Introduction
Acute lymphoblastic leukemia (ALL) is a common
malignancy in children worldwide. Genome-wide profiling studies,
including DNA microarray and next generation sequencing have
identified several genetic alterations associated with childhood
ALL (1–3) CREB-binding protein (CREBBP) gene
translocations have been observed in de novo ALL, and its
mutations are enriched in relapsed pediatric patients with ALL and
newly diagnosed patients with high hyperdiploid karyotype, with the
potential of recurrence (4–6). It
has been revealed that early loss of CREBBP confers murine
malignant stem cell properties on lymphoid progenitors (7). Furthermore, a previous study reported
that low CREBBP mRNA expression was associated with a high level of
minimal residual disease (MRD) at the end of induction therapy and
adverse long-term outcomes in pediatric patients with ALL, and
suggested that the negative effect of CREBBP on prognosis may be
improved with intensive chemotherapy (8). This clinical observation implies that
CREBBP levels may influence the sensitivity of leukemia cells to
chemotherapy.
The CREBBP gene encodes the cAMP response element
binding-binding protein, a ubiquitously expressed transcriptional
co-activator, which participates in regulating basic cellular
processes and functions as a tumor suppressor in cell cycle control
(9,10). The interaction between CREBBP and
E2F transcription factor 1 (E2F1), a member of the E2F family, has
been associated with prognosis in non-small cell lung carcinomas
(11). E2F family members are
considered to function as transcription factors, and thus have
critical roles in regulating cell cycle control (12). Gene microarray analyses have
detected E2F3 mRNA expression in diagnostic samples of ALL, while
other members are low or undetectable (13,14).
Furthermore, a previous study reported that overexpression of E2F3a
in 697 and Reh cells accelerated cell proliferation, promoted
leukemia cells to S and G2/M phases and enhanced
sensitivity to chemotherapeutic drugs (15).
Therefore, the present study aimed to investigate
the influence of CREBBP on chemotherapy sensitivity in leukemia
cells by regulating proliferation and cell cycle progression via
interaction with and regulation of E2F3a. In addition, the present
study suggested that targeting CREBBP may serve as a therapeutic
strategy for pediatric patients with ALL.
Materials and methods
Cell culture
The human leukemia cell lines, Jurkat and Reh, were
purchased from the Shanghai Institute of Biochemistry and Cell
Biology, Chinese Academy of Sciences. Cells were maintained in
RPMI-1640 (Thermo Fisher Scientific, Inc.) supplemented with 10%
FBS (cat. no. 16000036; Gibco; Thermo Fisher Scientific, Inc.) at
37°C in a 5% CO2 atmosphere. Cell lines underwent DNA
profiling of short tandem repeat analysis for authentication and
mycoplasma contamination testing by Shanghai GeneChem Co., Ltd.
Downregulating CREBBP expression via
short hairpin RNA (shRNA) transfection
The CREBBP lentivirus and scrambled shRNA control
were purchased from Shanghai GeneChem Co., Ltd. The targeting RNAi
sequences of sh-CREBBP and scrambled control were as follows:
5′-TATCAGAATAGGTATCATT-3′ and 5′-TTCTCCGAACGTGTCACGT-3′,
respectively. The shRNA-expressing recombinant plas- mids
(hU6-MCS-Ubiquitin-EGFP-IRES-puromycin-GV248, Shanghai GeneChem
Co., Ltd.; 20 µg), along with two helper plasmids, pGC-LV
pHelper1.0 (Shanghai GeneChem Co., Ltd.; 15 µg) and 2.0 (Shanghai
GeneChem Co., Ltd.; 10 µg) were transfected into 293T cells
(American Type Culture Collection) using transfection reagent (2 M
CaCl2; Shanghai GeneChem Co., Ltd.). Cells were cultured
to exponential phase in DMEM (Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS and transfected once they reached 70–80%
confluence. Lentivirus was collected 48 h post-transfection via
ultracentrifugation at 146,400 × g for 2 h at 4°C. Jurkat and Reh
cells were transduced at 37°C overnight with the concentrated virus
in the presence of polybrene (1 µg/ml; Shanghai GeneChem Co.,
Ltd.), at a final multiplicity of infection value of 50 and 100,
respectively. Subsequent experiments were performed 96 h after
transfection.
Upregulating CREBBP expression via
synergistic activation mediator (SAM)
The CRISPR/defective Cas9 (dCas9) synergistic
activation mediator system (16,17)
was used to overexpress CREBBP. dCas9-VP64-Puro (20 µg; cat. no.
