<0.01 and
***P<0.001 vs. OGD/R + CTRP6 + NC, #P<0.05,
##P<0.01 and ###P<0.001 vs. OGD/R +
CTRP6 + RhoA. CTRP6, C1q/tumor necrosis factor-related protein-6;
OGD/R, oxygen-glucose deprivation and reperfusion; NC, negative
control; RhoA, Ras homolog family member A; PTEN, phosphatase and
tensin homologue deleted on chromosome 10; TNF, tumor necrosis
factor; IL, interleukin; ROS, reactive oxygen species; MDA,
malondialdehyde; SOD, superoxide dismutase. |
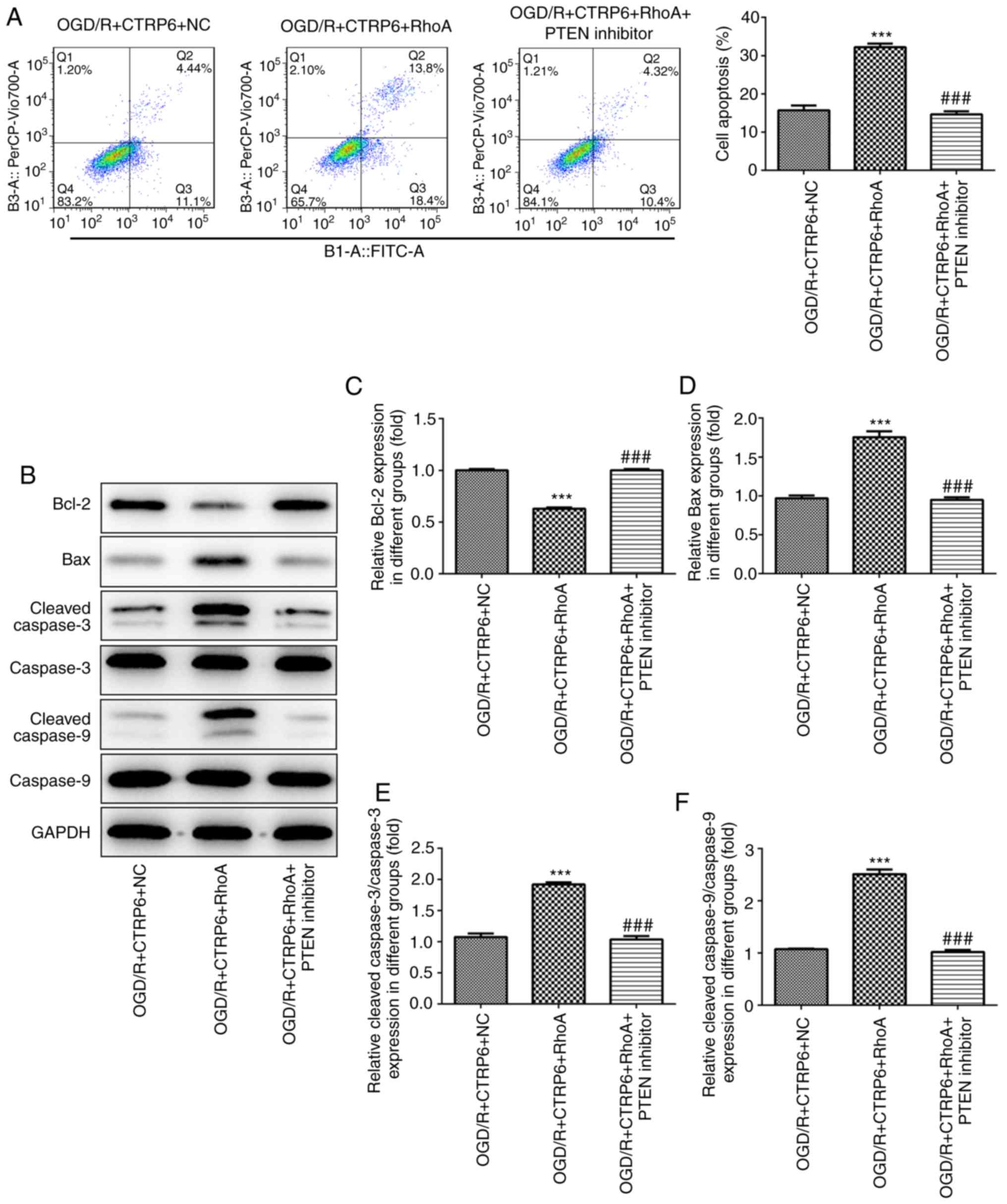 | Figure 8.RhoA overexpression abolishes, while
the PTEN inhibitor rescues, the protective effects of CTRP6 against
OGD/R-induced apoptosis. (A) Cell apoptosis was assessed by flow
cytometry and the ratio of apoptotic cells in each group was
calculated. (B) Representative immunoblot analysis for
apoptosis-related proteins. Relative protein expression of (C)
anti-apoptosis protein Bcl-2 and pro-apoptosis proteins (D) Bax,
(E) cleaved caspase-3 and (F) cleaved caspase-9 (n=3).
