Introduction
Porphyrias is a group of rare, mostly inherited
disorders that are each caused by a defect in a specific heme
biosynthetic enzyme (1). Acute
intermittent porphyria [AIP; Online Mendelian Inheritance in Man
(OMIM) ID, 176000] is caused by the partial deficiency of
hydroxymethylbilane synthase (HMBS; EC:4.3.1.8), an enzyme in the
heme biosynthetic pathway (2).
Recent updates for the management of acute attacks of porphyria
have described the clinical features of an AIP episode as including
abdominal pain, nausea, vomiting, constipation, dark urine,
hypertension, arrhythmia, psychosis, convulsions, peripheral motor
neuropathy and hyponatremia (3).
Abdominal pain, psychiatric disturbance and peripheral neuropathies
are the ‘classical triad’ of AIP (4).
With the advent of DNA technology, genetic analysis
has become the gold standard for the diagnosis of AIP (2). In total, >390 different mutations
responsible for AIP have been reported so far (1), most of them in Europe, particularly
in northern Europe (5).
At our department, a Chinese patient with AIP and
posterior reversible encephalopathy syndrome (PRES) was identified.
PRES is characterized by several neurological symptoms typically
corresponding to areas of vasogenic cerebral edema on MRI (6).
The patient of the present study was diagnosed with
AIP by genetic analysis and clinical presentation of PRES based on
typical symptoms and imaging results. PCR-based gene sequencing
identified a frameshift deletion (c.405-406delAA) in exon 8 of the
HMBS gene. Furthermore, the deletion variant truncated
(p.Glu135AspfsX74) the HMBS protein in the proband.
Materials and methods
Sample collection
All procedures were performed in accordance with the
ethical standards of the responsible Ethics committee of The Second
Military Medical University (Shanghai, China) on human
experimentation and with The Declaration of Helsinki from 1975 and
its revision from 2000. Blood samples were collected in EDTA from a
36-year-old Chinese female in May 2013, who was referred by her
physicians due to elevated D-aminolevulinic acid (ALA) and
porphobilinogen (PBG) levels, and due to having clinical symptoms
compatible with acute hepatic porphyria. The patient provided
informed consent for genetic testing and the study was approved by
the Ethics Committee of The Second Military Medical University
(Shanghai, China).
DNA isolation and mutation
analyses
Genomic DNA was extracted from lymphoblasts using
the QIAamp DNA Blood Mini kit (Qiagen, Inc.). Amplification of the
entire HMBS gene was performed in two parts as described previously
(7). Fragment 1 comprised the
promoter region through intron 3 [primers long-range (LR)1 and LR2]
and fragment 2 was comprised of exon 2 through exon 15 (primers LR3
and LR4), as presented in Table I.
Using the Extensor™ Hi-fidelity PCR master mix (Thermo Fisher
Scientific, Inc.), initial denaturation was performed at 94°C for 1
min. For the first 16 cycles, denaturation was performed at 94°C
for 30 sec, with annealing and extension at 67°C for 5 min. The
next 12 cycles included denaturation at 94°C for 30 sec, and
annealing and extension at 67°C for 5 min and 15 sec, respectively,
with 15-sec increments at each additional cycle. The final
extension was performed at 72°C for 10 min. PCR products were
analyzed by agarose gel electrophoresis to determine that the
long-range reactions were successful and to identify any gross gene
rearrangements.
 | Table I.Primers for PCR amplification and
sequencing. |
Table I.
Primers for PCR amplification and
sequencing.
| Primer | Oligonucleotide
sequence (5′-3′) |
|---|
| For long-range
PCR |
| LR1
(sense) |
TGCTCCCACTTCAGTTACTTGTCTTTA |
| LR2
(antisense) |
GACGCCCATCTCTAAACCTAATCAGGAC |
| LR3
(sense) |
AAGGGACCAGCCTTGGAGTATTTCCCCACTC |
| LR4
(antisense) |
CAAGGATAGAAGGGCGGTTGAGGTGTGC |
| For direct
sequencing |
| Exon
1 |
GAGACCAGGAGTCAGACTGT |
| Exon
2/3 |
CCCACTGACAACTGCCTTGGTCAAG |
| Exon
4 |
CCTAACCTGTGACAGTCT |
| Exon
5/6 |
AGACCTAGCATACTAGGG |
| Exon
7 |
AGGGTCAGGCCCCAAAGGGAAAGG |
| Exon
8 |
CGAGAGAATAGAGGTGAT |
| Exon
9 |
TTGTCTTTTTCCTTGGCTGC |
| Exon
10 |
GGGAAAGACAGACTCAGGCAGAG |
| Exon
11 |
CGGTAGCATCCCAAGGTCT |
| Exon
12 |
TAAGAAATCTTCCCTGC |
| Exon
13 |
CAGTGATGTCCTCAGGTCTG |
| Exon
14 |
ATCCCAGGTTTCTAGGTAG |
| Exon
15 |
AGACCATGCAGGCTACCATC |
|
|
CGTGACCTGTCGTCGTTG |
The amplified fragments were sequenced on the ABI
3500 Genetic Analyzer (Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocol. A list of the sense and antisense
primers used and sequencing parameters are provided in Table I. In brief, sequencing PCR was
performed as follows, following the manufacturer's instructions:
Denaturation at 95°C for 30 sec; annealing at 60°C for 30 sec; and
extension at 72°C for 60 sec for 30 cycles. After a final
denaturation for 3 min at 94°C, the PCR mixture was loaded on a 2%
agarose gel and air-dried for 15 min. The PCR product was excised
from the gel, purified with a QIAEX II gel extraction kit (Qiagen,
Inc.) and then subjected to capillary electrophoresis sequencing
using the ABI 3500 Genetic Analyzer (Thermo Fisher Scientific,
Inc.).
Generation and expression of HMBS
constructs in E. coli
The full-length human wild-type (WT) and mutant
(mut; c.405-406delAA) the synthesized HMBS complementary DNA
(Coding sequence of HMBS was download from NCBI database,
https://www.ncbi.nlm.nih.gov/nuccore/NM_001024382.2,
and cDNA was syntehsised by reverse transcription-PCR.) (8) was cloned into the PKK233-3 vector
(Ybscience, Inc.; http://www.ybio.net/) and designated as PKK-WT
(1.743±0.047 mg/ml) and PKK-mut (1.642±0.168 mg/ml), respectively.
