Introduction
Autophagy is a well-conserved intracellular process
involving the formation of double membrane structures and the
engulfing biological macromolecules, organelles or lipid droplets
for degradation (1–5). Moreover, autophagy is important for
cell survival, clearance of impaired cellular compartments and
lipid balances when challenged with various stressors, including
starvation and cholesterol or triglyceride overloading (1–5).
Impaired autophagic activities have been shown to increase lipid
accumulation, and thus are accompanied by severe lipotoxicity in
hepatocytes (4,6). Apoptosis, a major type of programmed
cell death, is an evolutionarily conserved process (7). In addition, by activating the
cascades of cysteine proteases, known as caspases, apoptosis plays
a key role in deciding cell fate when confronted with extrinsic and
intrinsic derived stressors (7).
Furthermore, apoptosis contributes to the development of various
disease states, including carcinogenesis (8), neurodegenerative disorders (9,10)
and diabetes (11,12). In addition, p62 (13), Beclin1 and bcl2 (14,15),
which are key molecules in autophagy, have been reported to act as
a bridge between autophagy and apoptosis; however, the relationship
between these factors is not fully understood.
2-Hydroxypropyl-β-cyclodextrin (HPβCD), a
chemically-modified water-soluble cyclodextrin derivative, has been
widely utilized as a drug delivery system (16–18),
as well as an efficient therapeutic strategy for neurodegenerative
diseases (19,20) and atherosclerosis regression
(19,21), due to its distinctive capability of
regulating cellular cholesterol transport and metabolism. Moreover,
the main target organs of HPβCD are the kidney, liver, lungs and
spleen (22). Approved by the Food
and Drug Administration (FDA), HPβCD is recognized as a relatively
innocuous therapeutic (18,22),
but its adverse effects have rarely been investigated. However, it
has been reported that a high dose HPβCD could impede autophagy
flux in fibroblasts (23) and in a
mouse model of Alzheimer's disease (AD) (24). Furthermore, HPβCD serves as a
potent molecule for inducing apoptosis in human leukemic cell lines
(25). Collectively, these
findings suggest potential negative effects of long-term
administration of HPβCD. Therefore, the present study examined the
dose-response effects of HPβCD on autophagy and apoptosis in a
liver cancer cell line (HepG2), which is widely used as a cellular
model for normal hepatocytes to assess the potential of chemical
hepatotoxicity (26). The present
study aimed to determine whether high dose HPβCD could impair the
autophagy flux and trigger apoptosis in HepG2 cells. Autophagy flux
blockage leads to autophagosome accumulation, which serves as a
platform for caspase-8 activation and may be responsible for the
activation of apoptosis (27).
Materials and methods
Reagents and materials
HPβCD, chloroquine (CQ), DMSO, SC 79,
3-Methyladenine (3-MA), Z-IETD-FMK and water-soluble cholesterol
(in methyl-β-cyclodextrin) were purchased from Sigma-Aldrich (Merck
KGaA) or APExBIO Technology LLC (Table SI). Details of the antibodies used
in the present study are shown in Table SII.
Cell culture
RPMI-1640 medium (Sigma-Aldrich; Merck KGaA)
supplemented with 10% FBS (Sigma-Aldrich; Merck KGaA) and 1%
penicillin-streptomycin (100 IU/ml penicillin and 100 mg/ml
streptomycin) was used as cell culture medium for HepG2 cells (The
Cell Bank of Type Culture Collection of the Chinese Academy of
Sciences). For further experiments, such as flow cytometry or
western blot analysis, HepG2 cells were plated at a density of
5×103 cells/well in six-well plates. When cells reached
~50% confluence, the medium was replaced and HepG2 cells were
treated with HPβCD with a combination of either cholesterol, SC 79
(Akt activator), CQ (lysosomal inhibitor), 3-MA (Class III PI3K
inhibitor), Z-IETD-FMK (selective caspase-8 inhibitor) for either
48 h to induce apoptosis or 24 h to detect autophagy flux at 37°C.
DMSO (0.5%) was added to cultures as solvent control. After
treatment, each well was washed twice with cold PBS and cells were
harvested.
To investigate the association between the
expression levels of phosphorylated (p)-AKT or p-mTOR and membrane
cholesterol levels, the cells were divided into four groups and
treated with either 0, 10, 20 or 20 mM HPβCD at 37°C for 12 h.
Then, the culture medium was removed, and the cells were washed
with PBS twice at room temperature. New culture medium was
subsequently added to each group and the fourth group of cells
treated with 20 mM HPβCD were further treated with 5 µg/ml free
cholesterol at 37°C for another 12 h.
Cell viability assay
A Cell Counting Kit-8 (CCK-8) colorimetric assay
(Dojindo Molecular Technologies, Inc.) was used to examine cell
viability, according to the manufacturer's protocol. Prior to the
CCK-8 colorimetric assay, HepG2 cells were plated in 96-well plates
at 2.5×103 cells/well in a volume of 80 µl cell culture
medium and treated with 0.2, 2 or 20 mM HPβCD for 0, 24, 48 or 72
h, respectively. Cells in 6-replicated wells were treated with 8 µl
CCK-8 and incubated at 37°C for 2 h. Absorbance was measured at a
wavelength of 450 nm using a microtiter plate reader (Tecan Safire
2; Tecan Group, Ltd.). The specific formula used to calculate cell
viability was described previously (28).
Monomeric red fluorescent protein
(mRFP)-green fluorescent protein (GFP-microtubule-associated
protein 1 light chain 3 (LC3) adenovirus transduction
mRFP-GFP-LC3 adenovirus, expressing a tandem
RFP-GFP-LC3B fusion protein, was provided by Hanbio Biotechnology
Co., Ltd. For mRFP-GFP-LC3 adenovirus transduction, HepG2 cells
were plated at a density of 5×103 cells/well in confocal
dishes. Upon reaching 30% confluence, 1 µl adenoviral particles
were added into each well, which were used to infect cells at ~10
multiplicity of infection at 37°C for 16 h. After the transduction
process, infected cells were treated with 20 mM HPβCD at 37°C for
24 h. Furthermore, 50 µg/ml CQ was utilized as a positive control
of autophagosomes accumulation. Then, cells were fixed with 4%
paraformaldehyde at room temperature for 30 min and subjected to
Zeiss LSM-710 confocal microscopy (magnification, ×630) to observe
GFP and mRFP staining.
Western blot analysis
HepG2 cell lysates were obtained by RIPA lysis
buffer (Beyotime Institute of Biotechnology) according to the
manufacturer's instructions, and protein concentrations were
determined using a standard bicinchoninic acid method. Then, 20 µg
protein/lane was separated by 12.5% SDS-PAGE. After blocking by 5%
BSA (Sigma-Aldrich; Merck KGaA) in TBS-0.1% Tween-20 at room
temperature for 2 h, PVDF (EMD Millipore) membranes were cut into
different strips according to the size of target proteins and the
strips were further incubated with appropriate primary antibodies
(1:2,000; Table SII) for ≥12 h
overnight at 4°C. The next day, the membranes were incubated with
either mouse or rabbit secondary antibodies (1:5,000; Table SII) for 2 h at room temperature.