GV418; Shanghai GeneChem Co., Ltd.) or single guide
(sg)RNA-MS2-P65-HSF1-Neo-GV468 lentiviral vectors (20 µg), along
with pGC-LV pHelper1.0 (15 µg) and 2.0 (10 µg) were transfected
into 293T cells (confluency, 70–80%), using transfection reagent (2
M CaCl2; Shanghai GeneChem Co., Ltd.). Lentivirus was
collected 48 h post-transfection via ultracentrifugation at 146,400
× g for 2 h at 4°C. Stable Jurkat and Reh cells were generated by
infecting with dCas9-VP64-Puro lentivirus at a final multiplicity
of infection value of 50 and 100, and screened with puromycin
dihydrochloride (3 µg/ml; Thermo Fisher Scientific, Inc.).
Subsequently, the stable cell lines were infected with CREBBP sgRNA
or mock vector lentivirus (cat. no. CON275; Shanghai GeneChem Co.,
Ltd.). The sgRNA sequence targeting the promoter of CREBBP was
designed by www.rgenome.net/cas-designer and the matching sequence
for CREBBP was 5′-CCACTTAATGAATTCGCTCG-3′. At 96 h
post-transfection, subsequent experiments were performed.
Reverse transcription-quantitative
(RT-q)PCR
Total RNA from Jurkat and Reh cells was extracted
using TRIzol® reagent (Thermo Fisher Scientific, Inc.).
Total RNA (1 µg) was incubated with random hexamers (Promega
Corporation) at 70°C for 5 min and then reverse transcribed into
cDNA at 37°C for 1 h using M-MLV Reverse Transcriptase and buffer
(Promega Corporation) with dNTPs (2.5 mM; Promega Corporation).
qPCR was performed in triplicate using TaqMan Gene Expression
Master Mix (cat. no. 4369016; Thermo Fisher Scientific, Inc.) and
ViiA 7 system (Thermo Fisher Scientific, Inc.). The Abelson (ABL)
gene was used as an internal control gene. The primers and probes
were synthesized by Sangon Biotech Co., Ltd. The primers used for
mRNA detection were as follows: Caspase 8 associated protein 2
(CASP8AP2) forward, 5′-CACTTGCCACTTCTACAAGTC-3′ and reverse,
5′-TGGCGGCTAAATATGCAAATG-3′; ABL forward, 5′-CCTTCAGCGGCCAGTAGC-3′
and reverse, 5′-GGACACAGGCCCATGGTAC-3′. The probes were
5′-FAM-TGTCAGAAAAGAGGGCCATCATTTAAA-TAMRA-3′ and
5′-FAM-CCATTTTTGGTTTGGGCTTCACACCATT-TAMRA-3′ for CASP8AP2 and ABL,
respectively. The following thermocycling conditions were used for
qPCR: Pre-denaturation at 95°C for 10 min; followed by 40 cycles of
denaturation at 95°C for 15 sec and annealing at 60°C for 1 min.
The relative expression of CASP8AP2 was calculated using the
2−∆∆Cq method (18) and
is shown as fold changes, as compared with control group. Gene
transcription levels were normalized to that of ABL and set to 1 in
the control group.
Western blotting
Protein lysates were prepared using RIPA lysis
buffer (Thermo Fisher Scientific, Inc.). Protein concentration was
determined using the Pierce bicinchoninic acid protein assay
(Thermo Fisher Scientific, Inc.), and 50 µg protein/lane was
separated on 4–15% Tris-glycine precast gels (Bio-Rad Laboratories,
Inc.). Proteins were transferred onto a PVDF membranes and blocked
with 5% non-fat milk at 25°C for 1 h, prior to incubation with
primary antibodies against CREBBP (cat. no. sc-369; 1:500; Santa
Cruz Biotechnology, Inc.), E2F3 (cat. no. sc-878; 1:500; Santa Cruz
Biotechnology, Inc.), CASP8AP2 (cat. no. YT6423; 1:500; ImmunoWay
Biotechnology, Inc.) or GAPDH (cat. no. 51332; 1:2,000; Cell
Signaling Technology, Inc.) overnight at 4°C. Membranes were washed
three times for 10 min with TBST buffer (cat. no. B1009; Applygen
Technologies, Inc.) and subsequently incubated with horseradish
peroxidase-conjugated goat anti-rabbit IgG or anti-mouse secondary
antibody (cat. nos. 7074 and 7076, 1:5,000; Cell Signaling
Technology, Inc.) for 1 h at room temperature. Blots were re-washed
and signals were visualized with super enhanced chemiluminescence
(ECL) detection reagent (Applygen Technologies, Inc.), then imaged
using Amersham Imager 600 (Cytiva) and analyzed with ImageQuant™ TL
software (version 7.0; Cytiva).