The constructs were confirmed by sequencing and transformed into
BL21 E. coli competent cells (Ybscience, Inc.), which were
cultured overnight in lysogeny broth medium (cat. no. ST156;
Beyotime Institute of Biotechnology) at 37°C. Inoculates were
induced with 1 µm isopropyl β-D-1-thiogalactopyranoside and
incubated for an additional 3 h. at 37°C After centrifugation at
6,000 × g for 1 min at room temperature, cells were resuspended in
1 ml bacterial lysis buffer (cat. no. C500003; Sangon Biotech Co.,
Ltd.) and lysed by freezing at −80°C for 30 min, followed by
thawing. Lysates were centrifuged at 12,000 × g for 5 min at 4°C,
and supernatants were collected and stored at −80°C in the dark
until further use. The time interval between transfection and
subsequent experiments was <1 week.
Western blot analysis
Denatured protein lysates were separated by 8%
SDS-PAGE and transferred on to a polyvinylidene difluoride membrane
(Beyotime Institute of Biotechnology). The lane of PKK223-1-1
contained 0.52 mg protein. The lane of PKK223-1-2 contained 0.56 mg
protein. The lane of PKK223-1-3 contained 0.62 mg protein. The lane
of HMBS mut-1-1 contained 0.46 mg protein. The lane of HMBS mut-1-2
contained 0.58 mg protein. The lane of HMBS mut-1-3 contained 0.48
mg protein. The lane of HMBS WT-5-1 contained 0.48 mg protein. The
lane of HMBS WT-5-2 contained 0.50 mg protein. The lane of HMBS
WT-5-3 contained 0.56 mg protein. Determination of protein
concentration using bicinchoninic acid Protein Assay kit (cat. no.
P0012s; Beyotime Institute of Biotechnology). After blocking in 5%
non-fat dry milk in Tris-buffered saline containing Tween-20 for 3
h at room temperature, the membrane was incubated with anti-HMBS
antibody (1:5,000; cat. no. ab129092; Abcam) at 4°C overnight,
followed by incubation with secondary horseradish peroxidase
(HRP)-conjugated anti-rabbit IgG (1:5,000; cat. no. Ab6721; Abcam)
for 2 h at room temperature. Signals were detected using the ECL
Chemiluminescence substrate reagent kit (Invitrogen; Thermo Fisher
Scientific, Inc.). Membranes were stripped and re-probed with
anti-GAPDH (1:1,000; cat. no. AG019-1; Beyotime Institute of
Biotechnology) and secondary HRP-conjugated anti-mouse IgG
(1:5,000; cat. no. A0261; Abcam) at room temperature for 1 h for
loading normalization.
HMBS enzyme activity and
thermostability
HMBS enzyme activity was determined in lysates by
measuring the conversion of PBG to uroporphyrin as previously
described (9). Lysates were
diluted 1:3 in phosphate buffer (pH 7.6) containing dithiothreitol,
MgCl2 and Triton X-100; 100 µl of this mixture was
pre-incubated with 1.8 ml of 0.1 M Tris-HCl (pH 8.1) for 3 min at
37°C, followed by incubation with 0.5 ml of 1 mm PBG substrate for
60 min at 37°C in the dark. The reaction was stopped with 350 µl
cold 40% trichloroacetic acid and oxidation of uroporphyrinogen to
uroporphyrin was performed under sunlight for 30 min. Uroporphyrins
were measured quantitatively by spectrofluorometry at an emission
wavelength of 405 nm. HMBS activity was expressed in the units pmol
uroporphyrin/mg protein/h using appropriate standards. All assays
were performed in triplicate and average values were expressed as
percent of WT activity. For enzyme thermostability studies, WT and
mutant lysates were assayed for HMBS activity after incubation at
65°C for 90 min.
Mapping of the missense mutation on
the human HMBS crystal structure
The 2.8-Å crystal structure of human HMBS (10) (Protein Data Bank ID: 3EQ1) was
viewed in PyMOL Molecular Graphics System (v2.0; Schrödinger, LLC).