Protein bands were visualized using High-sig ECL western blotting
substrate (Tanon Science and Technology, Co., Ltd.) and GAPDH was
used as the internal reference protein. Acquired bands were
quantified by ImageJ 1.52 software (National Institutes of Health).
Each independent experiment was replicated ≥3 times.
Flow cytometry for detecting apoptotic
cells
Flow cytometry was performed using a method
described previously (27). Both
cell culture medium and attached cells, were collected for
centrifugation at 1,332 × g at 25°C for 5 min. Then, cells were
washed twice with cold PBS, suspended in 100 µl PBS and incubated
with 3 µl Annexin V-FITC and 5 µl Propidium Iodide (PI) for 15 min
in the dark at room temperature. Following incubation, 300 µl
Annexin V binding buffer was added to each tube and detected by
flow cytometry (FACSCalibur; BD Biosciences). All Annexin
V+ cells were considered as apoptotic cells and data
were analyzed by CellQuest software (version 7.5.3; BD
Biosciences).
TUNEL assay
After treatment with 20 mM HPβCD at 37°C for 48 h,
200 HepG2 cells/mm2 were fixed with 4% paraformaldehyde
at 25°C for 15 min. Then, an in-situ cell death detection
kit, Fluorescein (Roche Applied Science) was used according to the
manufacturer's instructions. Subsequently, cell nuclei were
counterstained with 0.2 µg/ml DAPI at room temperature for 10 min
and mounted with glycerol gelatin (Sigma-Aldrich; Merck KGaA). An
Olympus IX-71 fluorescence microscope (magnification, ×40) was used
to acquire the images in ≥3 randomly selected fields of view.
Statistical analysis
Data are presented as the mean ± SEM of ≥3
experimental repeats. Comparisons among multiple groups were
performed using one-way ANOVA with Tukey's post hoc test. P<0.05
was considered to indicate a statistically significant difference.
All statistical analyses were performed using SPSS 21.0 software
(IBM Corp.).
Results
HPβCD leads to apoptosis in HepG2
cells
Cell viability was examined to determine whether
HPβCD treatment exerted any adverse effects on HepG2 cells. It was
found that 20 mM HPβCD significantly inhibited cell viability
compared with the 0.2 or 2 mM groups at 24, 48 and 72 h time points
(Fig. S1). Then, flow cytometry
was used to identify Annexin V-FITC/PI stained apoptotic cells.
Annexin V+PI− cells were considered as early
apoptotic and Annexin V+PI+ cells were
considered as late apoptotic (28). The results indicated that high
concentration (20 mM) of HPβCD treatment increased the proportion
of apoptotic cells compared with control and 2 mM HPβCD-treated
groups (Fig. 1A and C), which is
in line with the results of TUNEL assay (Fig. S2). Furthermore, HepG2 cells were
treated with a high dosage (20 mM) HPβCD for different durations.
It was found that the apoptotic rate was significantly higher after
48-h treatment compared with 24-h treatment (Fig. 1B and D). Collectively, the results
suggested that apoptosis caused by HPβCD treatment occurred in a
dose- and time-dependent manner.
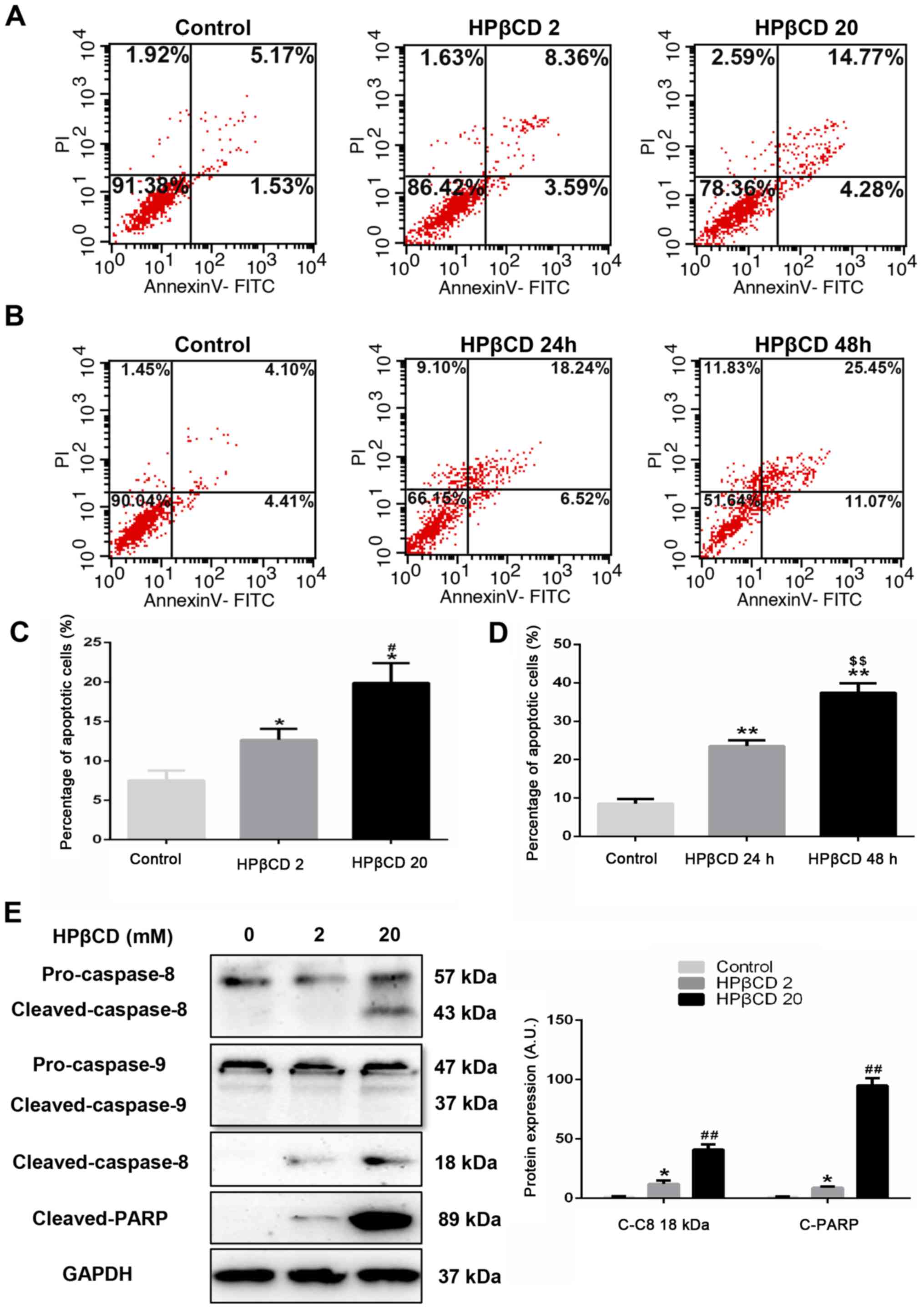 | Figure 1.HPβCD triggers apoptosis in HepG2
cells. Flow cytometry analysis of apoptotic HepG2 cells treated
with HPβCD using Annexin V-FITC/PI double staining; all Annexin
V-FITC positive cells were considered as apoptotic cells. HepG2
cells were treated with (A) 0, 2 or 20 mM HPβCD for 24 h or (B) 20
mM HPβCD for 0, 24 or 48 h and the percentage of apoptotic cells
were quantified for the treatment at different (C) doses or (D)
time. (E) Protein expression levels of cleaved-caspase-8 and PARP
after 48-h HPβCD (0, 2 or 20 mM) treatment. GAPDH was utilized as a
control. Data are representative of three independent experiments.