In vitro drug sensitivity assay
The in vitro sensitivity of daunorubicin
(DNR; Melone Pharmaceutical Co., Ltd), vincristine (VCR; Melone
Pharmaceutical Co., Ltd) and L-asparaginase (L-ASP; Kyowa Hakko
Kirin Co., Ltd.)-transfected Jurkat and Reh cells was determined
via the Cell Counting Kit-8 (CCK-8) assay. Transfected cells were
seeded into a 96-well plate (~10,000 cells/well) and treated with
or without gradient concentrations of each drug at 37°C for 24 h.
The gradient concentrations for DNR, VCR and L-ASP were 0.001–1000
µM, 0.001–1000 µM and 0.001–10 U/ml, respectively. All samples were
assessed in triplicate. After 24 h, CCK-8 reagent (10 µl per well;
Applygen Technologies, Inc.) was added to each well and incubated
at 37°C for 2 h. Following the CCK-8 incubation, the plates were
analyzed at a wavelength of 450 nm (A450) using a
microplate reader (Thermo Fisher Scientific, Inc.). The inhibitory
rate at each concentration was calculated as follows:
[1-(A450 of DNR exposed well/A450 of negative
control well)] ×100%. The IC50 value was calculated
using the curve fitting method (19), in order to determine cellular
sensitivity of each drug.
Cell proliferation assay and cell
cycle analysis
CCK-8 was used to assess cell viability. Cells were
seeded into a 96-well plate (2,000 cells/well) and incubated at
37°C in a humidified atmosphere containing 5% CO2 for
24, 48 or 72 h, respectively. CCK-8 (10 µl per well; Applygen
Technologies, Inc.) was subsequently added to the well at 0, 24, 48
and 72 h, respectively, and incubated for 2 h at 37°C. The plates
were analyzed at a wavelength A450 using a microplate
reader, and a spectrophotometer was used to measure the optical
density.
Flow cytometric analysis was performed to determine
changes in cell cycle distribution. Harvested cells were washed
twice with PBS, fixed with 70% cold ethanol overnight at 4°C and
digested with RNaseA (Nanjing KeyGen Biotech Co., Ltd.) at 37°C for
30 min. Cells were subsequently stained with 400 µl propidium
iodide (Nanjing KeyGen Biotech Co., Ltd.) and incubated at room
temperature for 30 min and the cell cycle phase fractions were
determined via flow cytometry (Canto II, BD Biosciences) using
Modfit LT software (version 3.1; Verity Software House, Inc.).
Immunofluorescence confocal
microscopy
Transfected cells (Jurkat or Reh) were seeded onto
poly-L-lysine (cat. no. P8120; Beijing Solarbio Science &
Technology Co., Ltd.) coated glass slides (20,000 cells/slide).
Cells were fixed with 4% paraformaldehyde (Sigma-Aldrich; Merck
KGaA) for 15 min at room temperature, washed three times with PBS
for 5 min, permeabilized with 0.1% Triton X-100 for 10 min and
blocked with 2% BSA (cat. no. 0332; Amresco, LLC) for 1 h at room
temperature. Subsequently, cells were incubated with primary
antibodies against CREBBP (cat. no. sc-369; 1:500; Santa Cruz
Biotechnology, Inc.) and E2F3 (cat. no. sc-56665; 1:500; Santa Cruz
Biotechnology, Inc.) overnight at 4°C. Following the primary
incubation, cells were incubated with goat anti-mouse (1:2,000;
cat. no. A32727) and goat anti-rabbit (1:2,000; cat. no. A32731;
both from Thermo Fisher Scientific, Inc.) secondary antibodies at
room temperature for 1 h. The nuclei were stained with DAPI (100
ng/ml; cat. no. ZLI-9557; OriGene Technologies, Inc.) at room
temperature for 15 min and fluorescence images were observed under
a spinning disk confocal microscope (PerkinElmer, Inc.), with a
×100 oil-immersion objective lens.