Mapping of the molecular mutations and graphical representations
were created on PyMOL Molecular Graphics System (v2.0; Schrödinger,
LLC) (10), which involved
comparing the structure of a protein simulated by the software with
that of a published protein.
Statistical analysis
Independent-samples t-tests were used as appropriate
to compare the thermostability between WT-HMBS and mutant protein.
The ratio of the enzyme activity prior to and after heating was
used as an index to describe the thermostability of HMBS. SPSS
version 21.0 (IBM Corp.) was used for analysis. P<0.05 was
considered to indicate statistical significance.
Results
Case report
The proband was a 36-year-old Chinese female. She
was treated at a local county hospital for severe acute abdominal
pain five years previously, for which she underwent a laparotomy
for intussusception, but the results were misdiagnoses. She then
had a recurrence of acute lower abdominal pain accompanied by
systemic seizures six months later; she recovered after one month
of conservative treatment. The acute abdominal pain recurred two
years ago, and after one week of this persistent pain, the patient
developed neurological and psychiatric symptoms, including blurred
vision, impaired speech, seizures and memory impairment. At one
month after the onset, the patient was referred to the Neurology
Department of The Second Affiliated Hospital of The Second Military
Medical University for further diagnosis. Medical examination
indicated that the patient's blood pressure was normal with good
heart and lung functions. Neurological examination revealed mental
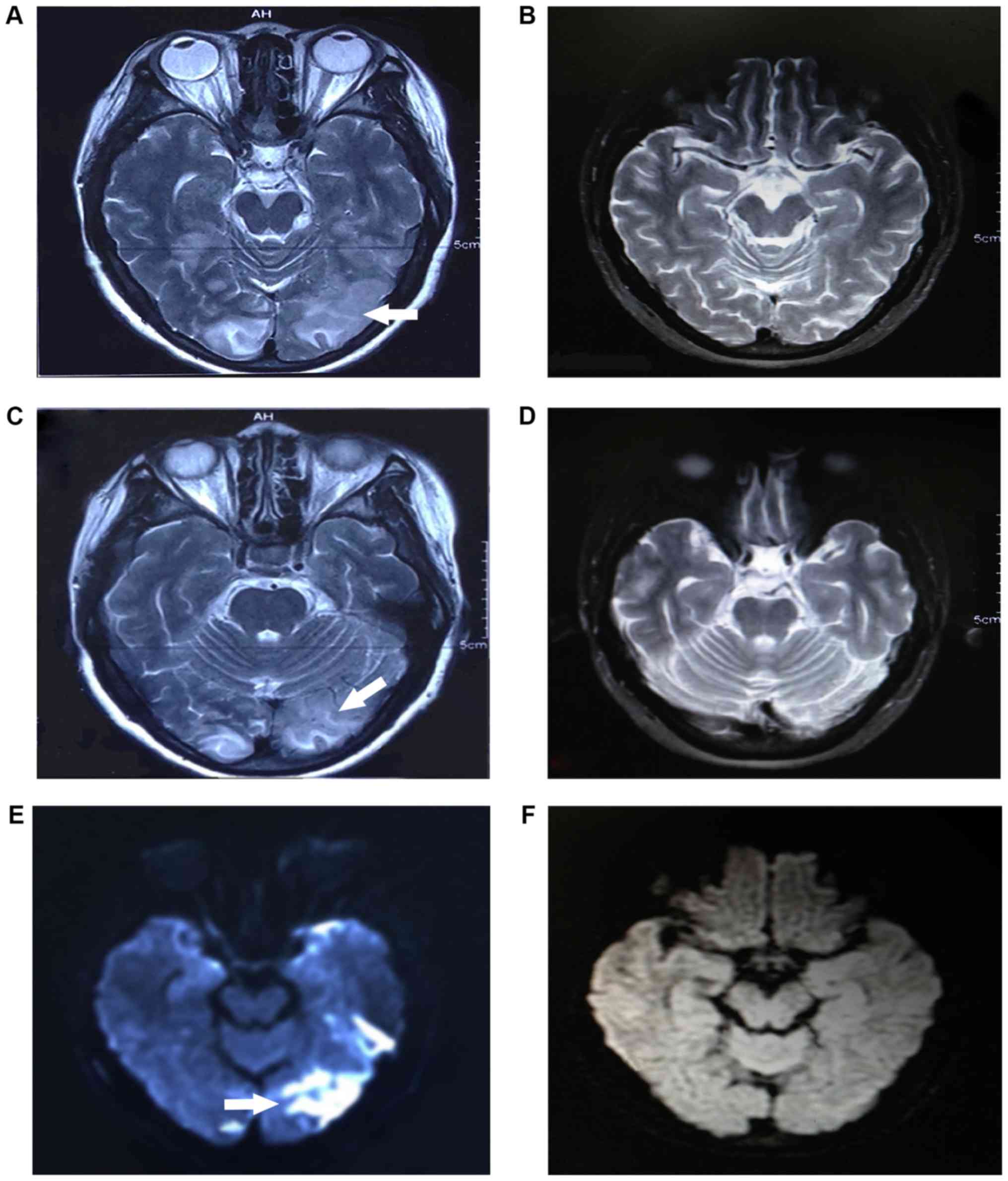
fatigue and limb paralysis. Brain MRI indicated intracranial
multifocal lesions in the cortical and subcortical white matter of
the left temporal, parietal, occipital and right occipital lobes.
After two weeks of treatment, MRI revealed partial regression of
the neurological abnormalities (Fig.
1). Laboratory tests indicated that the patient had
hypoproteinemia (25 g/l), hypoglycemia and mildly elevated alanine
aminotransferase and aspartate aminotransferase, while levels of
serum ceruloplasmin and blood electrolytes (potassium, sodium,
chloride, calcium and magnesium ions) were in the normal range. An
electroencephalogram revealed slower brain waves in the bilateral
cerebral hemispheres but no epileptic discharge. Chest X-rays were
normal. Pelvic ultrasound revealed intrauterine pregnancy, which
was the cause of changes in hormone levels. PBG was strongly
positive in the qualitative test (Watson-Schwartz test) (11,12)
of the patient's urine during the attack period, which became
negative after the episode. Repeated tests performed during the
remission period were weakly positive for PBG. Darkening of urine
after 30 min of exposure to sunlight confirmed the clinical
diagnosis of AIP. The patient had high levels of porphyrin
precursors in her urine. The proband was treated by intravenous
infusion of 300 g/day of glucose. Complete recovery was achieved
after 2 weeks of conservative treatment, and then the patient chose
to terminate the pregnancy.
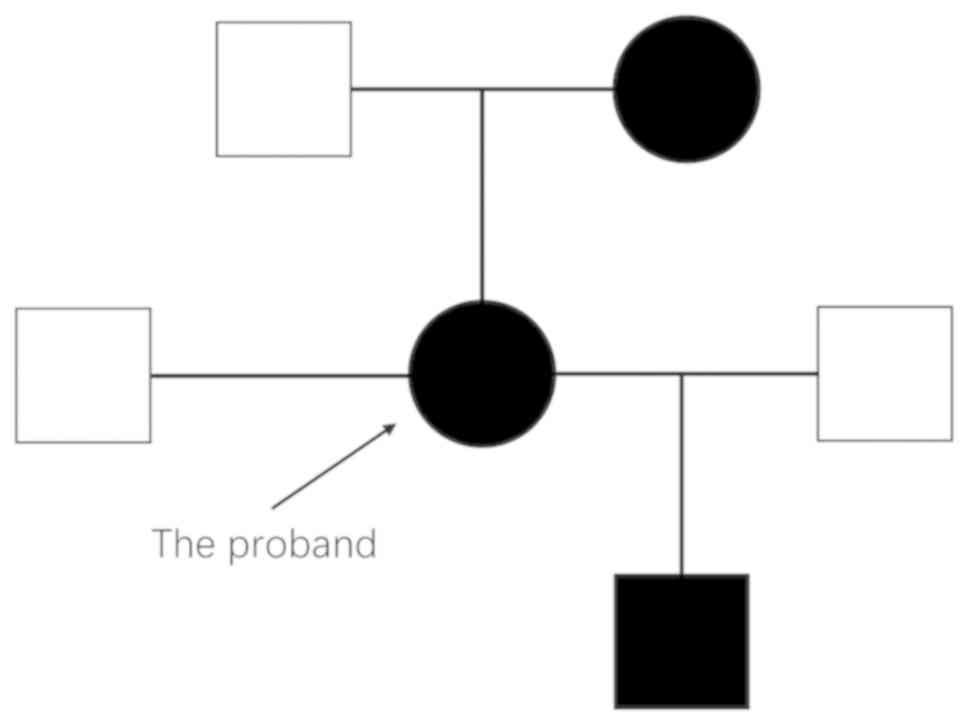
For genetic screening, blood samples of the patient
and her family members (the patient's parents and the patient's
son) were collected after informed consent. The patient's mother
developed symptoms of psychosis during her middle age and was
diagnosed with schizophrenia, which was managed with antipsychotic
drug treatment but was not diagnosed with AIP. The patient's son
(age, 18 years) had not developed any abdominal pain or nervous
system dysfunction.