*P<0.05 and **P<0.01 vs. control group; #P<0.05
and ##P<0.01 vs. HPβCD 2 group;
$$P<0.01 vs. HPβCD 24 h group. PI, propidium iodide;
HPβCD, 2-hydroxypropyl-β-cyclodextrin; C-C8, cleaved-caspase-8;
C-PARP, cleaved-PARP; PARP, poly ADP-ribose polymerase; HPβCD 2,
cells treated with 2 mM HPβCD; HPβCD 20, cells treated with 20 mM
HPβCD; A.U., arbitrary units. |
Subsequently, these results were assessed using
western blotting. Compared with the 2 mM HPβCD treatment group, it
was demonstrated that cleaved-poly ADP-ribose polymerase (PARP), a
major target of cleaved-caspase-3, was significantly upregulated by
20 mM HPβCD treatment (Fig. 1E).
Since apoptosis is induced mainly via extrinsic and intrinsic
pathways (7,8), cleaved-caspase-8 and
cleaved-caspase-9 expression levels in HepG2 cells with different
concentrations of HPβCD treatment were detected. However, the
results indicated that there was no significant difference in
cleaved-caspase-9 expression, while there was increased expression
of cleaved-caspase-8 at higher concentrations of HPβCD, which
suggested that the HPβCD-induced HepG2 cell apoptosis did not occur
through the intrinsic pathway (Fig.
1E).
HPβCD blocks autophagy flux in HepG2
cells
The expression levels of two important autophagy
protein markers, LC3 and p62, were detected in HepG2 cells treated
with different concentrations of HPβCD. Using the lysosomal
inhibitor CQ as a control, it was found that LC3B-II and p62
protein expression levels were significantly upregulated at a high
dose of HPβCD compared with controls and low doses of HPβCD-treated
cells (Fig. 2A), which suggested
that HPβCD blocked autophagy flux in HepG2 cells in a
dose-dependent manner. Then, cells were transfected with
mRFP-GFP-LC3 adenovirus expressing a tandem RFP-GFP-LC3B fusion
protein. As GFP signal is quenched in acidic conditions after
fusing with lysosomes, merged red and yellow dots represent
autolysosomes and autophagosomes, respectively (29). Using CQ as a positive control, an
obvious cluster of red dots was observed in negative control cells,
while HPβCD treatment almost completely inhibited the autophagy
flux, as shown by collections of yellow dots (Fig. 2B).
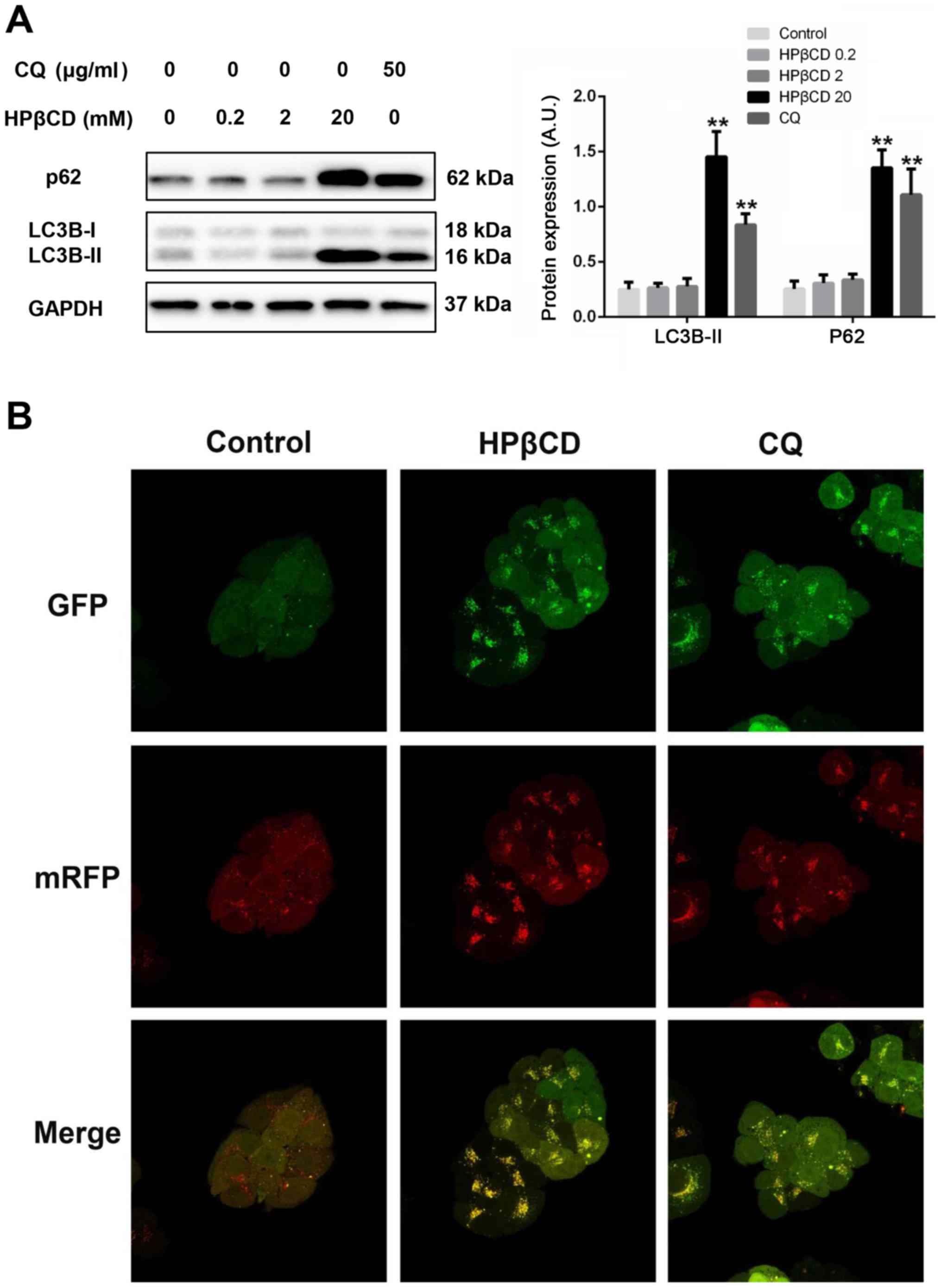 | Figure 2.HPβCD impairs autophagy flux in HepG2
cells. (A) Protein expression levels of p62 and LC3B-II after 24-h
HPβCD (0, 0.2, 2 and 20 mM) treatment. GAPDH was utilized as a
control. (B) Confocal microscopy images (magnification, ×630) of
HepG2 cells expressing a tandem GFP-RFP-LC3 fusion protein treated
with HPβCD (20 mM) and CQ (50 µg/ml) for 24 h. Merged yellow puncta
were considered as autophagosomes and merged red puncta were
considered as autolysosomes. **P<0.01 vs. control. HPβCD,
2-hydroxypropyl-β-cyclodextrin; LC3, microtubule-associated protein
1 light chain 3; CQ, chloroquine; GFP, green fluorescent protein;
mRFP, monomeric red fluorescent protein; HPβCD 0.2, cells treated
with 0.2 mM HPβCD; HPβCD 20, cells treated with 20 mM HPβCD; HPβCD
2, cells treated with 2 mM HPβCD; HPβCD 20, cells treated with 20
mM HPβCD; A.U., arbitrary units. |
AKT/mTOR axis contributes to
autophagosome accumulation and apoptosis in HPβCD-treated HepG2
cells
The protein expression levels of several proteins,
including ERK, AMP-activated protein kinase, AKT, mTOR and
Beclin-1, were then detected in HepG2 cells with HPβCD treatment.
