Introduction
Bone fractures are common musculoskeletal disorders
that are the result of trauma or fragility, and present as a
significant public health issue and socioeconomic burden (1). Despite advances in treatment options,
up to 10% of all fractures exhibit insufficient bone repair leading
to delayed or non-healing fractures. These fractures lead to
significant morbidity and a significant decrease in the quality of
life of patients who are affected (2,3).
Fracture healing is a complex process and causes of delayed bone
repair are manifold, and the underlying cellular and molecular
mechanisms are still unknown (4,5).
Understanding the biological and biomechanical aspects of the
repair process is crucial for clinicians to develop novel treatment
and management strategies for ensuring optimal repair of injured
bone.
Fracture repair is an intricately specialized and
complex postnatal regenerative process consisting of multiple
events that occur simultaneously and in succession, with the end
goal to restore the damaged/injured bone to pre-injury cellular
composition, structure, and a biomechanically sound state (4,6–8). The
fracture healing process can be simplified into overlapping
biological phases encompassing an early inflammatory response
phase, the cartilaginous calls and hard callus formation middle
phase, and the late bone remodeling phase. Each phase is governed
by the activities of respective cell types, including immune cells
(early phase), cartilage forming chondrocytes (early-middle
phases), bone resorbing osteoclasts (middle-late phases), and the
bone forming osteoblasts (middle-late phases) (6,8–11).
The early inflammatory phase occurs immediately post-fracture with
hematoma formation at the fracture site creating a microenvironment
that favors repair. Recruitment of repair cells, predominantly
mesenchymal stem cells, occurs and are induced to differentiate
into chondrocytes. During endochondral ossification, chondrocytes
form the semi-rigid soft or cartilaginous callus, which provides
mechanical support to the fracture ends. The cartilaginous callus
also acts as the template for the bony hard callus. As the
chondrocytes become hypertrophic, the cartilaginous callus becomes
mineralized and is invaded by the vasculature. This enables the
infiltration of osteoclasts, which functions to resorb the
mineralized cartilage, allowing osteoblasts to remodel it into hard
callus of immature woven bone. This ultimately leads to the
bridging of the fracture by woven bone and osteoblasts play a major
role in the initial formation of the woven bone matrix. The
transition from cartilaginous callus to hard callus and woven bone
formation represents the most active phase of osteogenesis. The
final phase is the remodeling phase in which the action of the
osteoblast and osteoclast remodels the immature hard callus of
woven bone into the stronger mature lamellar bone, yielding bone
that should structurally and mechanically match to the
pre-fractured bone (6,8–10).
The core of fracture healing is the remodeling
process, which involves osteoclastic bone resorption and
osteoblastic bone formation (6).
Maintaining a dynamic balance between the activities of osteoclasts
and osteoblasts is necessary for effective and adequate bone repair
(12). Thus, perturbations in
osteoclast-mediated bone/cartilage resorption and
osteoblast-mediated bone formation during the process of bone
renewal will negatively impact the rate and quality of fracture
repair compromising bone microarchitecture and biomechanical
properties (13,14). Previous studies have found that
blocking osteoclast formation and/or activity delays fracture
repair by impairing cartilaginous callus resorption and hard callus
remodeling (15,16). We have previously demonstrated that
the specific deletion of 3-phosphoinositide-dependent protein
kinase 1 (PDK1), a serine/threonine kinase and pivotal regulator of
PI3K-Akt signaling in osteoclasts, impairs receptor activator of
nuclear factor-κB (RANKL)-induced osteoclast formation and bone
resorption (17). Given the
importance of osteoclasts in fracture repair, we hypothesized that
the specific loss of PDK1 in osteoclasts will alter the
fracture healing process. In the present study, it was revealed,
using the tibial fracture model, that the specific deletion of
PDK1 in osteoclasts impeded the fracture healing process by
delaying the resorption of the cartilaginous callus and subsequent
remodeling of immature woven bone to structurally and mechanically
stronger lamellar bone.
Materials and methods
Materials
Calcein, Alizarin Red S, the tartrate-resistant acid
phosphatase (TRAP) staining kit and the Modified Safranin O-Fast
Green FCF (SO/FG) Cartilage staining kit was purchased from
Sigma-Aldrich (Merck KGaA). The TIANamp Genomic RNA Extraction kit
was purchased from Tiangen Biotech Co., Ltd., while TRIzol reagent
and the RevertAid First Strand cDNA Synthesis kit was obtained from
Thermo Fisher Scientific, Inc. ELISA for murine
glaprotein/osteocalcin (BGP/OC), procollagen I N-terminal
propeptide (PINP), TRACP-5b and C-terminal telopeptide of type I
collagen (CTX–I) were purchased from Cusabio Biotechnology Co.,
Ltd.
Animals and experimental groups
All mice were raised and maintained in the
Laboratory Animal Center of Guangxi Medical University (China).
Mice were individually housed at a controlled temperature (22-26°C)
and humidity (50-60%) in ventilated cages with a 12-h light/dark
cycle, with availability of standard chow and fresh-water ad
libitum. Osteoclast-specific knockout (KO) of the PDK1
gene was generated as described in our previous study. Male
transgenic mice expressing Cre recombinase, under the
transcriptional control of the Cathepsin K promoter
(CtsKCre) were mated with female homozygous
PDK1flox/flox mice to generate
CtsKCre-PDK1flox/+ mice. Subsequently,
CtsKCre-PDK1flox/+ mice were bred with
PDK1flox/+ mice to generate
CtsKCre-PDK1flox/flox mice (referred
to as PDK1 cKO) and
CtsKCre-PDK1+/+ control wild-type (WT)
mice. Genotyping was performed as previously described (17).
To generate the fracture model, 12-week-old WT and
cKO mice (n=36 per group; 72 mice in total) were anesthetized using
sevoflurane. Penicillin sodium (200 U/g) was administered
intraperitoneally to prevent infection following surgery. The left
and right lower limb were shaved, and the skin disinfected with
0.5% iodophor. A longitudinal incision was made into the skin along
the anterior-medial surface of the shaved lower limbs to expose the
tibialis anterior muscle of the middle and upper segment of the
tibia, which was then dissected. A total of 2, 0.5 mm Kirschner
wires were inserted into the upper third of the tibia to guide the
bone cut using small animal orthopedic scissors to create a
transverse fracture. After fracture-end reduction, a Kirschner wire
was surgically inserted from the tibial tubercle into the medullary
canal for intramedullary fixation and stabilization of the
fracture. The incision was sutured layer by layer. All mice were
placed on heating beds and monitored until they had awoken from
surgery. All mice were allowed to recover following surgery for 3–4
days with intraperitoneal administration of post-operative
analgesics for pain relief for the first 2–3 days. A total of 24
mice (24 WT and 24 PDK1 cKO) were subsequently divided into
4 experimental groups; 7-, 14-, 21- and 28-days post-fracture.
Prior to euthanasia, venous blood was extracted from the right
ventricle in each mouse under anesthesia, from the 4 experimental
groups, in order to analyze bone turnover markers using ELISA.
Following blood collection, mice were sacrificed using cervical
dislocation and the right tibias were dissected and processed for
X-ray, micro computed tomography (CT) and histological analyses.
The left tibias were removed, and the callus tissue was processed
for total RNA extraction followed by reverse
transcription-quantitative PCR (RT-qPCR) for analysis of gene
expression.
To examine callus formation and the conversion of
cartilage callus to hard callus, the remaining WT and cKO mice
(n=12 per group; 24 mice total) were subjected to Calcein
Green-Alizarin Red S double labeling. Calcein Green (25 mg/kg) and
Alizarin Red S (10 mg/kg) were administered intraperitoneally at
10- and 17-days post-surgery sequentially. Mice were sacrificed 21-
and 28-days following post-fracture surgery, after which the right
tibias were extracted for biomechanical testing and the left tibias
were extracted and processed for histological assessment of mineral
apposition rate (MAR).