Mutation analysis of the HMBS
gene
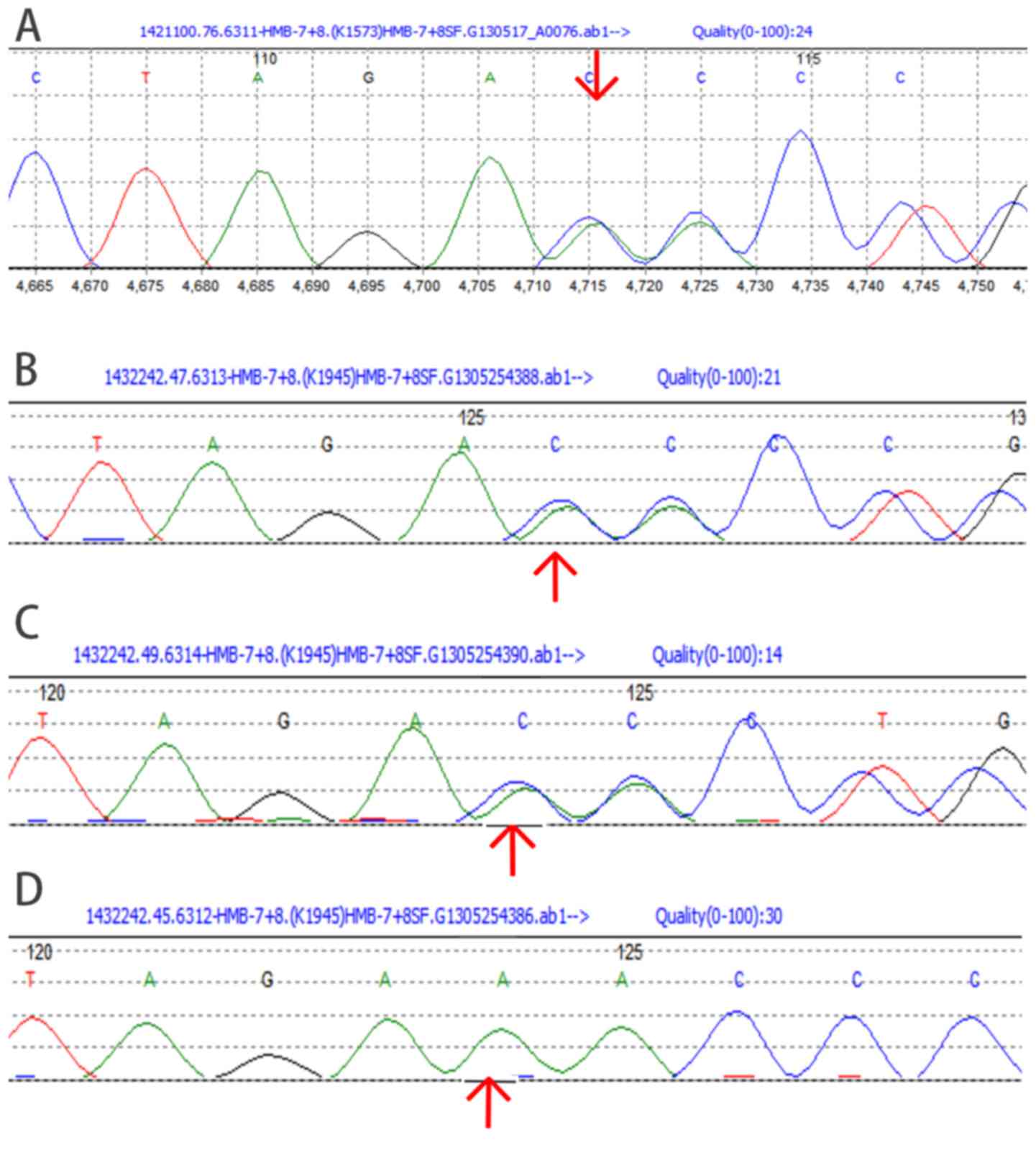
Direct sequencing of PCR-amplified genomic DNA,
including 15 exons and adjacent intronic areas, identified a
c.405-406delAA frameshift mutation in exon 8 of the HMBS gene
(Fig. 2). This was a heterozygous
mutation, identified in the proband, the proband's mother and the
proband's son, but not in the proband's father.
Enzyme activity and thermostability of
the mutated HMBS
HMBS enzyme activity of the mutant protein was 62.2%
of the activity of the expressed WT protein (71,449.00±21,708.96
vs. 44,461.00±14,412.49 U, t=1.794, P=0.147). The thermostability
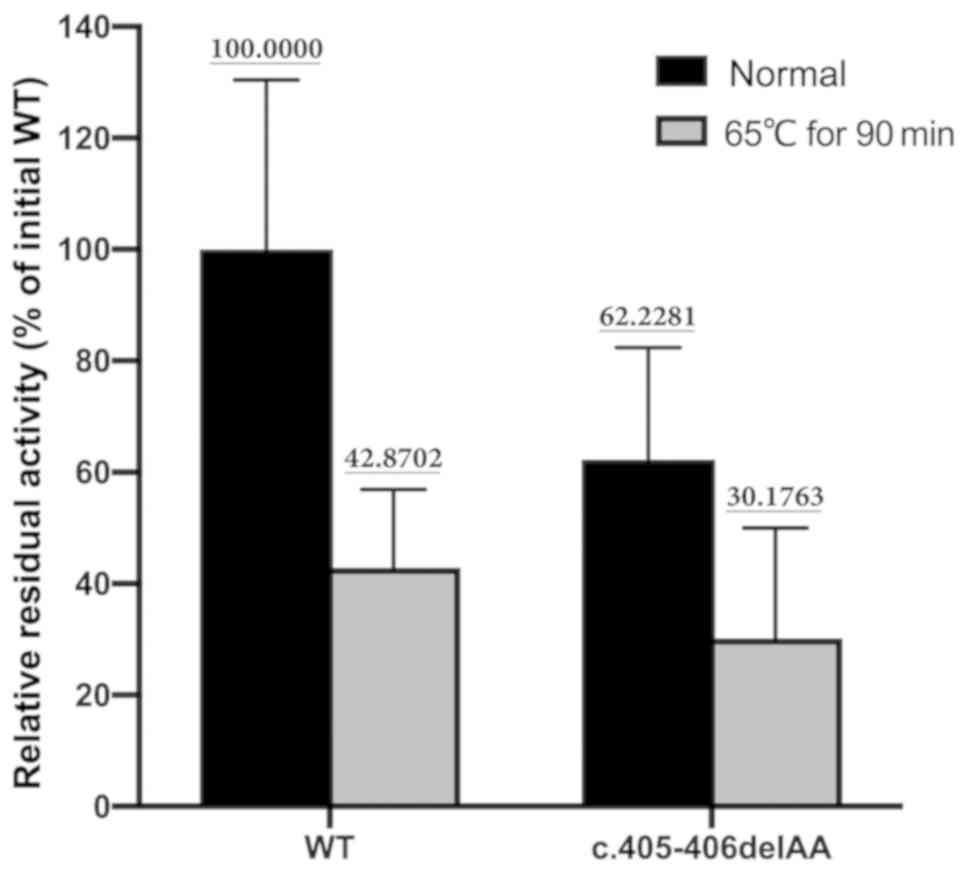
assay indicated that the mutant protein retained 48.5% of its
initial HMBS activity after incubation at 65°C for 90 min. On the
other hand, the WT enzyme retained 42.9% of its initial activity
after a 90-min incubation at 65°C (Fig. 3), and there was no statistically
significant difference in thermostability between the WT protein
and mutated protein (42.9 vs. 48.5%; P=0.703).