Co-immunoprecipitation (Co-IP)
assay
Cells were collected and lysed using Co-RIPA buffer
(Applygen Technologies, Inc.) on ice for 30 min. Cell lysates were
pre-cleared with protein A/G-Sepharose beads (EMD Millipore) at 4°C
for 1 h, prior to immunoprecipitation with primary antibodies
against CREBBP (4 µg/ml; cat. no. sc-7300), E2F3 (4 µg/ml; cat. no.
sc-56665) and IgG (4 µg/ml; cat. no. sc-2025; all from Santa Cruz
Biotechnology, Inc.) overnight at 4°C, with gentle rotation in a
rotation mixer (WH-986; Kylin-Bell Lab Instruments Co., Ltd.). The
protein-antibody complexes were incubated with protein
A/G-Sepharose beads for 4 h at 4°C, with rotation and then
centrifuged at 800 × g for 5 min at 4°C. Subsequently, the beads
were washed three times with cold Co-RIPA buffer and the bound
proteins were separated via 4–15% SDS-PAGE. The desired
co-immunoprecipitated proteins were detected using the Clean-Blot
IP Detection kit (Thermo Fisher Scientific, Inc.). The antibodies
used in the immunoblotting detection were the same as those used in
western blot analysis.
Chromatin immunoprecipitation (ChIP)
assay
Jurkat and Reh cells were used for ChIP analysis