X-ray analysis
X-rays of the right tibia and fibula were obtained
using a Faxitron MX20/DC2 (Faxitron Bioptics, LLC), equipped with
an FPX-2 imaging system (Teledyne DALSA), with a voltage of 5.0 kV
and an exposure time of 6.0 sec. Blinded post-mortem radiographs
were obtained for all samples in the 4 experimental groups and
graded based on fracture line blurring. The degree of fracture line
blurring was classified into three grades: clear, blurred and very
blurred, as previously described (18,19).
All assessments were observed, discussed and collated by three
independent researchers at the same time and recorded in detail,
and the final results were used as the basis for subsequent
downstream analysis.
MicroCT scanning
Tibial bone specimens were scanned using a
high-resolution high-throughput compact microCT system (SCANCO µCT
40; SCANCO Medical AG) and slices were analysed using the CT
Evaluation Program (v5.0A, Scanco Medical). Scans performed at a
voltage of 70 kV, current of 200 µA, and isotropic pixel size of 10
µm, with a 0.75-mm thick aluminum filter for beam-hardening
reduction. Morphometric analysis of callus bone volume percentage
[callus bone volume/total volume (BV/TV), %] and callus bone
mineral density (callus BMD, mg/cm3) were performed on
reconstructed three-dimensional (3D) images in a region of
interest, 1 cm centered around the fracture. The callus BMD
reflects the porosity of the measured callus and the density of
newly formed woven bone tissue.
Detection of bone turnover
markers
Prior to mice being euthanized on days 7, 14, 21 and
28 post-surgery, venous blood was extracted from the right
ventricle and serum was collected following centrifugation at 4°C,
1,500 × g for 20 min. Serum bone turnover markers, including BGP/OC
(CSB-E06917m), PINP (CSB-E12775m), TRACP-5b (CSB-E08492m), and
CTX–I (CSB-E12782m) were measured using the respective ELISA kits,
according to the manufacturers instructions. Serum samples were
diluted in PBS (1:400) accordingly. So that optical density (OD)
readings were within the range of the standard curve for the
respective biochemical marker. OD readings were measured at a
wavelength of 450 nm with a 650 nm reference filter, on a BioTek
Elx800 Absorbance Microplate Spectrophotometer (BioTek Instruments,
Inc.). The concentration of bone turnover in each group was
calculated based on the generated standard curve.
Histology and histomorphometry
Following microCT scanning, tibial bone specimens
were fixed in 4% paraformaldehyde for 48 h at 4°C and then
decalcified in 10% EDTA for 14 days at 4°C. Decalcified bone
tissues were embedded in paraffin, cut into 5-µm thick sections and
then stained with hematoxylin and eosin (H&E) at room
temperature (18 26°C) for 30 min, TRAP at 37°C for 1 h, and
Safranin O and Fast Green at room temperature (18 26°C) for 30 sec
and 20 min, respectively. Histological images in five randomly
selected fields were captured using an Olympus BX51
phase/fluorescence microscope (Olympus Corporation) at
magnification, ×100. TRAP-positive osteoclasts in the total callus
were counted by two independent investigators. The total callus
area (mm2), cartilage area relative to the total callus,
and hard callus area relative to total callus were calculated using
SPOT Advanced Software (SPOT Imaging; Diagnostic Instruments,
Inc.). Histomorphometry analysis of osteoclast parameters,
including TRAP-positive osteoclast surface per bone surface
(Oc.S/BS) and the number of TRAP-positve osteoclasts per trabecular
bone perimeter (N.Oc/B.Pm) were carried out on TRAP-stained
sections using the free Trap Histo Software (University of
Liverpool) as previously described (20). Dynamic histomorphometry was
performed on tibial bone samples from mice injected with Calcein
Green-Alizarin Red double fluorescent probes. Following fixation in
10% formaldehyde at 4°C for 3 days, undecalcified tibial bone
tissues were dehydrated in an ascending ethanol series (70, 95, and
twice with 100%), washed with xylene and subsequently embedded in
methyl methacrylate. The thin tissue sections (10 µm) were cut by a
Leica SM2500 microtome (Leica Microsystems GmbH), and images were
obtained using an Olympus BX51 phase/fluorescence microscope. The
callus bone apposition rate (MAR, µm/day) was quantified in the
following way: The distance between the fluorescence of Calcein
Green used as the initial baseline(to stain bone formation 10 days
after fracture) and the fluorescence of Alizarin Red used to reveal
novel bone formation (to stain new bone formation 17 days after
fracture) was measured and normalized by the time interval between
the two injections. Three serial sections were examined and the
mean value was analyzed, as previously described (20,21).
Biomechanical analysis
Biomechanical testing was conducted on tibial bone
specimens extracted from WT and cKO mice 21 and 28 days following
post-fracture surgery. Following removal of the soft tissue-free
full-length tibial shaft, the intramedullary nail was carefully
removed, and the two ends of the tibial bone were fixed with
matching fixtures, and then mounted on a torsion testing machine
(Instron) for biomechanical torsion testing of the callus. The
torsion angle rate was set to 0.2°/sec and the rotational torsion
testing was performed until failure. The maximum torque and
torsional stiffness were determined using Bluehill Universal
software (Instron). The maximum torque is the product of the force
when the specimen is broken and the moment arm, and the torsional
stiffness is a measure of the ability of the specimen to resist
torsional deformation.
RT-qPCR
Following removal of the intramedullary nails, the
RNA was extracted from the soft tissue-free tibial bone specimens
from each experimental group using TRIzol reagent. The tibial bone
specimens included normal bone tissues within 5 mm of the fracture
ends and all callus tissues. Complementary DNA was reverse
transcribed from 1–2 µg of extracted RNA using RevertAid First
Strand cDNA Synthesis kit under the following conditions: 37°C for
5 min, 42°C for 60 min and 70°C for 10 min. RT-qPCR was performed
using the 7500 Real-Time PCR system (Applied Biosystems; Thermo
Fisher Scientific, Inc.), with SYBR Green, cDNA template, and
specific primers for osteoblast [osteocalcin (OCN), alkaline
phosphatase (ALP), osteopontin (OPN) and type I collagen (COL1a2)]
and osteoclast marker genes [Cathepsin K (CTSK), matrix
metalloproteinase 9 (MMP-9), nuclear factor of activated t-cell
cytoplasmic 1 (NFATc1) and TRAP (ACP5)], presented in Table SI. The following cycling
conditions were used: initial denaturation at 95°C for 5 min; then
40 repeated cycles at 95°C for 5 sec, 60°C for 34 sec, 72°C for 15
sec; and final extension at 72°C for 1 min. mRNA levels were
quantified using the 2−ΔΔCq method (22), and normalized to the internal
reference gene β-actin.
Statistical analysis
All graphical data is presented as the mean ±
standard deviation (SD) of at least three independent biological
repeats unless otherwise stated. The unpaired Students t-test was
used to compare differences between two groups and one-way ANOVA
followed by the Tukeys post hoc test was used when comparing
multiple groups, using SPSS software (v22.0; IBM Corp.). P<0.05
was considered to indicate a statistically significant
difference.
Results
Specific deletion of PDK1 in
osteoclasts delays fracture healing
Previous studies have indicated that the process of
fracture healing in normal mice is as follows: Hematoma formation 3
days following the fracture, with new bone formation starting at
~day 7, this is followed by cartilaginous callus formation, which
peaks at ~day 10, followed by the transformation of the
cartilaginous callus to hard callus ~days 10–14. At ~day 21, the
surrounding callus begins to bridge the two ends of the fracture
leading to fracture healing, and by day 28, the fracture exhibits
standard clinical healing (23,24).