***P<0.001 vs. OGD/R + CTRP6 + NC, ###P<0.001 vs.
OGD/R + CTRP6 + RhoA. CTRP6, C1q/tumor necrosis factor-related
protein-6; OGD/R, oxygen-glucose deprivation and reperfusion; NC,
negative control; RhoA, Ras homolog family member A; PTEN,
phosphatase and tensin homologue deleted on chromosome 10; FITC,
fluorescein isothiocyanate. |
Discussion
To the best of our knowledge, the present study is
the first to document the pivotal role of CTRP6 in regulating
RhoA/Rock/PTEN and PI3K/Akt signaling-mediated inflammation,
oxidative stress and apoptosis in I/R injury. Consistent with
previously reported studies (6,8), the
present results verified the participation of inflammation,
oxidative stress and apoptosis in I/R injury. Due to the
significant decrease of CTRP6 level in OGD/R-treated PC12 cells,
the replenishment of CTRP6 may be effective in reducing cerebral
I/R damage. In accordance with the current hypothesis,
overexpression of CTRP6 before the induction of OGD/R led to a
reduction in inflammation, oxidative stress and apoptosis following
I/R injury in PC12 cells.
A recent report demonstrated that CTRP6 protected
cardiomyocytes from doxorubicin-induced apoptosis and activated the
Akt pathway in vitro (17).
In the present study, CTRP6 overexpression enhanced activation of
Akt and its upstream regulator PI3K, and inhibited the activation
of RhoA, Rock and PTEN in OGD/R-treated PC12 cells. Rock is a major
downstream effector of RhoA, while PTEN has been identified as a
Rock substrate as well as a negative regulator of the PI3K/Akt
pathway, which plays important roles in cell survival and apoptosis
(25–27). In previous years, an increasing
number of studies have confirmed that the RhoA/Rock/PTEN/PI3K/Akt
signaling network also plays a crucial role in I/R injury (21–23,28).
Therefore, it was inferred that replenishment of CTRP6 could
protect against OGD/R-induced injury via inhibiting the
RhoA/Rock/PTEN signaling pathway, thereby activating PI3K/Akt
signaling.
To validate the present hypothesis, RhoA was
overexpressed, with or without treatment with the PTEN inhibitor,
to observe the effect of CTRP6 on OGD/R-induced injury. As
expected, the protective effects of CTRP6 against OGD/R-induced
inflammation, oxidative stress and cell apoptosis were all
diminished by RhoA overexpression; however, the presence of the
PTEN inhibitor recovered the protective effects of CTRP6.
As an adipocytokine similar to other members of the
CTRP family, CTRP6 was shown to play a key role in obesity,
diabetes and vascular diseases (29–31);
however, the role of CTRP6, as well as that of other CTRPs, in
cerebral I/R injury remains elusive. Previous studies have
suggested the protective effects of CTRP1 and CTRP9 against
ischemia in the heart (20,32),
but the specific role of CTRPs in ischemic diseases is poorly
understood. The current study provided evidence that CTRP6 exerts
neuroprotective effects against I/R injury through the
RhoA/Rock/PTEN/PI3K/Akt signaling network. Further in vitro
and in vivo research is required to investigate the
functions of CTRP6 and other CTRP members in cerebral I/R
injury.