using the EZ-ChIP kit (EMD Millipore), according to manufacturer's
protocol. Each ChIP reaction contained chromatin collected from
1×107 cells and 2 µg anti-CREBBP antibody (cat. no.
sc-7300×; 1:500; Santa Cruz Biotechnology, Inc.) or normal negative
control IgG (cat. no. sc-2025; 1:500; Santa Cruz, Biotechnology,
Inc.). Following immunoprecipitation overnight at 4°C, the binding
sites (revealed by www.cbil.upenn.edu/tess) of CREBBP to target CASP8AP2
promoters were analyzed via PCR. The enriched DNA binding CREBBP
from Jurkat and Reh cells was amplified using Gotaq DNA polymerase
(Promega Corporation). The amplification conditions were as
follows: Pre-denaturation at 95°C for 3 min; followed by 40 cycles
of denaturation at 95°C for 20 sec, annealing at 60°C for 30 sec,
and extension at 72°C for 10 min. The following Primer sequences
were used in the ChIP assay to detect the CASP8AP2 promoter:
Forward, 5′-ACTCCAGTTTGGCGCCAACC-3′ and reverse,
5′-GACAACCCGGTTCCTTTCTG-3′.
Statistical analysis
IC50 curves were plotted and values were
determined via the non-linear curve fit of Y=100/ (1 +
10(LogIC50-X) × HillSlope), using GraphPad Prism
software 5.0 (GraphPad Software, Inc.). Differences between