Using the aforementioned fracture healing
characteristics as guidelines, the fracture healing process in
PDK1 cKO mice were examined using X-rays and microCT
imaging. At 7 days following fracture surgery, the fracture line
was clearly visible in both radiographs and microCT 3D images
(Fig. 1A and B). Some calcified
callus formation was observed in both WT and PDK1 cKO mice,
but no marked bone bridge between the two broken ends of fracture
was identified. Furthermore, no significant difference in
mineralized callus volume and BMD was observed between the WT and
PDK1 cKO mice (Fig. 1C and
D). At 14 days following fracture surgery, the fracture ends of
both WT and PDK1 cKO tibial bones showed distinct hard
callus formation with extension of the bone bridge that connects
the fracture ends (Fig. 1A and B).
However, compared with that in WT tibial bones, the fracture line
was visible in PDK1 cKO tibial bones, which we hypothesize
is a consequence of a slower hematoma reabsorption. In addition,
there was no significant difference in mineralized callus volume,
but a lower callus BMD was identified in WT mice (Fig. 1C and D). At ~21 days post-fracture
surgery, the calcified callus at the fracture ends of WT tibial
bones had largely been resorbed, with no visible fracture line
(Fig. 1A and B). The hard callus
bone surrounding the fracture exhibited signs of remodeling into
plate-like cortical bone (Fig.
1B). On the other hand, large amounts of calcified callus were
observed in the fracture ends of PDK1 cKO tibial bones with
apparent bridging callus between fracture ends similar to that
observed in WT tibial bones on day 14 (Fig. 1A and B). Mineralized callus volume
was significantly higher in WT tibial bones as compared with that
in PDK1 cKO bones at day 21 (Fig. 1C). As with day 14, callus BMD at
day 21 in WT tibial bones were lower compared with that in
PDK1 cKO tibial bones (Fig.
1D). Lastly, at 28 days following post-fracture surgery, the
callus at the fracture site of WT tibial bone had completely healed
with complete cortical bone remodeling (Fig. 1A and B). On the other hand,
PDK1 cKO tibial bones exhibited callus characteristics
similar to those in WT tibial bones observed on day 21 with
calcified callus resorption, and cortical bridge remodeling
(Fig. 1B). Mineralized callus
volume was also significantly higher in WT mice compared with that
in PDK1 cKO mice (Fig. 1C),
whereas callus BMD was significantly lower in WT mice (Fig. 1D). The results observed at days 21
and 28 indicates that delayed healing, slower callus resorption and
cortical bridge remodeling of the tibial bone fracture is observed
in PDK1 cKO mice. The detailed score of the blurry degree of
fracture line is shown in Table
SII. The delayed callus resorption and cortical bridge
remodeling is likely to be the consequence of reduced osteoclast
formation and activity, as a result of loss of PDK1.
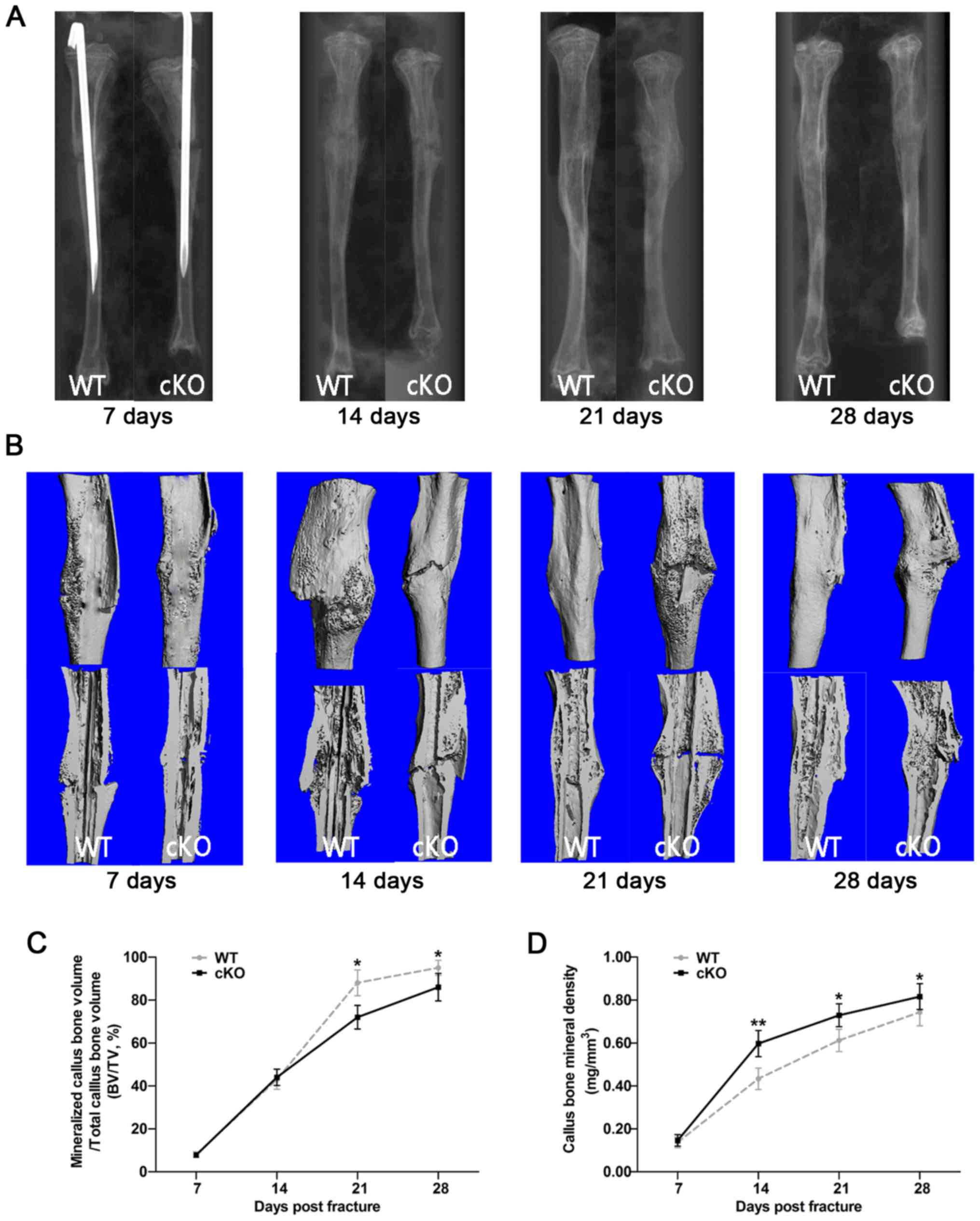 | Figure 1.Deletion of the PDK1 gene in