In conclusion, the present study demonstrated that
CTRP6 acts as a novel regulator of cerebral I/R-induced
inflammation, oxidative stress and cell apoptosis through
inhibition of the RhoA/Rock/PTEN pathway and activation of PI3K/Akt
signaling. Therefore, enhancing CTRP6 production may be of value
for the prevention and/or treatment of cerebral I/R injury.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from the
Key Medical Discipline Development Programs of Beijing Hospitals
Authority (grant no. ZYLX201832).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
XG and YL conceived and designed the study; YL and
JS performed the experiments and acquired the data; LG analyzed and
interpreted the data; and YL and XG drafted the manuscript and
revised it critically for important intellectual content. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Prabhakaran S, Ruff I and Bernstein RA:
Acute stroke intervention: A systematic review. JAMA.
313:1451–1462. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Palomares SM and Cipolla MJ: Vascular
protection following cerebral ischemia and reperfusion. J Neurol
Neurophysiol. 2011(pii): S1–004. 2011.PubMed/NCBI
|
|
3
|
Thompson BJ and Ronaldson PT: Drug
delivery to the ischemic brain. Adv Pharmacol. 71:165–202. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sanderson TH, Reynolds CA, Kumar R,
Przyklenk K and Huttemann M: Molecular mechanisms of
ischemia-reperfusion injury in brain: Pivotal role of the
mitochondrial membrane potential in reactive oxygen species
generation. Mol Neurobiol. 47:9–23. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gong L, Tang Y, An R, Lin M, Chen L and Du
J: RTN1-C mediates cerebral ischemia/reperfusion injury via ER
stress and mitochondria-associated apoptosis pathways. Cell Death
Dis. 8:e30802017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang DD, Zou MJ, Zhang YT, Fu WL, Xu T,
Wang JX, Xia WR, Huang ZG, Gan XD, Zhu XM and Xu DG: A novel
IL-1RA-PEP fusion protein with enhanced brain penetration
ameliorates cerebral ischemia-reperfusion injury by inhibition of
oxidative stress and neuroinflammation. Exp Neurol. 297:1–13. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu H, Tang C, Tai LW, Yao W, Guo P, Hong
J, Yang X, Li X, Jin Z, Ke J and Wang Y: Flurbiprofen axetil
attenuates cerebral ischemia/reperfusion injury by reducing
inflammation in a rat model of transient global cerebral
ischemia/reperfusion. Biosci Rep. 38(pii): BSR201715622018.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Majid A: Neuroprotection in stroke: Past,
present, and future. ISRN Neurol. 2014:5157162014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yu ZH, Cai M, Xiang J, Zhang ZN, Zhang JS,
Song XL, Zhang W, Bao J, Li WW and Cai DF: PI3K/Akt pathway
contributes to neuroprotective effect of Tongxinluo against focal
cerebral ischemia and reperfusion injury in rats. J Ethnopharmacol.
181:8–19. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang Q, An R, Tian X, Yang M, Li M, Lou
J, Xu L and Dong Z: β-Caryophyllene pretreatment alleviates focal
cerebral ischemia-reperfusion injury by activating PI3K/Akt
signaling pathway. Neurochem Res. 42:1459–1469. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang JF, Zhang L, Shi LL, Zhao ZH, Xu H,
Liang F, Li HB, Zhao Y, Xu X, Yang K and Tian YF: Parthenolide
attenuates cerebral ischemia/reperfusion injury via Akt/GSK-3β
pathway in PC12 cells. Biomed Pharmacother. 89:1159–1165. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schaffler A and Buechler C: CTRP family:
Linking immunity to metabolism. Trends Endocrinol Metab.
23:194–204. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wong GW, Krawczyk SA, Kitidis-Mitrokostas
C, Revett T, Gimeno R and Lodish HF: Molecular, biochemical and
functional characterizations of C1q/TNF family members:
Adipose-tissue-selective expression patterns, regulation by
PPAR-gamma agonist, cysteine-mediated oligomerizations,
combinatorial associations and metabolic functions. Biochem J.
416:161–177. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ohashi K, Shibata R, Murohara T and Ouchi
N: Role of anti-inflammatory adipokines in obesity-related
diseases. Trends Endocrinol Metab. 25:348–355. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim MJ, Lee W, Park EJ and Park SY:
C1qTNF-related protein-6 increases the expression of interleukin-10
in macrophages. Mol Cells. 30:59–64. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lei H, Wu D, Wang JY, Li L, Zhang CL, Feng
H, Fu FY and Wu LL: C1q/tumor necrosis factor-related protein-6
attenuates post-infarct cardiac fibrosis by targeting RhoA/MRTF-A
pathway and inhibiting myofibroblast differentiation. Basic Res
Cardiol. 110:352015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zheng WF, Zhang SY, Ma HF, Chang XW and
Wang H: C1qTNF-related protein-6 protects against
doxorubicin-induced cardiac injury. J Cell Biochem.