Effects of deletion mutation on
protein stability and structure of the mutant HMBS protein
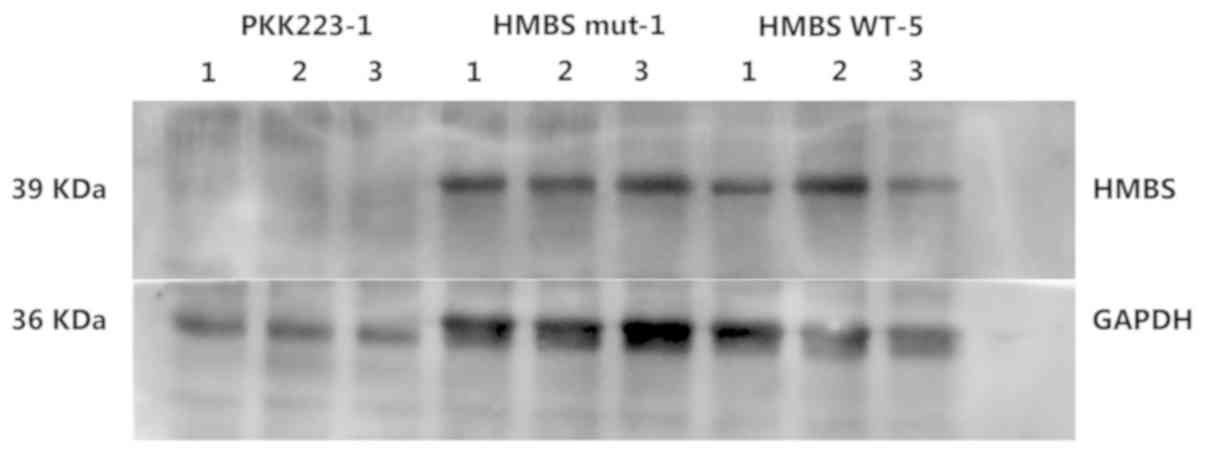
Western blot analysis indicated that the expression
of the mutant protein (Glu135AspfsX74) was similar to that of the
WT protein (Fig. 4). However, the
secondary structure of the truncated protein was notably different
from that of the WT. The mutant protein exhibited a significant
reduction in α-helix and β-sheets and folding of the peptide chains
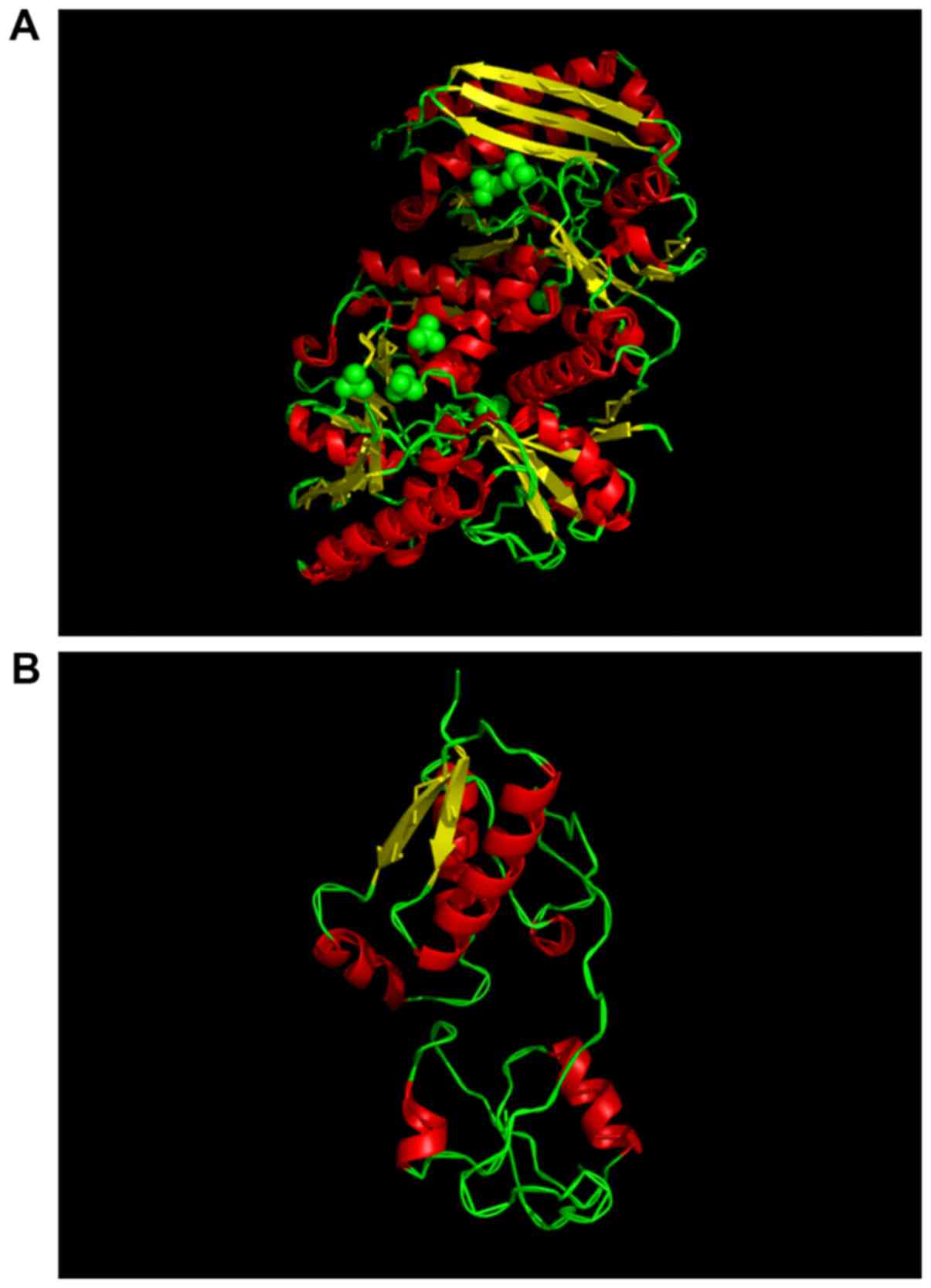
(Fig. 5).