IC50 values for each chemotherapeutic drug, with or
without affecting CREBBP expression, were compared using the
extra-sum-of-squares F test within GraphPad Prism. SPSS software
16.0 (SPSS, Inc.) was used for comparison of the gene or protein
expression level, cell counts and cell cycle fraction, and
two-tailed independent-samples t-test was used to compare
differences between two groups. Data are presented as the mean ±
standard error of the mean, and all experiments were performed in
triplicate. P<0.05 was considered to indicate a statistically
significant difference.
Results
Effect of CREBBP on drug sensitivity
in leukemia cells
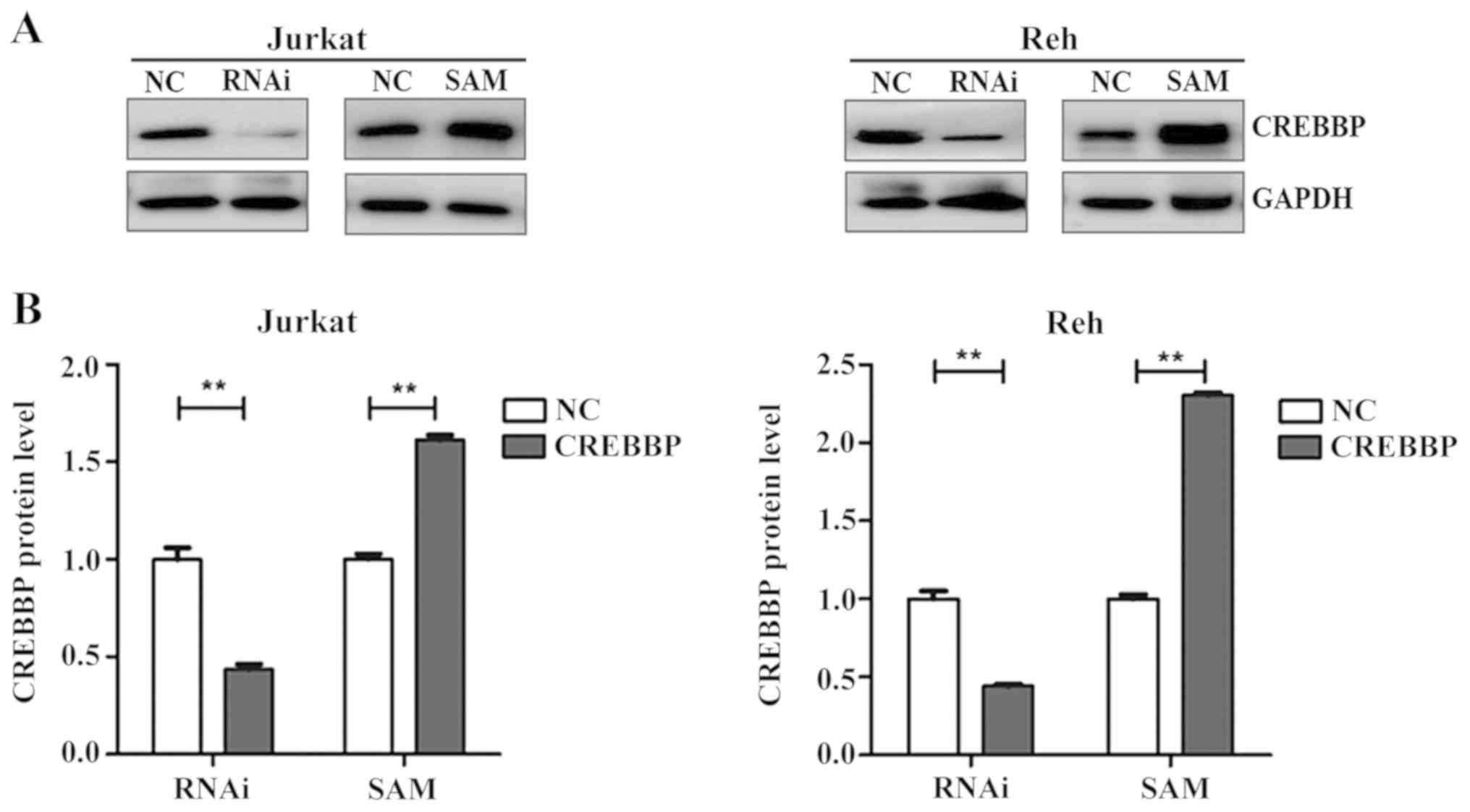
CREBBP was successfully downregulated (P<0.01 and
P<0.01 for Jurkat and Reh, respectively) and overexpressed
(P<0.01 and P<0.01 for Jurkat and Reh, respectively) in
Jurkat and Reh cells following treatment with RNAi and SAM for 96
h, respectively (Fig. 1). The
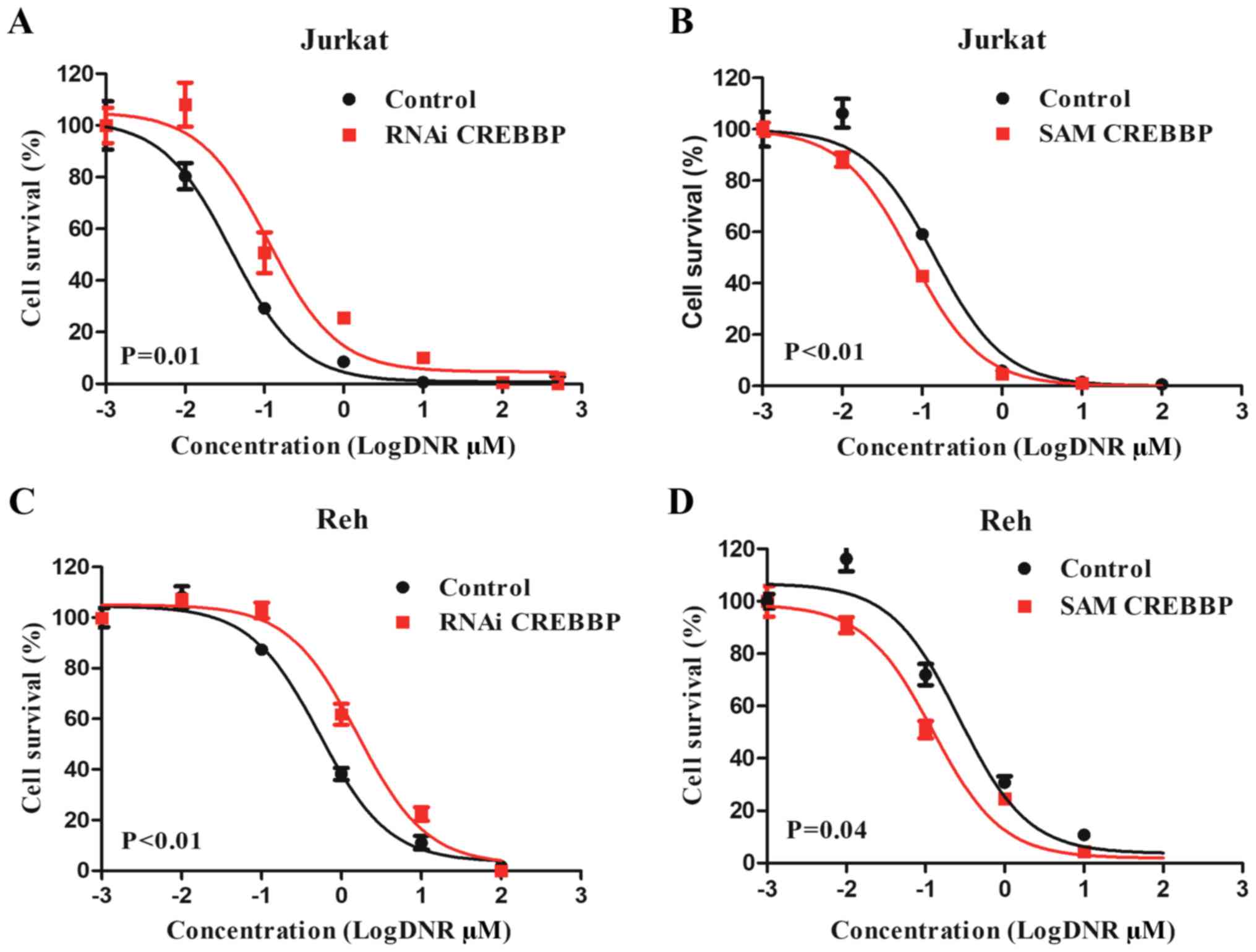
effect of CREBBP on sensitivity of leukemia cells to DNR, VCR and
L-ASP was assessed. The results demonstrated that the
IC50 values of DNR were significantly increased in