osteoclasts delays fracture union and repair. (A) Representative
X-ray and (B) three-dimensional microCT images of the fractured
tibia in WT and PDK1 cKO mice at 7, 14, 21 and 28 days
post-fracture. (C and D) Morphometric analysis of the changes in
mineralized callus BV/TV, and callus bone mineral density were
measured. Data are presented as mean ± SD. *P<0.05,
**P<0.01.BV, bone volume; TV, tissue volume; WT, wild-type; cKO,
conditional knockout; PDK1 cKO,
CtskCre-PDK1flox/flox; PDK1,
3-phosphoinositide-dependent protein kinase 1. |
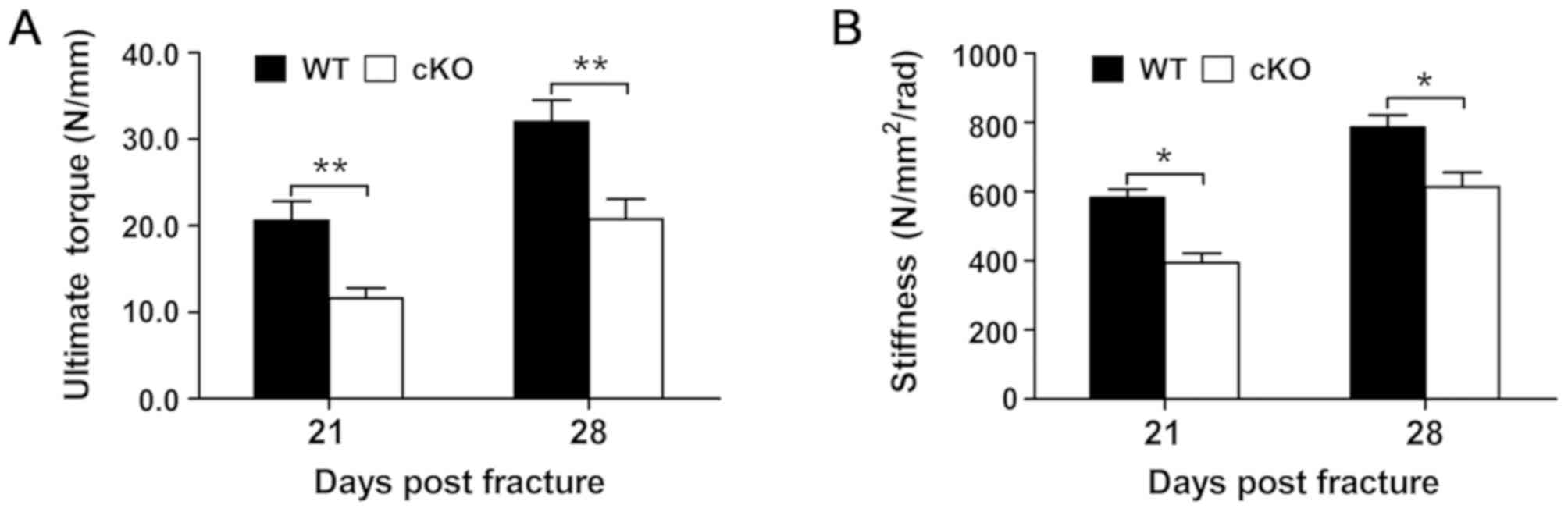
Loss of PDK1 in osteoclasts
compromises the biomechanical quality of the fracture callus
The biomechanical properties of the fracture callus
was assessed at days 21 and 28 following fracture surgery and the
results revealed that the fracture callus from PDK1 cKO
tibial bones were markedly weaker compared with that in WT mice at
both days 21 and 28 following fracture surgery. The maximum torque
and torsional stiffness were significantly decreased in PDK1
cKO tibial bones when compared with that in WT tibial bones
(Fig. 2A and B). These
biomechanical results indicate that the specific loss of
PDK1 in osteoclasts negatively impacts the bone-callus
microarchitecture during the fracture healing process, leading to
compromised biomechanical quality.
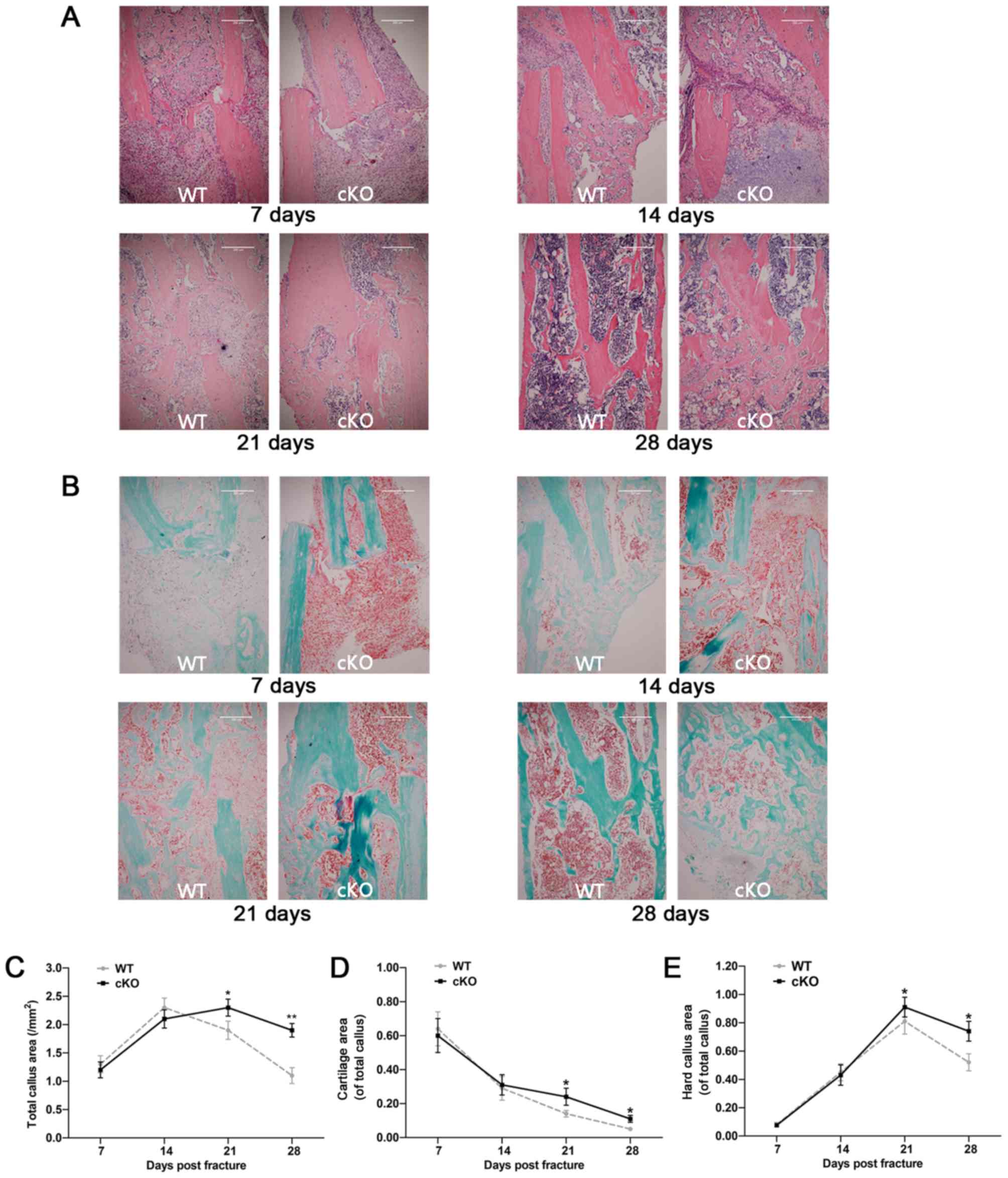
Loss of PDK1 in osteoclasts delays
callus resorption and remodeling
H&E and SO/FG staining was performed on tibial
bone sections to visualize general bone tissue and cartilage
microarchitecture, respectively, during the fracture healing
process. As shown in Fig. 3A and
B, the progression of fracture healing was delayed in
PDK1 cKO mice. At 7 days post-fracture surgery, large
amounts of undifferentiated mesenchyme and some proliferating
chondrocytes accumulated at the broken end of the fracture in both
WT and PDK1 cKO tibial bones. Most of the callus at this
timepoint, in both experimental groups were organized hematoma,
with some degree of cartilaginous callus. By day 14 post-fracture,
large numbers of hypertrophic chondrocytes were present at the
broken ends of the fracture, and transition from cartilaginous
calluses to hard calluses were observed in both WT and PDK1
cKO groups (Fig. 3A and B). At 7-
and 14- days post-fracture, no significant differences in the area
of cartilaginous callus, hard callus and total callus were observed
between the WT and PDK1 groups (Fig. 3C-E). At 21-days post-fracture, few
chondrocytes were observed in the broken ends of the fracture in WT
tibial bones. The hard callus in the WT tibial bone fracture site
were gradually reabsorbed transitioning from immature woven to more
mature lamellar bone structure. A large number of osteoblasts and
osteocytes in the WT tibial bones were observed (Fig. 3A and B). By day 28, most of the
hard callus in the WT tibial bones had been resorbed with the
observation of mature lamellar cortical bone and medullary cavity.