The results suggested that only the AKT/mTOR axis, a master pathway
regulating autophagy (30,31), was downregulated, which was
reversed via free cholesterol (FC) replenishment (Figs. 3A and S3). Inhibition of the AKT/mTOR axis
caused substantial formation of new autophagosomes, which serves as
platforms for caspase-8 activation and subsequent apoptotic
cascades (27,32). It was found that SC 79, a potent
AKT activator, significantly reduced the expression of LC3B-II
protein, which is an indicator for the level of autophagosomes
(Fig. 3C and E) (29). Furthermore, compared with 20 mM
HPβCD-treated cells, significantly decreased cleaved-caspase-8 and
PARP protein expression levels (Fig.
3C and E), as well as decreased levels of apoptotic cells, were
observed (Fig. 3B and D) after SC
79 treatment.
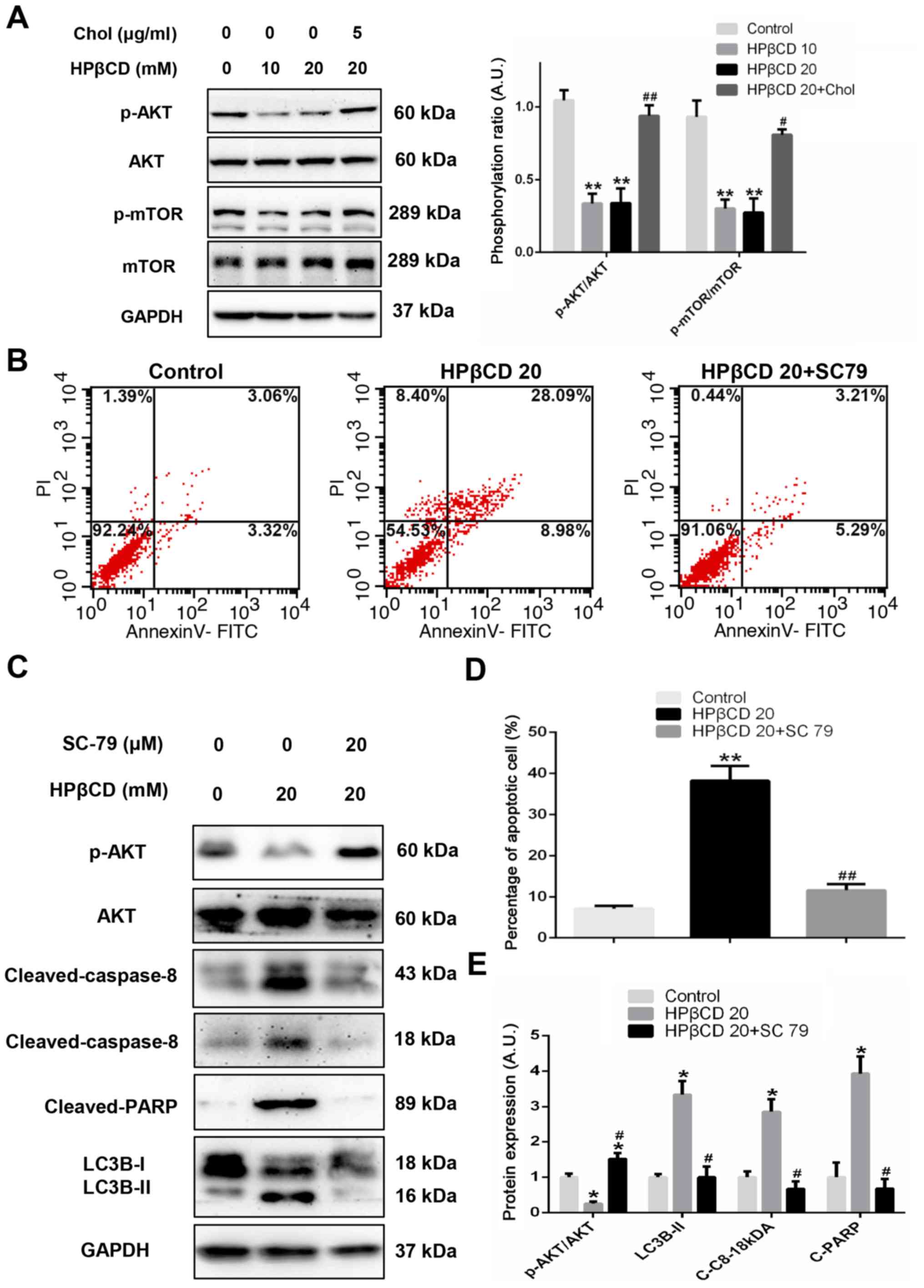 | Figure 3.AKT/mTOR pathway is involved in the
regulation of autophagy and apoptosis after HPβCD treatment in
HepG2 cells. (A) Expression levels of p-AKT and mTOR in HepG2 cells
treated with 0, 10 or 20 mM HPβCD or 5 µg/ml chol + 20 mM HPβCD for
12 h. The non-specific band may have resulted from the non-specific
binding of the primary antibody for p-mTOR. (B) Flow cytometry
analysis of apoptotic cells treated with 20 mM HPβCD or 20 µM SC 79
+ 20 mM HPβCD for 48 h and (D) the percentage of apoptotic cells
were quantified. (C) Protein expression levels of
cleaved-caspase-8, cleaved-PARP, p-AKT and LC3B-II after 20 mM
HPβCD or 20 µM SC 79 + 20 mM HPβCD treatment for 48 h were (E)
quantified. GAPDH was utilized as a control. Data are
representative of three independent experiments. *P<0.05 and
**P<0.01 vs. control; #P<0.05 and
##P<0.01 vs. HPβCD 20. Chol, cholesterol; p-,
phosphorylated; HPβCD, 2-hydroxypropyl-β-cyclodextrin; LC3,
microtubule-associated protein 1 light chain 3; C-C8,
cleaved-caspase-8; C-PARP, cleaved-PARP; PARP, poly ADP-ribose
polymerase; PI, propidium iodide; HPβCD 10, cells treated with 10
mM HPβCD; HPβCD 20, cells treated with 20 mM HPβCD; A.U., arbitrary
units. |
Autophagosome accumulation is
positively associated with caspase-8 activation
To further assess the results, the concentration of
HPβCD was increased to a higher level, and cells were also
incubated with 3-MA, CQ or Z-IETD-FMK. Flow cytometry results
demonstrated that higher concentration of HPβCD treatment
significantly increased the proportion of apoptotic cells compared