RNAi-treated Jurkat cells (0.04 µM vs. 0.11 µM; P=0.01; Fig. 2A), and significantly decreased in
SAM-treated Jurkat cells (0.13 µM vs. 0.07 µM; P<0.01; Fig. 2B). Similar effects were observed in
RNAi-treated Reh cells (0.53 µM vs. 1.59 µM; P<0.01; Fig. 2C) and SAM-treated Reh cells (0.27
µM vs. 0.12 µM; P=0.04; Fig. 2D).
However, no significant effects were observed between the
IC50 values of VCR and L-ASP and CREBBP expression
(Figs. S1 and S2). Taken together, these results
suggested that leukemia cells with low CREBBP expression may
be more resistant to DNR.
Effect of CREBBP on cell proliferation
and cell cycle progression
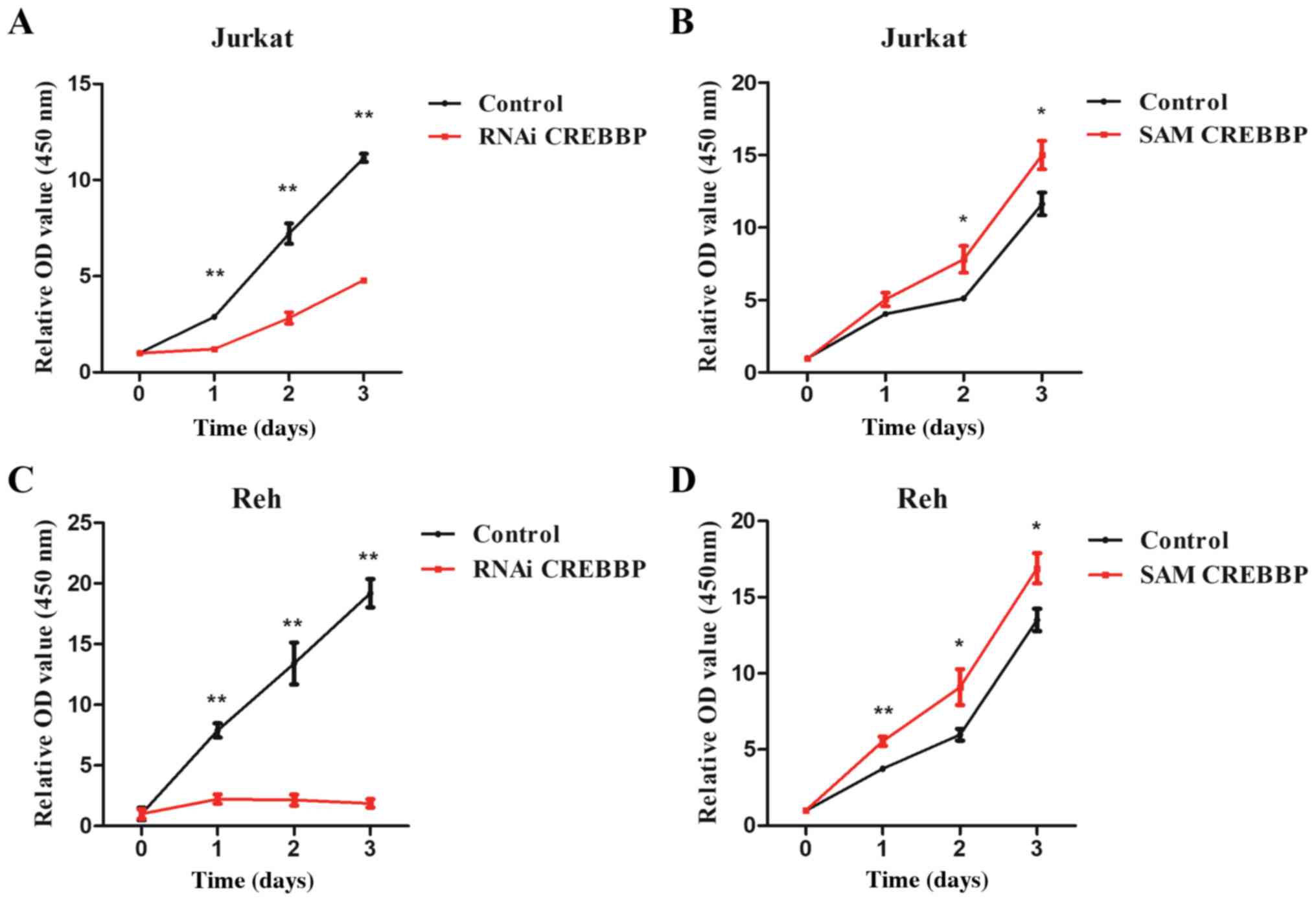
The CCK-8 assay demonstrated that downregulation of
CREBBP significantly inhibited cell proliferation at 24 h
(P<0.01 and P<0.01 for Jurkat and Reh, respectively), 48 h
(P<0.01 and P<0.01 for Jurkat and Reh, respectively) and 72 h
(P<0.01 and P<0.01 for Jurkat and Reh, respectively)
(Fig. 3A and C), whereas
overexpression of CREBBP significantly promoted proliferation of
Jurkat (P=0.04 and P=0.04 for 48 and 72 h, respectively) and Reh
(P<0.01, P=0.04 and P=0.03 for 24, 48 and 72 h, respectively)
cells (Fig. 3B and D).