On the other hand, hypertrophic chondrocytes were the most abundant
cells observed in the remaining cartilaginous callus and the hard
callus at the fracture site were mostly immature woven trabecular
bone in the PDK1 cKO group at 21 days post-fracture.
Transition from immature woven bone to lamellar bone started at day
28 in the PDK1 cKO group, with evidence of residual
cartilaginous callus and large amounts of irregular trabecular bone
within the medullary space. No cortical bone structure was observed
(Fig. 3A and B). The cartilaginous
callus, hard callus and total callus area in the cKO group were
markedly greater compared with that in the WT group at both 21- and
28-days following fracture surgery (Fig. 3C-E). Together, these results
suggest that the loss of PDK1 in osteoclasts significantly
delays callus resorption and bone remodeling during the fracture
healing process.
Osteoclast deletion of PDK1 reduces
osteoclast activity but not osteoblast bone formation activity
TRAP staining was also performed to examine
osteoclast parameters in the fracture callus during the fracture
healing process. From day 7 to 21 post-fracture, abundant numbers
of TRAP+ve multinucleated osteoclasts were observed in
the fracture ends of WT tibial bones, which were predominantly
localized to the hypertrophic zone of the cartilaginous callus and
the trabecular surface of the hard callus (Fig. 4A). Morphometric quantitation of the
N.Oc/B.Pm and Oc.S/BS (%) further showed that peak osteoclast
formation and activity occurred between days 14 and 21 (Fig. 4B and C). By day 28, the number and
activity of TRAP+ve osteoclasts were significantly
reduced, coinciding with the end of the fracture healing process.
On the other hand, the number and activity of TRAP+ve
osteoclasts in the fracture callus of PDK1 cKO tibial bones
were significantly lower at all timepoints when compared with that
in WT tibial bones (Fig.
4A-C).
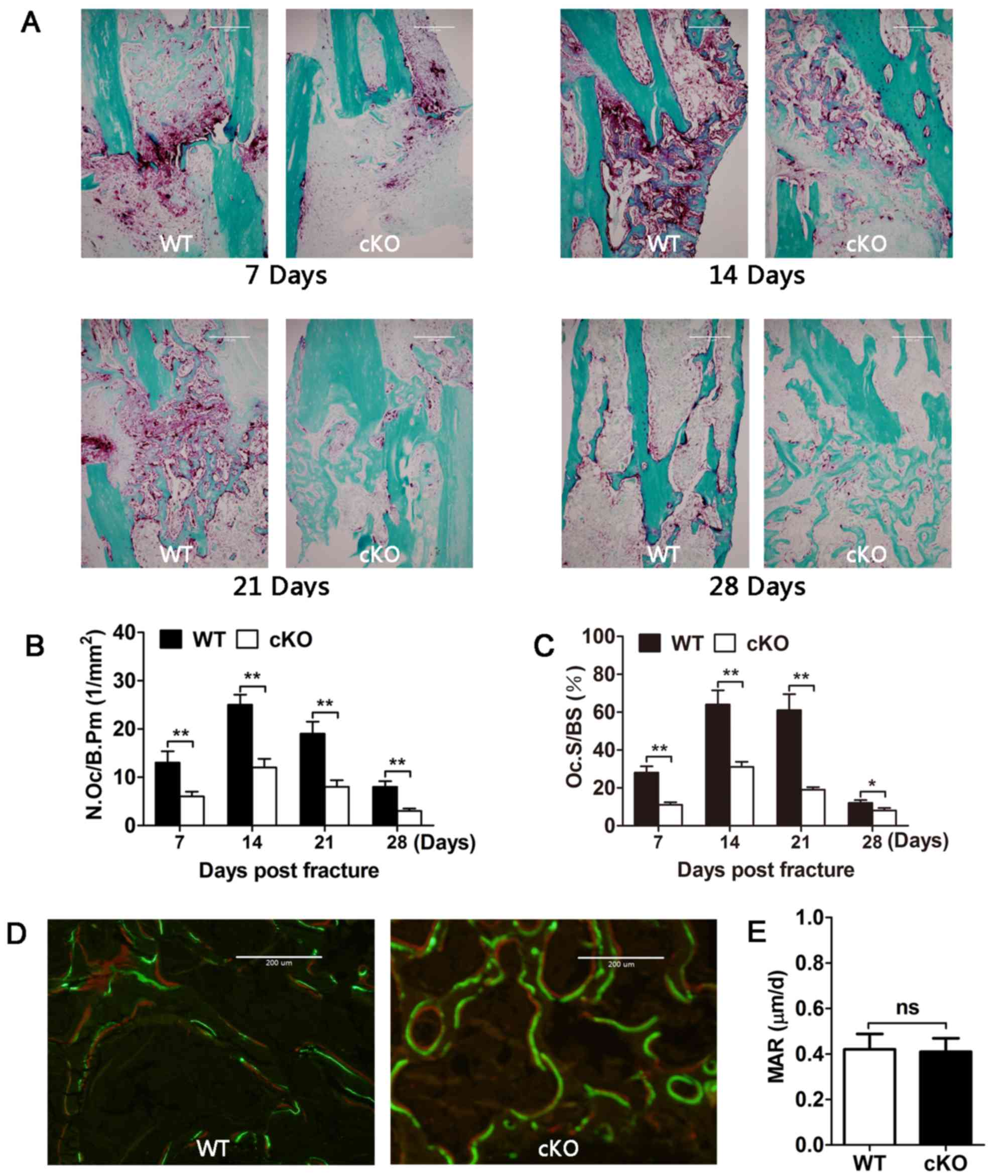 | Figure 4.PDK1 is required for osteoclast
formation and bone resorption, but not osteoblast bone formation.
(A) Representative TRAP stained sections of tibial fracture callus
in WT and PDK1 cKO mice 7, 14, 21- and 28-days
post-fracture. (B and C) Quantitative histomorphometric analysis of
the N.Oc/B.Pm and Oc.S/BS in the fracture callus were performed. (D
and E) Representative Calcein-Alizarin Red S double stained
sections of tibial fracture callus and associated dynamic
histomorphometric measurement of bone mineral apposition rate in WT
and PDK1 cKO mice. Fluorescence staining of irregular
trabecular bone in new callus. Green fluorescence represents
calcein and red fluorescence represents Alizarin Red S. Data are
presented as mean ± SD. *P<0.05, **P<0.01. ns, no significant
difference; cKO, conditional knockout; PDK1 cKO,
CtskCre-PDK1flox/flox; WT, wild-type;
PDK1, 3-phosphoinositide-dependent protein kinase 1; TRAP,
tartrate-resistant acid phosphatase; Oc.S/BS, osteoclast surface
per bone surface; N.Oc/B.Pm, number of osteoclasts per bone
perimeter. |
The MAR results showed no significant difference
between WT and PDK1 cKO mice (Fig. 4D and E), indicating that
osteoblast-mediated bone formation was not affected during the
fracture healing process. Analysis of serum levels of bone turnover
markers measured at days 7, 14, 21 and 28 showed a significant
reduction in bone resorption markers, such as TRAP and CTX–I in
PDK1 cKO mice (Fig. 5A and
B). No change in bone formation markers of BGP and PINP were
observed between WT and PDK1 cKO mice (Fig. 5C and D). Consistent with these
serum results, RT-qPCR analysis of gene expression in callus
tissues further showed a reduction in osteoclast marker genes,
including CTSK, MMP-9, TRAP and NFATc1 (Fig. 6A-D). As with previous results, no
change in the mRNA expression levels of osteoblast marker genes,
such as OCN, ALP, OPN and COL1a2 was observed (Fig. 6E-H). It appears that osteoblast
bone formation and osteogenesis was not affected. Collectively,
these results provide further evidence that reduced osteoclast
formation and activity, and not osteoblast activity, was
responsible for the delayed cartilaginous callus resorption and
bone remodeling during the fracture healing process.