Discussion
AIP is an inherited metabolic disease characterized
by the partial deficiency of HMBS (1). In the present study, a novel
frameshift deletion (c.405-406delAA) in exon 8 of the HMBS gene
leading to a truncated (p.Glu135AspfsX74) protein was identified in
a Chinese pedigree (Fig. 6). The
structure of the mutated protein was significantly different from
the WT and its enzyme activity was lower than that of the WT
protein with similar thermostability. As no previous records of the
mutation were present in the OMIM database, it was inferred that
this was a novel mutation identified in a Chinese family.
As exemplified in the present case, AIP is easily
and frequently misdiagnosed in the clinic (1). In spite of the presentation with
clinical symptoms of AIP, including acute abdominal pain,
neuropathy and psychiatric disorders, the patient of the present
study underwent laparotomy for intussusceptions. Following the
onset of the recent episode of disease activity, AIP was diagnosed
based on the typical clinical manifestations, urine PBG test and
genetic testing.
In humans, HMBS is the third enzyme of the heme
biosynthetic pathway that has a high degree of conservation
(2). Human HMBS consists of three
domains: Domains 1 and 2 have five b-sheets and three a-helices;
and domain 3 has a flattened b-sheet geometry. The functional core
of HMBS enzyme activity is located in domain 1 (13). It has the DPM cofactor in the
native reduced conformation with an ordered sulphate ion hydrogen
bound to Arg26 and Ser28 at the proposed
substrate-binding site (13).
Although domain 3 is distant from domain 1, its interaction with
domain 1 may modulate conformational fluctuations associated with
the enzyme action (13). In the
present study, mutations led to the truncation of the HMBS protein.
It is worth noting that western blot analysis demonstrated that the
truncated mutant proteins had similar molecular weights to those of
WT proteins. It was demonstrated that the mutation was the deletion
of two bases AA, and after the base deletion, the primary structure
of the protein changed. The translation was terminated prematurely,
but it was not possible to determine what changes occurred in the
tertiary structure of the protein, and thus no significant
difference was observed in the western blotting results. However,
the present study was unable to verify the protein structure
changes. Therefore, the structural changes caused by this mutation
will be investigated in further studies. The mutant protein is
p.Glu135AspfsX74. This means that the missing AA resulted in the
conversion of amino acid residues at position 135 from Glu to Asp,
and premature termination occurred at position 74 after this
position. ACC is not a stop codon so this mutation results in
frameshift mutations and produces different amino acid residues in
subsequent sequences. According to the prediction analysis of the
truncated protein, the mutant protein has 300 amino acid residues.
Compared to the WT protein, the mutant protein does lose ~60
residues, primarily affecting the domain 3 regions. However,
structural prediction revealed that the b-folding and the a-coiling
of the mutant protein were reduced, which may be due to the loss of
domain 3 affecting the correct folding of the domain 1 and 2 region
peptide chains. It was concluded that the function of the mutated
protein was reduced to ~60% of the WT protein because the
functional core of the domain 1 region is not affected by the
mutation, but the deletion of domain 3 results in a conformational
change in domain 1, leading to a decrease in overall enzyme
function.
Although PBG and ALA accumulation is hypothesized to
be the trigger of AIP (1), their
exhaustive pathological role remains elusive. In the present study,
the focus was on the neurological manifestations of AIP. Due to the
presence of the typical symptoms (headache, visual disturbances and
seizures) and characteristic MRI, the patient was clinically
diagnosed with PRES.
Brain lesions in patients with acute porphyria are
not rare. However, only a few studies have reported on brain MRI
features of patients with AIP. MRIs performed early during an
attack have demonstrated reversible cortical changes consistent
with PRES (14). PRES refers to a
reversible subcortical vasogenic brain edema in patients with acute
neurological symptoms (15),
including seizures, headaches and visual disturbances, which are
the three major clinical manifestations of AIP.
Since the first report on PRES in 1996, numerous
studies describing the disease have been published, but the exact
pathogenesis has remained to be defined. There are currently two
widely accepted hypotheses regarding the pathogenesis of PRES: i)
Excessive brain perfusion caused by an increase in blood pressure
that breaks the cerebral blood flow autoregulation threshold; ii)
disruption of the blood-brain barrier (BBB) by vascular endothelial
dysfunction due to toxic substances (15). The first theory is the core
mechanism leading to PRES, as the posterior cerebrum lacks
sympathetic innervation and is sensitive to blood pressure
fluctuations (16). Since there
was no significant increase in blood pressure in the patient of the
present study, it was inferred that PRES was caused by other
mechanisms.
Neurovisceral symptoms are one of the typical
manifestations of AIP. Autonomic testing has demonstrated
parasympathetic and sympathetic dysfunction during an acute attack
of AIP (14). Considering the lack
of sympathetic innervation in the posterior region of cerebrum, it
was concluded that sympathetic dysfunction in AIP was the
pathological basis for the initiation of PRES. Parasympathetic, as
well as sympathetic dysfunction, makes the arteries in the
posterior region more susceptible to vasodilation and
hyperperfusion, leading to homeostatic dysregulation of cerebral
blood flow (15).