120:10748–10755. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Murayama MA, Kakuta S, Inoue A, Umeda N,
Yonezawa T, Maruhashi T, Tateishi K, Ishigame H, Yabe R, Ikeda S,
et al: CTRP6 is an endogenous complement regulator that can
effectively treat induced arthritis. Nat Commun. 6:84832015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao D, Feng P, Sun Y, Qin Z, Zhang Z, Tan
Y, Gao E, Lau WB, Ma X, Yang J, et al: Cardiac-derived CTRP9
protects against myocardial ischemia/reperfusion injury via
calreticulin-dependent inhibition of apoptosis. Cell Death Dis.
9:7232018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kambara T, Ohashi K, Shibata R, Ogura Y,
Maruyama S, Enomoto T, Uemura Y, Shimizu Y, Yuasa D, Matsuo K, et
al: CTRP9 protein protects against myocardial injury following
ischemia-reperfusion through AMP-activated protein kinase
(AMPK)-dependent mechanism. J Biol Chem. 287:18965–18973. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhong Y, Yu C and Qin W: LncRNA SNHG14
promotes inflammatory response induced by cerebral
ischemia/reperfusion injury through regulating miR-136-5p /ROCK1.
Cancer Gene Ther. 26:234–247. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lu L, Yue S, Jiang L, Li C, Zhu Q, Ke M,
Lu H, Wang X, Busuttil RW, Ying QL, et al: Myeloid Notch1
deficiency activates the RhoA/ROCK pathway and aggravates
hepatocellular damage in mouse ischemic livers. Hepatology.
67:1041–1055. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chang J, Yao X, Zou H, Wang L, Lu Y, Zhang
Q and Zhao H: BDNF/PI3K/Akt and Nogo-A/RhoA/ROCK signaling pathways
contribute to neurorestorative effect of Houshiheisan against
cerebral ischemia injury in rats. J Ethnopharmacol. 194:1032–1042.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang S and Kim HM: The RhoA-ROCK-PTEN
pathway as a molecular switch for anchorage dependent cell
behavior. Biomaterials. 33:2902–2915. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li G, Liu L, Shan C, Cheng Q, Budhraja A,
Zhou T, Cui H and Gao N: RhoA/ROCK/PTEN signaling is involved in
AT-101-mediated apoptosis in human leukemia cells in vitro and in
vivo. Cell Death Dis. 5:e9982014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Papakonstanti EA, Ridley AJ and
Vanhaesebroeck B: The p110delta isoform of PI 3-kinase negatively
controls RhoA and PTEN. EMBO J. 26:3050–3061. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lauriol J, Keith K, Jaffre F, Couvillon A,
Saci A, Goonasekera SA, McCarthy JR, Kessinger CW, Wang J, Ke Q, et
al: RhoA signaling in cardiomyocytes protects against
stress-induced heart failure but facilitates cardiac fibrosis. Sci
Signal. 7:ra1002014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lei X, Seldin MM, Little HC, Choy N,
Klonisch T and Wong GW: C1q/TNF-related protein 6 (CTRP6) links
obesity to adipose tissue inflammation and insulin resistance. J
Biol Chem. 292:14836–14850. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang M, Tang X, Li L, Liu D, Liu H, Zheng
H, Deng W, Zhao X and Yang G: C1q/TNF-related protein-6 is
associated with insulin resistance and the development of diabetes
in Chinese population. Acta Diabetol. 55:1221–1229. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chi L, Hu X, Zhang W, Bai T, Zhang L, Zeng
H, Guo R, Zhang Y and Tian H: Adipokine CTRP6 improves PPARү
activation to alleviate angiotensin II-induced hypertension and
vascular endothelial dysfunction in spontaneously hypertensive
rats. Biochem Biophys Res Commun. 482:727–734. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yuasa D, Ohashi K, Shibata R, Mizutani N,
Kataoka Y, Kambara T, Uemura Y, Matsuo K, Kanemura N, Hayakawa S,
et al: C1q/TNF-related protein-1 functions to protect against acute
ischemic injury in the heart. FASEB J. 30:1065–1075. 2016.
View Article : Google Scholar : PubMed/NCBI
|