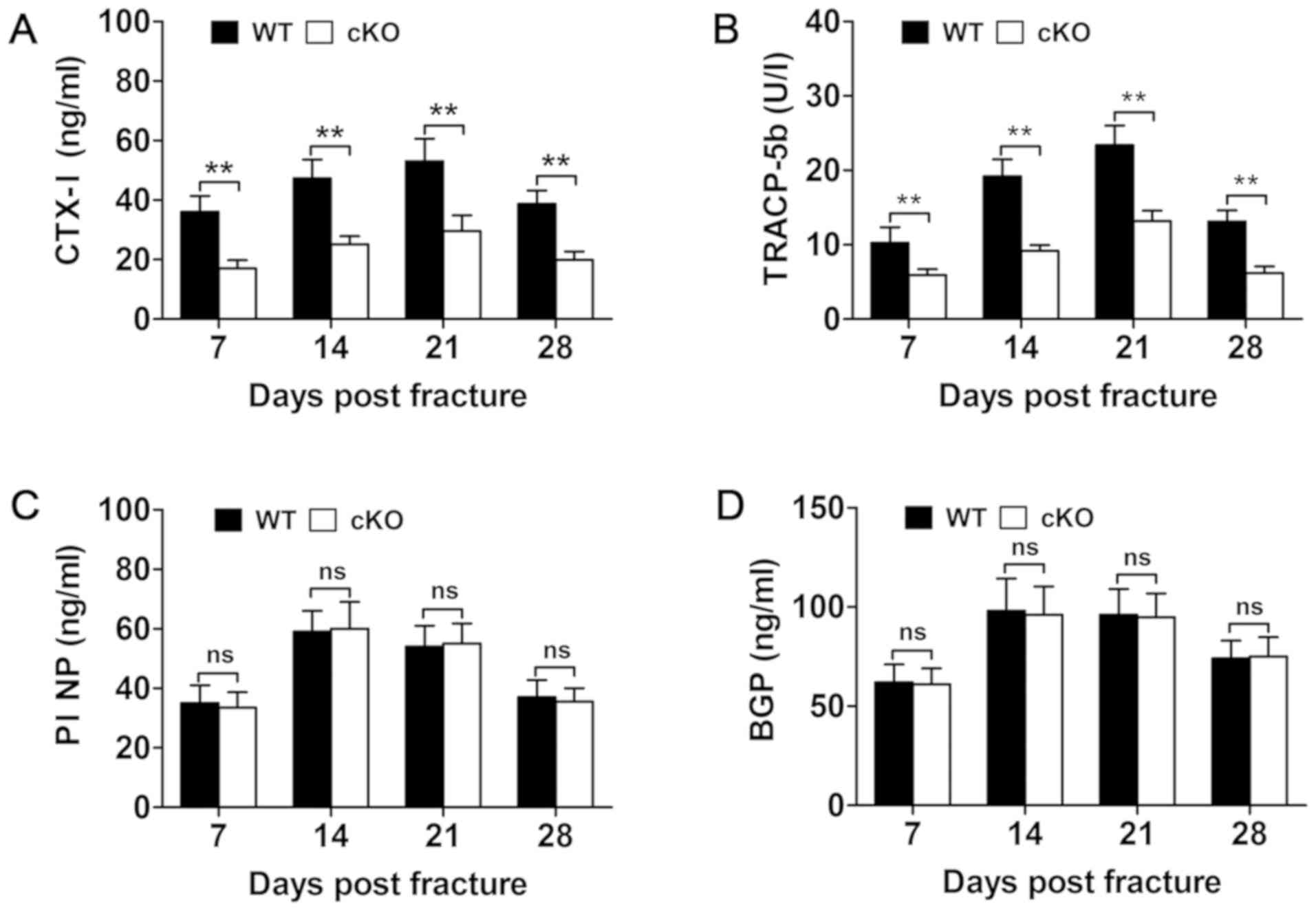 | Figure 5.Osteoclast-specific deletion of
PDK1 impairs osteoclasts bone resorption, but not osteoblast
bone formation. Changes in serum biochemical indices of bone
resorption, including (B) TRACP5b and (A) CTX–I, and bone
formation, such as (D) BGP/osteocalcin and (C) PINP at 7, 14, 21
and 28 days post-fracture in WT and PDK1 cKO mice were
measured using ELISA. Data presented as mean ± SD. **P<0.01. ns,
no significant difference; cKO, conditional knockout; PDK1
cKO, CtskCre-PDK1flox/flox; WT,
wild-type; PDK1, 3-phosphoinositide-dependent protein kinase 1;
TRAP, tartrate-resistant acid phosphatase; CTx-I, C-terminal
telopeptide of type I collagen; BGP, glaprotein; PINP, procollagen
I N-terminal propeptide. |
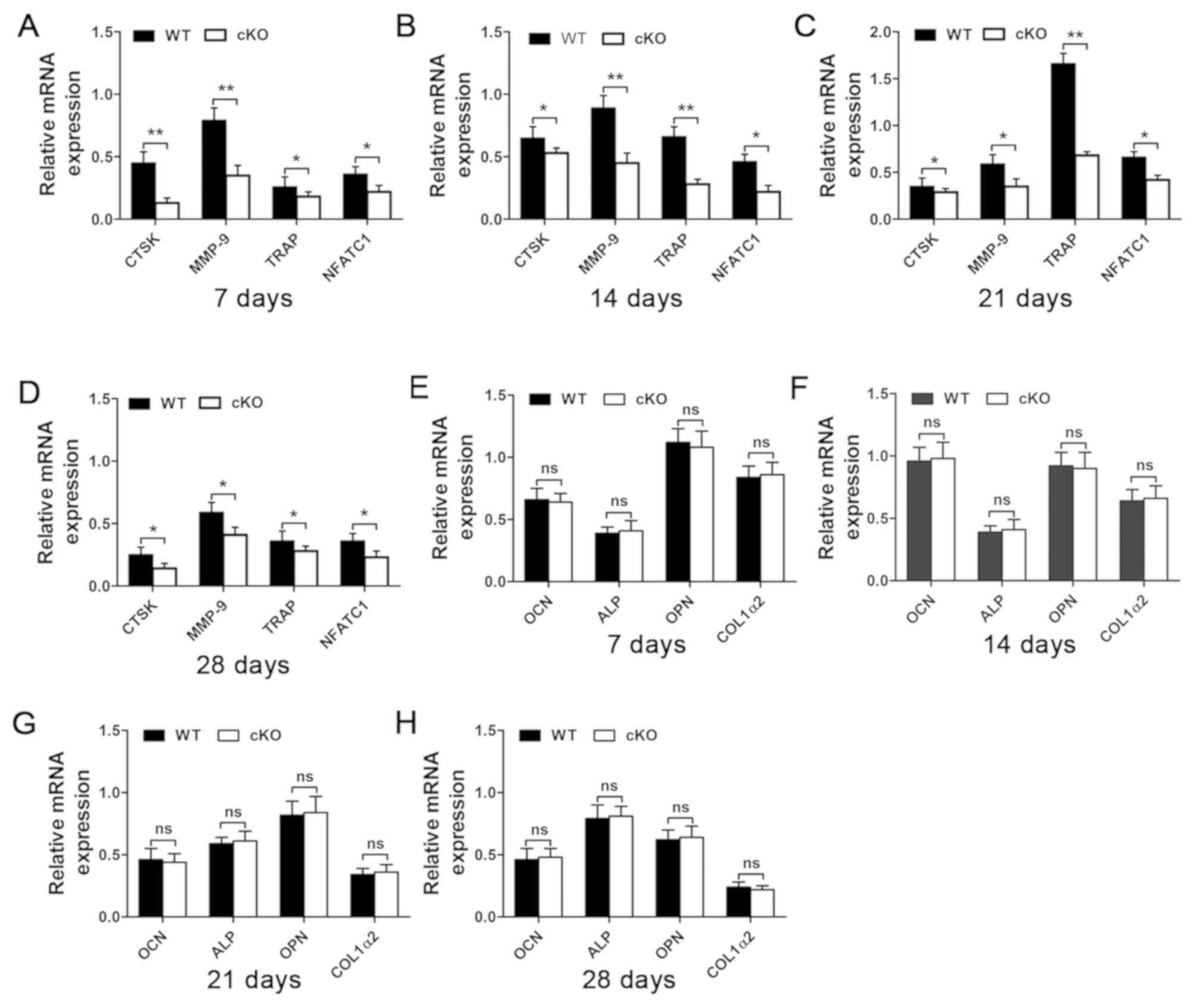 | Figure 6.Osteoclasts marker gene expression
level is downregulated following the deletion of PDK1 gene.