with controls and 20 mM HPβCD-treated cells (Fig. 4A and C). Moreover, when incubated
with CQ, the percentage of 20 mM HPβCD-treated apoptotic cells
significantly increased and the protein expression levels of
LC3B-II, cleaved-PARP and cleaved-caspase-8 were significantly
upregulated compared with cells treated with 20 mM HPβCD alone
(Fig. 4A-D). However, after
incubation with 3-MA, a Class III PI3K inhibitor, apoptotic cells
were significantly reduced and the protein expression levels of
LC3B-II, cleaved-PARP and cleaved-caspase-8 were significantly
downregulated compared with 20 mM HPβCD-treated cells (Fig. 4A-D). Therefore, the results
suggested that HPβCD-induced apoptosis was positively associated
with LC3B-II. In addition, it was found that Z-IETD-FMK, a
selective caspase-8 inhibitor, almost completely blocked caspase-8
expression and apoptosis was significantly reduced compared with 20
mM HPβCD-treated cells (Fig.
4A-D), which may serve as evidence for autophagosomal
membrane-mediated caspase-8 activation.
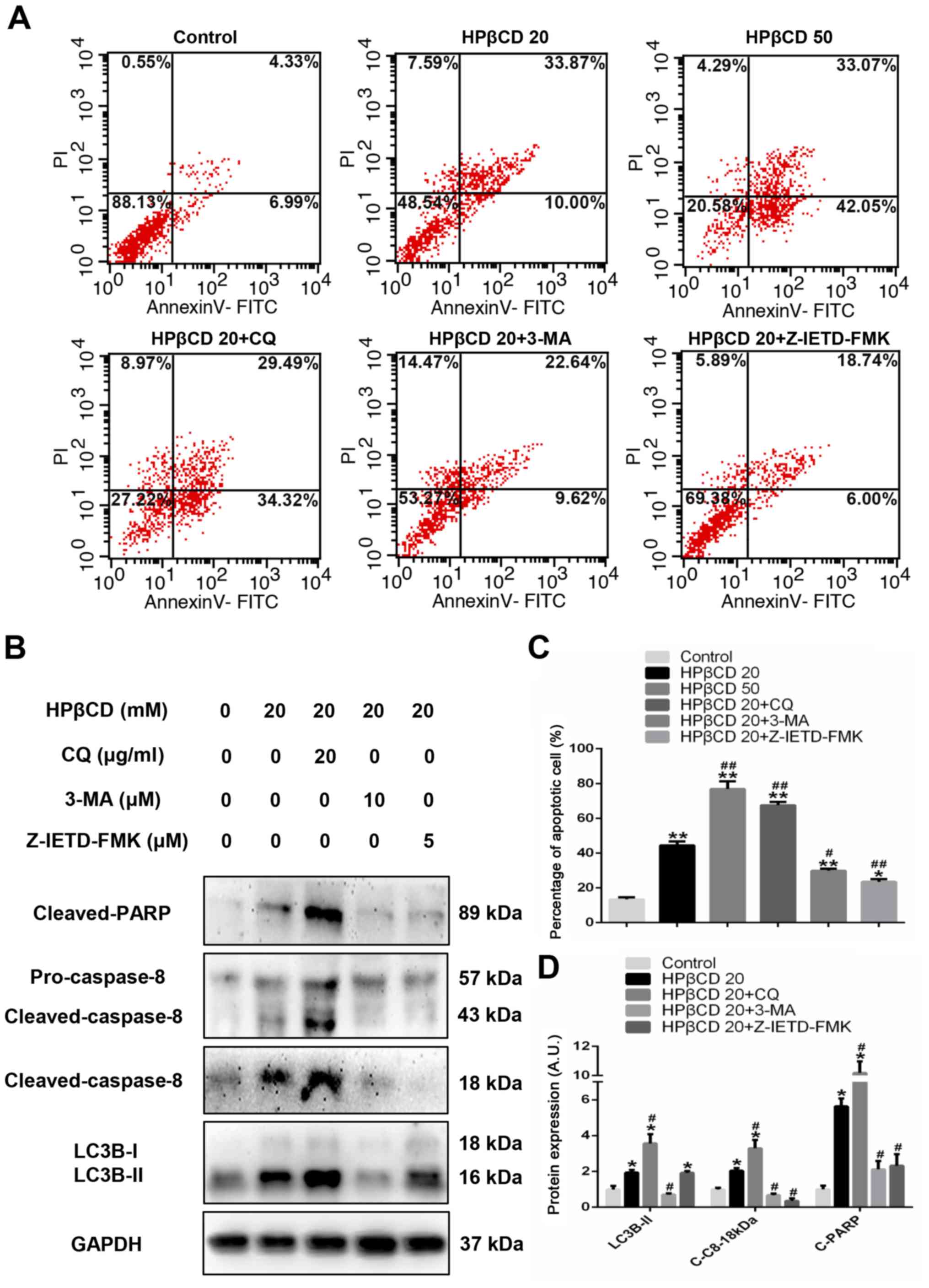 | Figure 4.HPβCD triggers apoptosis by
activating the autophagosome/caspase-8 axis in HepG2 cells. (A)
Flow cytometry results of apoptotic cells treated with HPβCD (20 or
50 mM) and HPβCD (20 mM) + 3-MA (10 µM), CQ (20 µg/ml) or
Z-IETD-FMK (5 µM) for 48 h. (C) Results from three replicated
experiments were quantified. (B) Representative western blot image
and (D) protein expression levels of cleaved-caspase-8,
cleaved-PARP and LC3B-II with HPβCD (20 mM) and HPβCD (20 mM) +3-MA
(10 µM), CQ (50 µg/ml) or Z-IETD-FMK (5 µM) for 48 h. *P<0.05
and **P<0.01 vs. control; #P<0.05 and
##P<0.01 vs. HPβCD 20. HPβCD,
2-hydroxypropyl-β-cyclodextrin; LC3, microtubule-associated protein
1 light chain 3; C-C8, cleaved-caspase-8; C-PARP, cleaved-PARP;
PARP, poly ADP-ribose polymerase; PI, propidium iodide; CQ,
chloroquine; 3-MA, 3-Methyladenine; HPβCD 20, cells treated with 20
mM HPβCD; HPβCD 50, cells treated with 50 mM HPβCD; A.U., arbitrary
units. |
Discussion
The present results suggested that high dosage of
HPβCD inactivated the AKT/mTOR pathway, which facilitated the
initiation of autophagy and blocked autophagy flux in the late
stage, and thus led to massive autophagosomes accumulation in HepG2
cells. Therefore, it was speculated that the membrane of gradually
stacked autophagosomes may served as platforms for the cleavage of
caspase-8, which lead to downstream caspase-cascades and ultimately
apoptosis.