A systematic review reported that AIP complicated
with PRES is more common in female patients (17). In the present case, the common
causes of PRES, including hypertension and hyponatremia, were not
present. It may be hypothesized that pregnancy may be an important
cause of AIP attacks and PRES, as it alters hormone secretion, the
internal environment and sympathetic function. At the same time,
pregnancy may increase the metabolic burden of patients. All of
these factors may be the core reasons for the induction of AIP and
PRES in the present case.
In fact, through the experiments in the present
study, it was confirmed that the mutated HMBS protein had ~60%
activity and the patient's HMBS mutation was heterozygous. This
means that under normal circumstances, the HMBS activity in the
patient may reach ~80% of the normal level (50% contributed from
the WT allele and 30% from the mutant allele). In AIP, however, a
50% decrease in HMBS activity is typically indicative of the
disease (18). Under these
conditions, patients usually do not have severe AIP attacks.
Therefore, it is hypothesized that the onset of the episode of AIP
the present case was associated with a marked increase in
hemoglobin demand due to pregnancy. The patient's mutated HMBS was
not able to meet the increased hemoglobin synthesis burden,
resulting in an abnormal increase in ALA and PBG and leading to
this episode of AIP.
The identified HMBS mutation, c.405-406delAA, causes
a frameshift and leads to premature termination of the protein.
Most likely, this mutation triggers nonsense-mediated decay (NMD)
and decreases HMBS mRNA expression, resulting in AIP. NMD is a
translation-dependent mRNA surveillance mechanism that helps to
maintain the quality of gene expression (19). The function of NMD is to eliminate
the production of truncated proteins. A premature termination codon
that triggers NMD may give rise to disease by precluding the
production of the full-length protein. Therefore, it was speculated
that the premature termination of the HMBS protein may trigger NMD,
resulting in a decrease in the production of full-length HMBS
protein, which in turn induces AIP. This is a potential mechanism
for the onset of AIP in this patient.
Abnormal accumulation of ALA and PBG is the major
pathological process in AIP (1)
and a potential mechanism of PRES (17). ALA is neurotoxic and affects the
binding affinity of g-aminobutyric acid (GABA) to its receptors,
but not the binding of serotonin or dopamine (14). Studies have indicated that most or
all of the ALA and PBG that are toxic to the central nervous system
are produced by the metabolism of cells localized in the brain, and
the BBB was not, or was only slightly permeable to ALA and PBG
(20,21). Hu et al (22) reported that disruption of the 5-ALA
transport mechanism is a key factor in ALA neurotoxicity. In the
present study, the patient had an AIP attack and PRES. It was
speculated that an underlying mechanism affects the permeability of
the BBB and induces vasogenic edema in the brain tissue, leading to
PRES. At the same time, changes in the permeability of the BBB also
lead to changes in the intracranial concentration of ALA, causing
neurotoxicity of ALA.
Inhibition of GABA receptors increased vascular
endothelial growth factor (VEGF) expression in tumor cells
(23). VEGF is an important
mediator of vascular permeability, and changes in BBB permeability
due to changes in VEGF expression are also important causes of PRES
(24). Increased levels of
circulating VEGF induces brain edema seen in PRES (24). Thus, it was speculated that
accumulating ALA may act on GABA receptors of astrocytes and alter
VEGF expression. This possible mechanism has remained to be
confirmed by experimental studies and will require further
exploration in the future.
ALA also reversibly inhibits energy-dependent
Na+/K+ ATPase in the brain, which may
subsequently lead to the development of cellular edema (14). In addition, the production of
nitric oxide (NO) may also decrease during the onset of AIP and
cause vasoconstriction (25). NO
synthase is a hemoprotein and the relative deficiency of heme
during AIP may reduce the production of NO (26).
Currently, there are no established criteria and
guidelines for the diagnosis and classification of PRES. Although
not 100% specific, imaging findings combined with clinical symptoms
are acceptable for the diagnosis of PRES. The T2-weighted imaging
(T2WI) or T2-fluid-attenuated inversion recovery (FLAIR)
hyperintensities in the posterior region of the brain is a typical
imaging pattern of PRES, which indicates the manifestation of
vasogenic edema, observed in >95% of patients with PRES
(16). However, cytotoxic edema is
also observed on diffusion-weighted imaging in certain patients
(27). In the case of the present
study, T2WI or T2-FLAIR hyperintensities were detected in the
posterior region of the brain and significant diffusion restriction
was observed, indicating vasogenic and cytotoxic edema in this
patient. These results confirmed the inference that PRES in this
patient was mainly due to toxic metabolites. It is noteworthy to
mention that the presence of extensive vasogenic edema or diffusion
restriction is associated with an unfavorable clinical outcome
(28). However, contrary to
certain studies (16), the patient
of the present study recovered completely. It may therefore be
hypothesized that lesions caused by restricted diffusion may not be
irreversible.
AIP diagnosis during or following an acute attack is
usually based on clinical symptoms combined with PBG excretion.
Over the years, genetic analysis has become the most reliable
method to confirm AIP in symptomatic patients, and asymptomatic
patients who do not always have elevated levels of ALA and PBG, as
well as in patients with the non-erythroid variant of AIP with a
normal level of urinary ALA and PBG (2).
There are several limitations to the present study.
First, due to the experimental limitations, results on the ALA
concentration in patient samples could not be obtained. The ALA
concentration is considered necessary for the diagnosis of AIP.
However, AIP was diagnosed by the Watson-Schwartz test, which has
been proven feasible in the diagnosis of AIP. In addition, it was
not possible to isolate RNA from the patient to assess HMBS mRNA
expression levels, although it was speculated that NMD-induced mRNA
degradation of HMBS may be a potential pathogenesis of AIP.
Complementary experiments will be performed in future studies to
clarify the pathogenesis associated with the mutant HMBS.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YY, XC and ZY contributed to the conception and
design of the study. HW and HP provided important medical decisions
in the clinical treatment of patient, contributed the acquisition
and analysis of clinical data and wrote the Case Report section. WS
and BH performed the experimental study. YY and XC wrote the first
draft of the manuscript. ZY and BH wrote sections of the
manuscript. All authors contributed to manuscript revision and they
read and approved the submitted version.
Ethics approval and consent to
participate
All procedures followed were in accordance with the
ethical standards of the Ethics Committee of The Second Military
Medical University, and with The Helsinki Declaration from 1975 and
its revision from 2000. The patient provided informed consent for
genetic testing and the study was approved by the ethics committee
of Second Military Medical University (Shanghai, China).