The expression of (A-D) osteoclast marker genes, including CTSK,
MMP-9, TRAP and NFATc1, and (E-H) osteoblast marker
genes, including OCN, ALP, OPN and COL1α2 at 7, 14, 21- and 28-days
post-fracture were examined using reverse
transcription-quantitative PCR. *P<0.05, **P<0.01. ns, no
significant difference; cKO, conditional knockout; PDK1 cKO,
CtskCre-PDK1flox/flox; WT, wild-type;
PDK1, 3-phosphoinositide-dependent protein kinase 1; TRAP,
tartrate-resistant acid phosphatase; CTSK, Cathepsin K; MMP-9,
matrix metalloproteinase 9; NFATc1, nuclear factor of activated
t-cell cytoplasmic 1; OCN, osteocalcin; ALP, alkaline phosphatase;
OPN, osteopontin; COL1α2, type I collagen. |
Discussion
Bone fracture healing is a complex, dynamic and
multi-staged process necessary for the restoration of damaged bone
to a functionally and biomechanically normal state. Both the bone
resorbing osteoclasts and the bone forming osteoblasts play
indispensable roles in the fracture healing process (6,9,10,25,26).
In our previous study, it was revealed, using cellular and
biochemical methodologies, that the specific deletion of
PDK1 in osteoclasts impairs RANKL-induced osteoclast
formation and bone resorption via the suppression of the
Akt-GSK3β-NFATc1 signaling pathway (17). Thus, we hypothesized that the
specific loss of PDK1 in osteoclasts will affect fracture
healing. In the present study, using the PDK1 cKO mice and
the tibial fracture model, it was revealed that the specific
deletion of PDK1 in osteoclasts impeded the fracture healing
process by delaying the resorption of the cartilaginous callus
during the middle phase (14–21 days post-fracture), thereby
hindering the remodeling of immature woven bone to mature lamellar
bone during the late phase (21–28 days post-fracture).
As the present data has shown, osteoclasts play an
important role in fracture healing process. Osteoclasts are
predominantly involved in the middle and late phases of the
fracture healing process and are typically responsible for the
resorption and remodeling of cartilaginous soft callus for
replacement with hard callus (16,27).
On the other hand, immune cells and chondrocytes play a more
dominant role during the early phase of fracture repair (4,6,28).
As such, and despite significant decreases in osteoclasts in the
fracture site of PDK1 cKO tibial bones, significant
differences were not observed in fracture repair between WT and
PDK1 cKO mice during the early phase (7–14 days
post-fracture) of inflammation, hematoma development and
cartilaginous callus formation. Callus volume and cartilaginous
callus/hard callus relative to total callus, were similar between
WT and PDK1 cKO mice during the early phase of fracture
repair.
The middle and late phases (14–28 days
post-fracture) of cartilaginous callus resorption, and remodeling
of woven bone to lamellar bone are important processes during
fracture repair. Previous genetic and pharmacological drug studies
have shown that inhibition of osteoclast formation and/or bone
resorption activity delays fracture healing by disrupting cartilage
dissolution and remodeling, and increases fracture callus size
(15,16,27,29,30).
On the other hand, increased osteoclast formation accelerated
fracture healing (31). In the
present study, X-rays and microCT 3D images from PDK1 cKO
mice revealed distinct fracture lines and increased callus size
from 14–28 days post-fracture. Histological assessments of tibial
bone sections from PDK1 cKO mice further showed markedly
decreased total numbers of TRAP+ve osteoclasts near the
chondro-osseous junctions at the fracture site, as well as the
number of active osteoclasts on the bone surface, which was
associated with delayed cartilaginous callus resorption and woven
bone remodeling. Reduced osteoclast formation and activity was
further confirmed by gene expression analyses of osteoclast marker
genes, including NFATc1, TRAP, CTSK and MMP-9. Both CTSK and MMP-9
are important proteases required for the resorption process. In
particular, it has been reported that MMP-9 is crucially involved
in soft callus remodeling and hypertrophic chondrocyte apoptosis,
with the lack of MMP-9 leading to delayed fracture healing
(32,33).
The amount of cartilaginous callus in fracture site
of PDK1 cKO tibial bones were comparable to WT mice in the
early phase of fracture repair; however, there was considerably
more cartilaginous callus that was not reabsorbed (greater overall
callus bone volume and area with respect to total callus area) in
the middle phase in PDK1 cKO tibial bones, suggesting that
the impediment of fracture repair was not due to the initial size
of the formed cartilaginous callus but rather the resorption and
remodeling of the cartilaginous callus to hard callus. This delay
became more apparent during the late phase (28 days post-fracture)
of fracture healing, with the total callus volume in the
PDK1 cKO group being significantly larger compared with that
in the WT group. The larger callus area, bone volume and BMD
observed at the middle and late phases of fracture repair was not
due to increased osteoblast activity, as osteoblast MAR, serum bone
formation markers (PINP and BGP), and osteoblast marker genes (OCN,
ALP, OPN, COL1a2) were not affected by the loss of PDK1 in
osteoclasts. By this time most of the cartilaginous callus had been
remodeled to hard callus in the PDK1 cKO group; however,
morphologically abundant immature woven trabecular bone was
observed in the medullary of the tibial bones with no apparent
cortical shell formation. In comparison, in the WT group, most of
the immature woven trabecular bone had been remodeled to lamellar
bone with cortical shell formation largely complete, displaying
overall normal bone architecture. These results were similar to
previous studies which examined the effects of bisphosphonates on
fracture healing and repair (14,34).
Biomechanical analysis of bone torsional stiffness and maximum
torsion torque of the fracture callus at the middle and late phases
of fracture repair further indicate that bone remodeling during the
fracture healing process was impaired. Immature woven bone is
structurally and mechanically weaker compared with that in mature
lamella bone, and the maximum torsion torque value and stiffness
strength value are associated with the quality and quantity of new
bone.
Our previous cellular and biochemical study provided
evidence for the importance of PDK1 in regulating osteoclast
formation and bone resorption function in vitro and in vivo
(17). The present study further
demonstrated, using the tibial bone fracture model in PDK1
cKO mice, that the loss of PDK1, specifically in osteoclasts,
impedes fracture healing and repair by delaying cartilaginous
callus resorption and remodeling into hard callus in the middle
phase and subsequent remodeling of immature woven bone into
lamellar bone in the late phase. The impairment in cartilage
resorption and bone remodeling was solely due to the decrease of
osteoclast formation and activity, as osteoblast bone formation was
not affected. These results not only enhance our understanding of
the physiological role of PDK1 in osteoclasts, but also
provides important clinical implications for the use of
anti-resorptive agents, such as bisphosphonates for the treatment
of osteolytic conditions. Such anti-resorptive therapies may
detrimentally delay fracture healing and repair, and these
hypotheses will require validation in future studies.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 81860402), Guangxi Natural
Science Foundation of China (grant no. 2017GXNSFAA198073), Guangxi
Key Research and Development Project (grant no. GuikeAB17195001),
High Level Innovation Team and Excellence Scholars Program of
Guangxi High Education Institutions and Guangxi Medical High-level
Key Talents Training ‘139’ Program Training Project.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors contributions
DX, QuZ and YB performed the experiments, and wrote
the manuscript; BC and QiZ analyzed the data; GZ and SZ conceived
and designed the study, and reviewed and edited the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The animal experiments were approved by the Animals
Ethics Committee of Guangxi Medical University and the Guide for
the Care and Use of Laboratory Animals (approval no.