Due to its ability to enhance efflux from cell
membranes and endo-lysosomal trafficking, HPβCD is a promising
therapeutic strategy for treating cholesterol metabolism-related
diseases such as Niemann-Pick type C (19) and atherosclerosis regression
(21). As a derivative of the
cyclodextrin family, HPβCD can directly alter the properties of
lipid bilayers by extracting lipids, including cholesterol and
phospholipids from biological membranes (33). Lipid rafts, highly ordered membrane
domains that are enriched in cholesterol and gangliosides (34), are essential for the transduction
of various cell signaling cascades, such as the activation of the
PI3K/AKT and Fas/CD95 signaling pathways. In addition, the amount
of cellular lipid rafts is reported to be positively associated
with cholesterol levels, because cholesterol promotes the formation
of lipid rafts (35,36). For instance, increasing membrane
cholesterol by adding free cholesterol to prostate cancer cells
increased the amount of cellular lipid rafts and, thus, upregulated
the phosphorylation of AKT (35),
while reducing the membrane cholesterol of glioblastoma cells by
inhibiting sterol carrier protein 2, which is one of the main
cholesterol transporters targeting the plasma membrane (PM),
reduced cellular lipid rafts and subsequently, downregulated the
phosphorylation levels of AKT (36).
The present results suggested that HPβCD treatment
directly inhibited the AKT/mTOR pathway in HepG2 cells, which was
significantly restored after the replenishment of extra FC.
Moreover, these results were in line with the results from studies
by Oh et al (37,38). Direct alterations of the
cholesterol levels of the biological membrane by HPβCD may also
cause disruption in the fusion of autophagosomes and lysosomes,
thus hindering the autophagy flux in the late stage (23). In the brain tissue of AD model mice
or C57BL wild-type mice treated with HPβCD for 2 weeks, a large
number of immature autophagosomes are observed (24). In addition, it has been shown that
high dose of HPβCD treatment blocked the autophagy flux in
fibroblasts (23). Moreover, these
findings were identified in the present study, as indicated by
increased protein expression levels of LC3-BII and p62, and the
accumulation of merged yellow puncta in HepG2 cells. Collectively,
due to its modulation of lipid rafts in the PM and disruption of
basal lysosome membrane lipid properties, HPβCD inhibited the
AKT/mTOR pathway that potentiated the formation of autophagosomes,
while the downstream infusion with lysosomes was blocked, thus
leading to massive accumulation of autophagosomes in cytoplasm,
which may be a risk factor for the development of various diseases,
especially in the long-term (39–41).
Caspase-8 plays a vital role in the activation of
caspase-cascades for extrinsic signaling pathways (42,43),
and oligomerization is a crucial step for caspase-8 activation
(44). It has been reported that
the autophagosomal membrane acts as a platform for the
oligomerization of caspase-8 (32,27).
Furthermore, either knockdown of p62 (27) or LC3 (32) is shown to significantly reduce the
activity of apoptosis. In addition, pharmaceutically inhibiting
autophagosome formation using 3-MA, a Class III PI3K inhibitor,
attenuated caspase-8 activation in 293T cells (45). However, induction of autophagy by
SKI–I, a pansphingosine kinase inhibitor, facilitated the
activation of caspase-8 and subsequent caspase-cascades, which is
further enhanced when treated with lysosomal inhibitors, including
bafilomycin A1, CQ and ammonium chloride (27). In the present study, it was
identified that SC 79 and 3-MA significantly mitigated
HPβCD-induced autophagosome accumulation and caspase-8 activation
in HepG2 cells, which was via the activation of Class I PI3K and
inhibition of the Class III PI3K pathway. However, it was
demonstrated that when incubated with CQ, HPβCD further facilitated
the activation of caspase-8, which may be due to the effect of CQ
alkalizing the acid condition of lysosomes, and that of HPβCD
disrupting the lysosomal membrane property. Caspase-8 is the
initiator caspase-of the extrinsic apoptotic pathway and is
commonly activated by cell surface death receptors (42,46,47).
The present results suggested that Z-IETD-FMK, a selective
inhibitor for caspase-8, inhibited the apoptotic activities induced
by HPβCD treatment, thus suggesting that autophagosomal
membrane-mediated caspase-8 self-activation may be the pivotal
mechanism for HPβCD-induced programmed cell death in HepG2
cells.
Moreover, Song et al (48) reported that HPβCD could promote the
nuclear translocation of Transcription Factor EB (TFEB), the main
regulator of lysosomal function and autophagy, in fibroblasts with
a lysosomal storage disorder, which restored its lysosome-autophagy
system and enhanced the clearance of ceroid lipopigment deposits.
Furthermore, TFEB is downstream of the AKT/mTOR pathway and is
negatively regulated by the phosphorylation levels of mTOR
(49). It was discovered that the
activation of TFEB by HPβCD treatment in a study by Song et
al (48) was consistent with
inhibition of the AKT/mTOR pathway in the present study. Song et
al (48) revealed that HPβCD
treatment did not lead to the activation of apoptotic pathways.