Patient consent for publication
The patient, her parents and her child provided
informed consent for publication of this manuscript.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Stölzel U, Doss MO and Schuppan D:
Clinical guide and update on porphyrias. Gastroenterology.
157:365–381.e4. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pischik E and Kauppinen R: An update of
clinical management of acute intermittent porphyria. Appl Clin
Genet. 8:201–214. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Edel Y and Mamet R: Porphyria: What is it
and who should be evaluated? Rambam Maimonides Med J. 9:2018.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Walterfang M, Bonnot O, Mocellin R and
Velakoulis D: The neuropsychiatry of inborn errors of metabolism. J
Inherit Metab Dis. 36:687–702. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Phillips JD: Heme biosynthesis and the
porphyrias. Mol Genet Metab. 128:164–177. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vilas-Boas S and Corte-Real A: Posterior
reversible encephalopathy syndrome and azathioprine. Eur J Case Rep
Intern Med. 6:0010322019.PubMed/NCBI
|
|
7
|
De Siervi A, Rossetti MV, Parera VE,
Astrin KH, Aizencang GI, Glass IA, Batlle AM and Desnick RJ:
Identification and characterization of hydroxymethylbilane synthase
mutations causing acute intermittent porphyria: Evidence for an
ancestral founder of the common G111R mutation. Am J Med Genet.
86:366–375. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sun G, Wang C, Zhen J, Zhang G, Xu Y and
Deng Z: Cloning and sequencing of KIR2DL1 Framework Gene cDNA and
identification of a novel allele. Zhonghua Yi Xue Yi Chuan Xue Za
Zhi. 33:694–697. 2016.(In Chinese). PubMed/NCBI
|
|
9
|
Unzu C, Sampedro A, Mauleón I, Alegre M,
Beattie SG, de Salamanca RE, Snapper J, Twisk J, Petry H,
González-Aseguinolaza G, et al: Sustained enzymatic correction by
rAAV-mediated liver gene therapy protects against induced motor
neuropathy in acute porphyria mice. Mol Ther. 19:243–250. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen B, Solis-Villa C, Erwin AL, Balwani
M, Nazarenko I, Phillips JD, Desnick RJ and Yasuda M:
Identification and characterization of 40 novel hydroxymethylbilane
synthase mutations that cause acute intermittent porphyria. J
Inherit Metab Dis. 42:186–194. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Corbett MB: The Watson-Schwartz Test.
JAMA. 195:4811966. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Elder GH and Sandberg S: Identifying acute
porphyria in patients with acute polyneuropathy or encephalopathy.
Nat Clin Pract Neurol. 4:648–649. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gill R, Kolstoe SE, Mohammed F, Al D-Bass
A, Mosely JE, Sarwar M, Cooper JB, Wood SP and Shoolingin-Jordan
PM: Structure of human porphobilinogen deaminase at 2.8 A: The
molecular basis of acute intermittent porphyria. Biochem J.
420:17–25. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Simon NG and Herkes GK: The neurologic
manifestations of the acute porphyrias. J Clin Neurosci.
18:1147–1153. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fugate JE and Rabinstein AA: Posterior
reversible encephalopathy syndrome: Clinical and radiological
manifestations, pathophysiology, and outstanding questions. Lancet
Neurol. 14:914–925. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ollivier M, Bertrand A, Clarençon F,
Gerber S, Deltour S, Domont F, Trunet S, Dormont D and Leclercq D:
Neuroimaging features in posterior reversible encephalopathy
syndrome: A pictorial review. J Neurol Sci. 373:188–200. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zheng X, Liu X, Wang Y, Zhao R, Qu L, Pei
H, Tuo M, Zhang Y, Song Y, Ji X, et al: Acute intermittent
porphyria presenting with seizures and posterior reversible
encephalopathy syndrome: Two case reports and a literature review.
Medicine (Baltimore). 97:e116652018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Arora S, Young S, Kodali S and Singal AK:
Hepatic porphyria: A narrative review. Indian J Gastroenterol.
35:405–418. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kurosaki T and Maquat LE:
Nonsense-mediated mRNA decay in humans at a glance. J Cell Sci.
129:461–467. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yasuda M, Gan L, Chen B, Yu C, Zhang J,
Gama-Sosa MA, Pollak DD, Berger S, Phillips JD, Edelmann W and
Desnick RJ: Homozygous hydroxymethylbilane synthase knock-in mice
provide pathogenic insights into the severe neurological
impairments present in human homozygous dominant acute intermittent
porphyria. Hum Mol Genet. 28:1755–1767. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Solis C, Martinez-Bermejo A, Naidich TP,
Kaufmann WE, Astrin KH, Bishop DF and Desnick RJ: Acute
intermittent porphyria: studies of the severe homozygous dominant
disease provides insights into the neurologic attacks in acute
porphyrias. Arch Neurol. 61:1764–1770. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hu Y, Shen H, Keep RF and Smith DE:
Peptide transporter 2 (PEPT2) expression in brain protects against
5-aminolevulinic acid neurotoxicity. J Neurochem. 103:2058–2065.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fava G, Marucci L, Glaser S, Francis H, De
Morrow S, Benedetti A, Alvaro D, Venter J, Meininger C, Patel T, et
al: gamma-Aminobutyric acid inhibits cholangiocarcinoma growth by
cyclic AMP-dependent regulation of the protein kinase
A/extracellular signal-regulated kinase 1/2 pathway. Cancer Res.
65:11437–11446. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen Z, Shen GQ, Lerner A and Gao B:
Immune system activation in the pathogenesis of posterior
reversible encephalopathy syndrome. Brain Res Bull. 131:93–99.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Olivier P, Van Melkebeke D, Honoré PJ,
Defreyne L and Hemelsoet D: Cerebral vasospasm in acute porphyria.
Eur J Neurol. 24:1183–1187. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Thachil J: Nitric oxide and the clinical
manifestations of acute porphyria. Intern Med J. 38:732–735. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gao B, Lyu C, Lerner A and McKinney AM:
Controversy of posterior reversible encephalopathy syndrome: What
have we learnt in the last 20 years? J Neurol Neurosurg Psychiatry.
89:14–20. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schweitzer AD, Parikh NS, Askin G, Nemade
A, Lyo J, Karimi S, Knobel A, Navi BB, Young RJ and Gupta A:
Imaging characteristics associated with clinical outcomes in
posterior reversible encephalopathy syndrome. Neuroradiology.
59:379–386. 2017. View Article : Google Scholar : PubMed/NCBI
|