201805021).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kanis JA, Odén A, McCloskey EV, Johansson
H, Wahl DA and Cooper C; IOF Working Group on Epidemiology and
Quality of Life, : A systematic review of hip fracture incidence
and probability of fracture worldwide. Osteoporos Int.
23:2239–2256. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tzioupis C and Giannoudis PV: Prevalence
of long-bone non-unions. Injury. 38 (Suppl 2):S3–S9. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hak DJ, Fitzpatrick D, Bishop JA, Marsh
JL, Tilp S, Schnettler R, Simpson H and Alt V: Delayed union and
nonunions: Epidemiology, clinical issues, and financial aspects.
Injury. 45 (Suppl 2):S3–S7. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Einhorn TA: The cell and molecular biology
of fracture healing. Clin Orthop Relat Res. 355S (Suppl):S7–S21.
1998. View Article : Google Scholar
|
|
5
|
Claes L, Recknagel S and Ignatius A:
Fracture healing under healthy and inflammatory conditions. Nat Rev
Rheumatol. 8:133–143. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schindeler A, McDonald MM, Bokko P and
Little DG: Bone remodeling during fracture repair: The cellular
picture. Semin Cell Dev Biol. 19:459–466. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hadjiargyrou M, Lombardo F, Zhao S, Ahrens
W, Joo J, Ahn H, Jurman M, White DW and Rubin CT: Transcriptional
profiling of bone regeneration. Insight into the molecular
complexity of wound repair. J Biol Chem. 277:30177–30182. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gerstenfeld LC, Cullinane DM, Barnes GL,
Graves DT and Einhorn TA: Fracture healing as a post-natal
developmental process: Molecular, spatial, and temporal aspects of
its regulation. J Cell Biochem. 88:873–884. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Marsell R and Einhorn TA: The biology of
fracture healing. Injury. 42:551–555. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Einhorn TA and Gerstenfeld LC: Fracture
healing: Mechanisms and interventions. Nat Rev Rheumatol. 11:45–54.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ferguson C, Alpern E, Miclau T and Helms
JA: Does adult fracture repair recapitulate embryonic skeletal
formation? Mech Dev. 87:57–66. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xin C, Zhi-Ming H, Yasuaki S, Tomoo T and
Akira Y: Quantitative study of osteoclastic related factors in the
process of bone reconstruction. Hua Xi Kou Qiang Yi Xue Za Zhi.
24:164–165. 2006.(In Chinese). PubMed/NCBI
|
|
13
|
Lin HN and OConnor JP: Osteoclast
depletion with clodronate liposomes delays fracture healing in
mice. J Orthop Res. 35:1699–1706. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cao Y, Mori S, Mashiba T, Westmore MS, Ma
L, Sato M, Akiyama T, Shi L, Komatsubara S, Miyamoto K, et al:
Raloxifene, estrogen, and alendronate affect the processes of
fracture repair differently in ovariectomized rats. J Bone Miner
Res. 17:2237–2246. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Flick LM, Weaver JM, Ulrich-Vinther M,
Abuzzahab F, Zhang X, Dougall WC, Anderson D, OKeefe RJ and Schwarz
EM: Effects of receptor activator of NFkappaB (RANK) signaling
blockade on fracture healing. J Orthop Res. 21:676–684. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gerstenfeld LC, Sacks DJ, Pelis M, Mason
ZD, Graves DT, Barrero M, Ominsky MS, Kostenuik PJ, Morgan EF and
Einhorn TA: Comparison of effects of the bisphosphonate alendronate
versus the RANKL inhibitor denosumab on murine fracture healing. J
Bone Miner Res. 24:196–208. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xiao D, Zhou Q, Gao Y, Cao B, Zhang Q,
Zeng G and Zong S: PDK1 is important lipid kinase for RANKL-induced
osteoclast formation and function via the regulation of the
Akt-GSK3β-NFATc1 signaling cascade. J Cell Biochem. Feb
12–2020.(Epub ahead of print). View Article : Google Scholar
|
|
18
|
Morshed S: Current options for determining
fracture union. Adv Med. 2014.doi:10.1155/2014/708574. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mi M, Jin H, Wang B, Yukata K, Sheu TJ, Ke
QH, Tong P, Im HJ, Xiao G and Chen D: Chondrocyte BMP2 signaling
plays an essential role in bone fracture healing. Gene.
512:211–218. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
van t Hof RJ, Rose L, Bassonga E and
Daroszewska A: Open source software for semi-automated
histomorphometry of bone resorption and formation parameters. Bone.
99:69–79. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Abstracts of the Academy of Pediatric
Physical Therapy Poster Presentations at the Combined Sections
Meeting. Pediatr Phys Ther. 31:e31–e69. 2019. View Article : Google Scholar
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Inada M, Matsumoto C and Miyaura C: Animal
models for bone and joint disease. Ovariectomized and
orchidectomized animals. Clin Calcium. 21:164–170. 2011.(In
Japanese). PubMed/NCBI
|
|
24
|
Frost HM and Jee WS: On the rat model of
human osteopenias and osteoporoses. Bone Miner. 18:227–236. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ghiasi MS, Chen J, Vaziri A, Rodriguez EK
and Nazarian A: Bone fracture healing in mechanobiological
modeling: A review of principles and methods. Bone Rep. 6:87–100.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Loi F, Córdova LA, Pajarinen J, Lin TH,
Yao Z and Goodman SB: Inflammation, fracture and bone repair. Bone.
86:119–130. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kamimura M, Mori Y, Sugahara-Tobinai A,
Takai T and Itoi E: Impaired fracture healing caused by deficiency
of the immunoreceptor adaptor protein DAP12. PLoS One.
10:e01282102015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shefelbine SJ, Augat P, Claes L and Beck
A: Intact fibula improves fracture healing in a rat tibia osteotomy
model. J Orthop Res. 23:489–493. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
He LH, Liu M, He Y, Xiao E, Zhao L, Zhang
T, Yang HQ and Zhang Y: TRPV1 deletion impaired fracture healing
and inhibited osteoclast and osteoblast differentiation. Sci Rep.
7:423852017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
McDonald MM, Dulai S, Godfrey C, Amanat N,
Sztynda T and Little DG: Bolus or weekly zoledronic acid
administration does not delay endochondral fracture repair but
weekly dosing enhances delays in hard callus remodeling. Bone.
43:653–662. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ota N, Takaishi H, Kosaki N, Takito J,
Yoda M, Tohmonda T, Kimura T, Okada Y, Yasuda H, Kawaguchi H, et
al: Accelerated cartilage resorption by chondroclasts during bone
fracture healing in osteoprotegerin-deficient mice. Endocrinology.
150:4823–4834. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Takahara M, Naruse T, Takagi M, Orui H and
Ogino T: Matrix metalloproteinase-9 expression, tartrate-resistant
acid phosphatase activity, and DNA fragmentation in vascular and
cellular invasion into cartilage preceding primary endochondral
ossification in long bones. J Orthop Res. 22:1050–1057. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Colnot C, Thompson Z, Miclau T, Werb Z and
Helms JA: Altered fracture repair in the absence of MMP9.
Development. 130:4123–4133. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li J, Mori S, Kaji Y, Kawanishi J, Akiyama
T and Norimatsu H: Concentration of bisphosphonate (incadronate) in
callus area and its effects on fracture healing in rats. J Bone
Miner Res. 15:2042–2051. 2000. View Article : Google Scholar : PubMed/NCBI
|