However, the present results suggested that HPβCD blocked autophagy
flux and induced caspase-8-mediated apoptosis in HepG2 cells. In
the future, investigations using numerous other different cell
models are required to determine whether HPβCD could induce similar
effects, as only the HepG2 cell line was investigated in the
present study. Therefore, to the best of our knowledge, the present
study was the first to demonstrate that, accompanied by the
downregulation of the AKT/mTOR pathway, a master pro-survival and
autophagy regulating signaling axis, high doses of HPβCD treatment
impaired autophagy flux and induced autophagosome
caspase-8-mediated apoptosis in hepatocytes; the specific mechanism
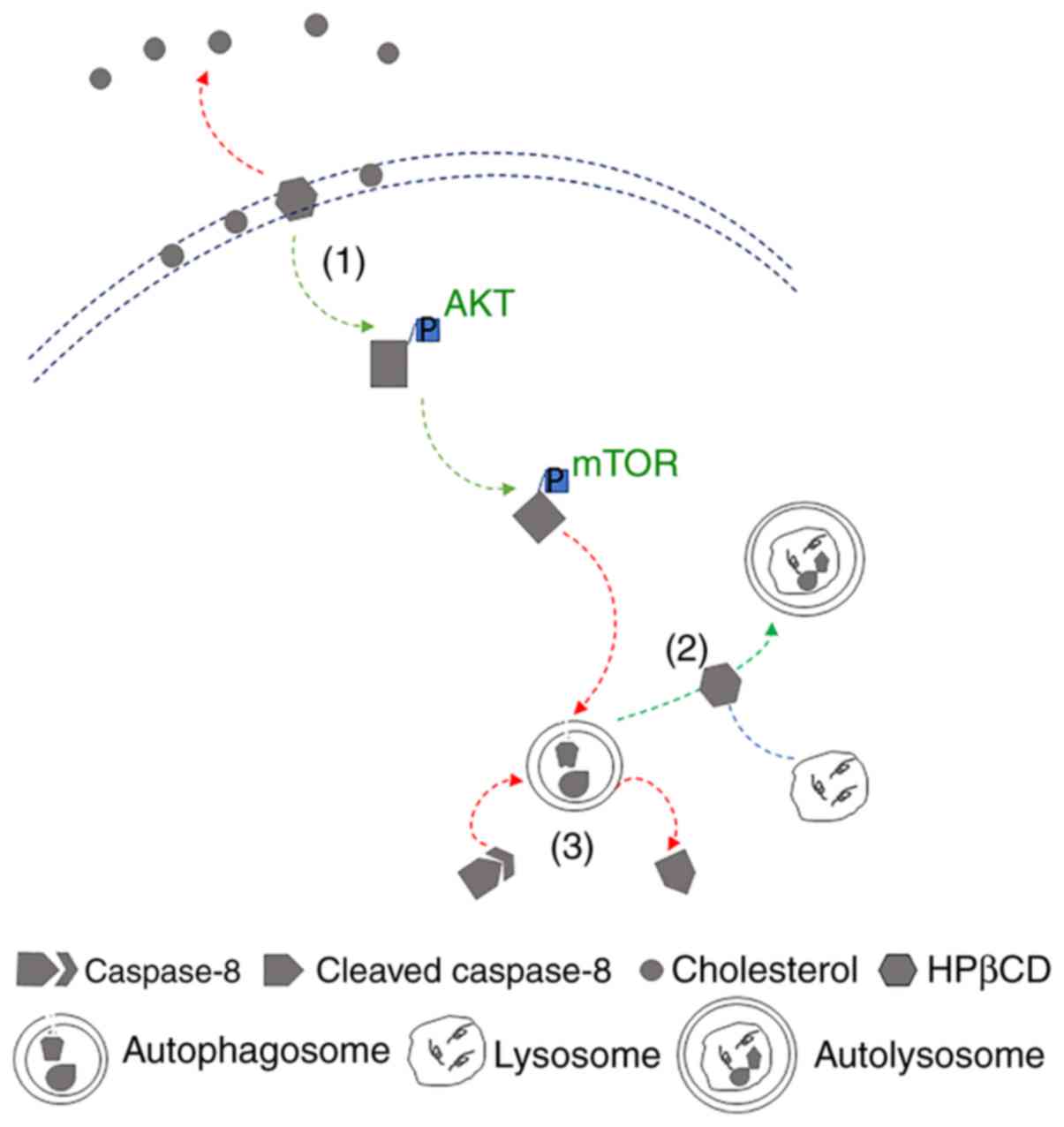
of which is depicted in Fig. 5.
While HPβCD has been intensively studied and approved by the FDA,
its potential adverse effects regarding autophagy and apoptosis
should be considered, especially when administrated at high doses
or long-term use for patients with disfunctions of kidney, which is
the main organ for the clearance of HPβCD (18,22).
The present study provided novel evidence of the significance of
the side effects of HPβCD administration with regard to particular
conditions. For instance, HPβCD may worsen the status of patients
with non-alcoholic fatty liver disease or impede the recovery of
patients who have undergone liver transplantation.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by Shanghai
Municipal Commission of Health and Family Planning (grant no.
201540191).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HZ and GW conceived the study, carried out the
experimental design and data interpretation, and prepared and
revised the manuscript. HS performed the experiments. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
FC
|
free cholesterol
|
|
LC3
|
microtubule-associated protein 1 light
chain 3
|
|
PM
|
plasma membrane
|
|
PARP
|
poly ADP-ribose polymerase
|
|
HPβCD
|
2-hydroxypropyl-β-cyclodextrin
|
References
|
1
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ueno T and Komatsu M: Autophagy in the
liver: Functions in health and disease. Nat Rev Gastroenterol
Hepatol. 14:170–184. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xu K, Yang Y, Yan M, Zhan J, Fu X and
Zheng X: Autophagy plays a protective role in free cholesterol
overload-induced death of smooth muscle cells. J Lipid Res.
51:2581–2590. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Singh R, Kaushik S, Wang Y, Xiang Y, Novak
I, Komatsu M, Tanaka K, Cuervo AM and Czaja MJ: Autophagy regulates
lipid metabolism. Nature. 458:1131–1135. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu K and Czaja MJ: Regulation of lipid
stores and metabolism by lipophagy. Cell Death Differ. 20:3–11.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
He Q, Mei D, Sha S, Fan S, Wang L and Dong
M: ERK-dependent mTOR pathway is involved in berberine-induced
autophagy in hepatic steatosis. J Mol Endocrinol. 57:251–260. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vaux DL, Haecker G and Strasser A: An
evolutionary perspective on apoptosis. Cell. 76:777–779. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Williams GT: Programmed cell death:
Apoptosis and oncogenesis. Cell. 65:1097–1098. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Radi E, Formichi P, Battisti C and
Federico A: Apoptosis and oxidative stress in neurodegenerative
diseases. J Alzheimers Dis. 42 (Suppl 3):S125–S152. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mizuno Y, Mochizuki H, Sugita Y and Goto
K: Apoptosis in neurodegenerative disorders. Intern Med.
37:192–193. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Eizirik DL and Darville MI: Beta-cell
apoptosis and defense mechanisms: Lessons from type 1 diabetes.
Diabetes. 50 (Suppl 1):S64–S69. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chandra J, Zhivotovsky B, Zaitsev S,
Juntti-Berggren L, Berggren PO and Orrenius S: Role of apoptosis in
pancreatic beta-cell death in diabetes. Diabetes. 50 (Suppl
1):S44–S47. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Moscat J and Diaz-Meco MT: p62 at the
crossroads of autophagy, apoptosis, and cancer. Cell.
137:1001–1004. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Levine B, Sinha SC and Kroemer G: Bcl-2
family members: Dual regulators of apoptosis and autophagy.
Autophagy. 4:600–606. 2008. View Article : Google Scholar
|
|
15
|
Wang J: Beclin 1 bridges autophagy,
apoptosis and differentiation. Autophagy. 4:947–948. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Albers E and Muller BW: Cyclodextrin
derivatives in pharmaceutics. Crit Rev Ther Drug Carrier Syst.
12:311–337. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brewster ME and Loftsson T: The use of
chemically modified cyclodextrins in the development of
formulations for chemical delivery systems. Pharmazie. 57:94–101.
2002.PubMed/NCBI
|
|
18
|
Stella VJ and He Q: Cyclodextrins. Toxicol
Pathol. 36:30–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Coisne C, Tilloy S, Monflier E, Wils D,
Fenart L and Gosselet F: Cyclodextrins as emerging therapeutic
tools in the treatment of cholesterol-associated vascular and
neurodegenerative diseases. Molecules. 21:17482016. View Article : Google Scholar
|
|
20
|
Ottinger EA, Kao ML, Carrillo-Carrasco N,
Yanjanin N, Shankar RK, Janssen M, Brewster M, Scott I, Xu X,
Cradock J, et al: Collaborative development of
2-hydroxypropyl-β-cyclodextrin for the treatment of Niemann-Pick
type C1 disease. Curr Top Med Chem. 14:330–339. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zimmer S, Grebe A, Bakke SS, Bode N,
Halvorsen B, Ulas T, Skjelland M, De Nardo D, Labzin LI, Kerksiek
A, et al: Cyclodextrin promotes atherosclerosis regression via
macrophage reprogramming. Sci Transl Med. 8:333ra3502016.
View Article : Google Scholar
|
|
22
|
Gould S and Scott RC:
2-Hydroxypropyl-beta-cyclodextrin (HP-beta-CD): A toxicology
review. Food Chem Toxicol. 43:1451–1459. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tamura A and Yui N:
β-Cyclodextrin-threaded biocleavable polyrotaxanes ameliorate
impaired autophagic flux in Niemann-Pick type C disease. J Biol
Chem. 290:9442–9454. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang DS, Stavrides P, Kumar A, Jiang Y,
Mohan PS, Ohno M, Dobrenis K, Davidson CD, Saito M, Pawlik M, et
al: Cyclodextrin has conflicting actions on autophagy flux in vivo
in brains of normal and Alzheimer model mice. Hum Mol Genet.
26:843–859. 2017.PubMed/NCBI
|
|
25
|
Yokoo M, Kubota Y, Motoyama K, Higashi T,
Taniyoshi M, Tokumaru H, Nishiyama R, Tabe Y, Mochinaga S, Sato A,
et al: 2-Hydroxypropyl-β-Cyclodextrin acts as a novel anticancer
agent. PLoS One. 10:e01419462015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Huang T, Huang Y, Huang Y, Yang Y, Zhao Y
and Martyniuk CJ: Toxicity assessment of the herbicide acetochlor
in the human liver carcinoma (HepG2) cell line. Chemosphere.
243:1253452020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Young MM, Takahashi Y, Khan O, Park S,
Hori T, Yun J, Sharma AK, Amin S, Hu CD, Zhang J, et al:
Autophagosomal membrane serves as platform for intracellular
death-inducing signaling complex (iDISC)-mediated caspase-8
activation and apoptosis. J Biol Chem. 287:12455–12468. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fu Z, Cheng X, Kuang J, Feng H, Chen L,
Liang J, Shen X, Yuen S, Peng C, Shen B, et al: CQ sensitizes human
pancreatic cancer cells to gemcitabine through the lysosomal
apoptotic pathway via reactive oxygen species. Mol Oncol.
12:529–544. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy (3rd edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Laplante M and Sabatini DM: mTOR signaling
in growth control and disease. Cell. 149:274–293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Manning BD and Toker A: AKT/PKB signaling:
Navigating the network. Cell. 169:381–405. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pan JA, Ullman E, Dou Z and Zong WX:
Inhibition of protein degradation induces apoptosis through a
microtubule-associated protein 1 light chain 3-mediated activation
of caspase-8 at intracellular membranes. Mol Cell Biol.
31:3158–3170. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mahammad S and Parmryd I: Cholesterol
depletion using methyl-β-cyclodextrin. Methods Mol Biol.
1232:91–102. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mollinedo F and Gajate C: Lipid rafts as
major platforms for signaling regulation in cancer. Adv Biol Regul.
57:130–146. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhuang L, Kim J, Adam RM, Solomon KR and
Freeman MR: Cholesterol targeting alters lipid raft composition and
cell survival in prostate cancer cells and xenografts. J Clin
Invest. 115:959–968. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu R, Li J, Zhang T, Zou L, Chen Y, Wang
K, Lei Y, Yuan K, Li Y, Lan J, et al: Itraconazole suppresses the
growth of glioblastoma through induction of autophagy: Involvement
of abnormal cholesterol trafficking. Autophagy. 10:1241–1255. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Oh HY, Lee EJ, Yoon S, Chung BH, Cho KS
and Hong SJ: Cholesterol level of lipid raft microdomains regulates
apoptotic cell death in prostate cancer cells through EGFR-mediated
Akt and ERK signal transduction. Prostate. 67:1061–1069. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Oh HY, Leem J, Yoon SJ, Yoon S and Hong
SJ: Lipid raft cholesterol and genistein inhibit the cell viability
of prostate cancer cells via the partial contribution of
EGFR-Akt/p70S6k pathway and down-regulation of androgen receptor.
Biochem Biophys Res Commun. 393:319–324. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kuwano K, Araya J, Hara H, Minagawa S,
Takasaka N, Ito S, Kobayashi K and Nakayama K: Cellular senescence
and autophagy in the pathogenesis of chronic obstructive pulmonary
disease (COPD) and idiopathic pulmonary fibrosis (IPF). Respir
Investig. 54:397–406. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kizilarslanoglu MC and Ulger Z: Role of
autophagy in the pathogenesis of Alzheimer disease. Turk J Med Sci.
45:998–1003. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li J and Yuan J: Caspases in apoptosis and
beyond. Oncogene. 27:6194–6206. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lavrik IN, Golks A and Krammer PH:
Caspases: Pharmacological manipulation of cell death. J Clin
Invest. 115:2665–2672. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pan JA, Fan Y, Gandhirajan RK, Madesh M
and Zong WX: Hyperactivation of the mammalian degenerin MDEG
promotes caspase-8 activation and apoptosis. J Biol Chem.
288:2952–2963. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Nagata S: Apoptosis by death factor. Cell.
88:355–365. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Keller N, Ozmadenci D, Ichim G and Stupack
D: Caspase-8 function, and phosphorylation, in cell migration.
Semin Cell Dev Biol. 82:105–117. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Song W, Wang F, Lotfi P, Sardiello M and
Segatori L: 2-Hydroxypropyl-β-cyclodextrin promotes transcription
factor EB-mediated activation of autophagy: Implications for
therapy. J Biol Chem. 289:10211–10222. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Raben N and Puertollano R: TFEB and TFE3:
Linking lysosomes to cellular adaptation to stress. Annu Rev Cell
Dev Biol. 32:255–278. 2016. View Article : Google Scholar : PubMed/NCBI
|