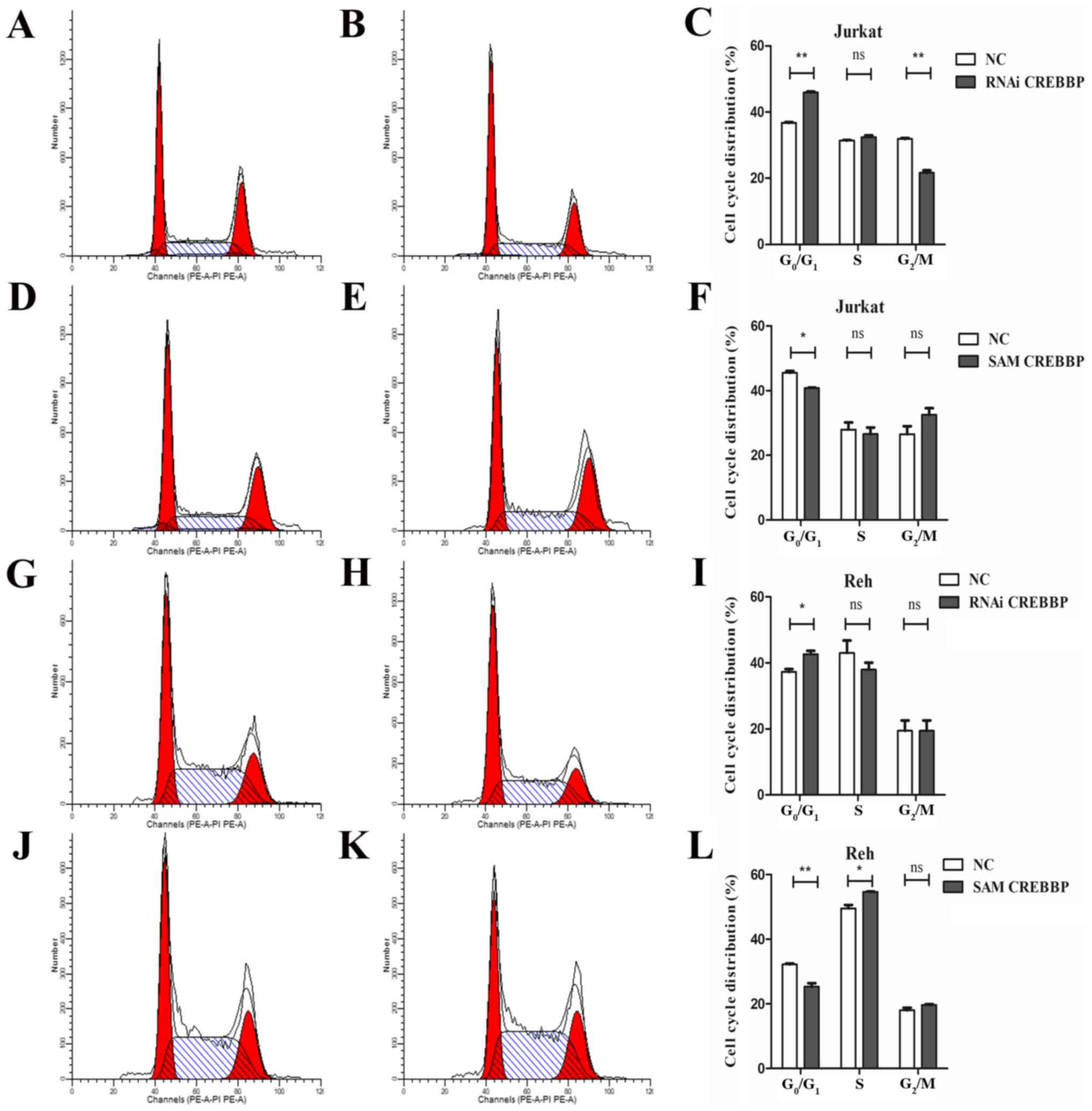
Furthermore, downregulation of CREBBP arrested cell cycle in the
G0/G1 phase, while overexpression of CREBBP
decreased the proportion of cells in the
G0/G1 phase (Fig. 4). Furthermore, the proportion of
G2/M cells was decreased in CREBBP downregulation Jurkat
(P<0.01) and the proportion of S phase cells was increased in
CREBBP overexpression Reh (P=0.04). No significant effect on S
phase in CREBBP downregulation Jurkat and Reh cells and
G2/M phase in CREBBP overexpression Jurkat and Reh cells
was observed. Overall, these results indicated that CREBBP plays an
essential role in G1/S transition and cell
proliferation.
CREBBP interacts and regulates the
transcription factor E2F3a
The E2F transcription factor family serves a key
role in cell cycle regulation (12). Our previous study reported that
overexpression of E2F3a enhances the sensitivity of 697 and Reh
cells to DNR (15). In the present
study, immunofluorescence confocal microscopy analysis demonstrated
that CREBBP and E2F3a co-localized in the nuclei of Jurkat and Reh
cells (Fig. 5A). The interaction
between CREBBP and E2F3a was confirmed via Co-IP analysis (Fig. 5B). Furthermore, western blot
analysis demonstrated that E2F3a protein expression was associated
with CREBBP expression (Fig. 5C and
D). E2F3a was downregulated in RNAi-treated Jurkat and Reh
cells (P<0.01 and P<0.01 for Jurkat and Reh, respectively),
and upregulated in SAM-treated Jurkat and Reh cells (P<0.01 and
P<0.01 for Jurkat and Reh, respectively) (Fig. 5C and D). Collectively, these
results suggested that CREBBP may influence the cell cycle via the
interaction and regulation of E2F3a.
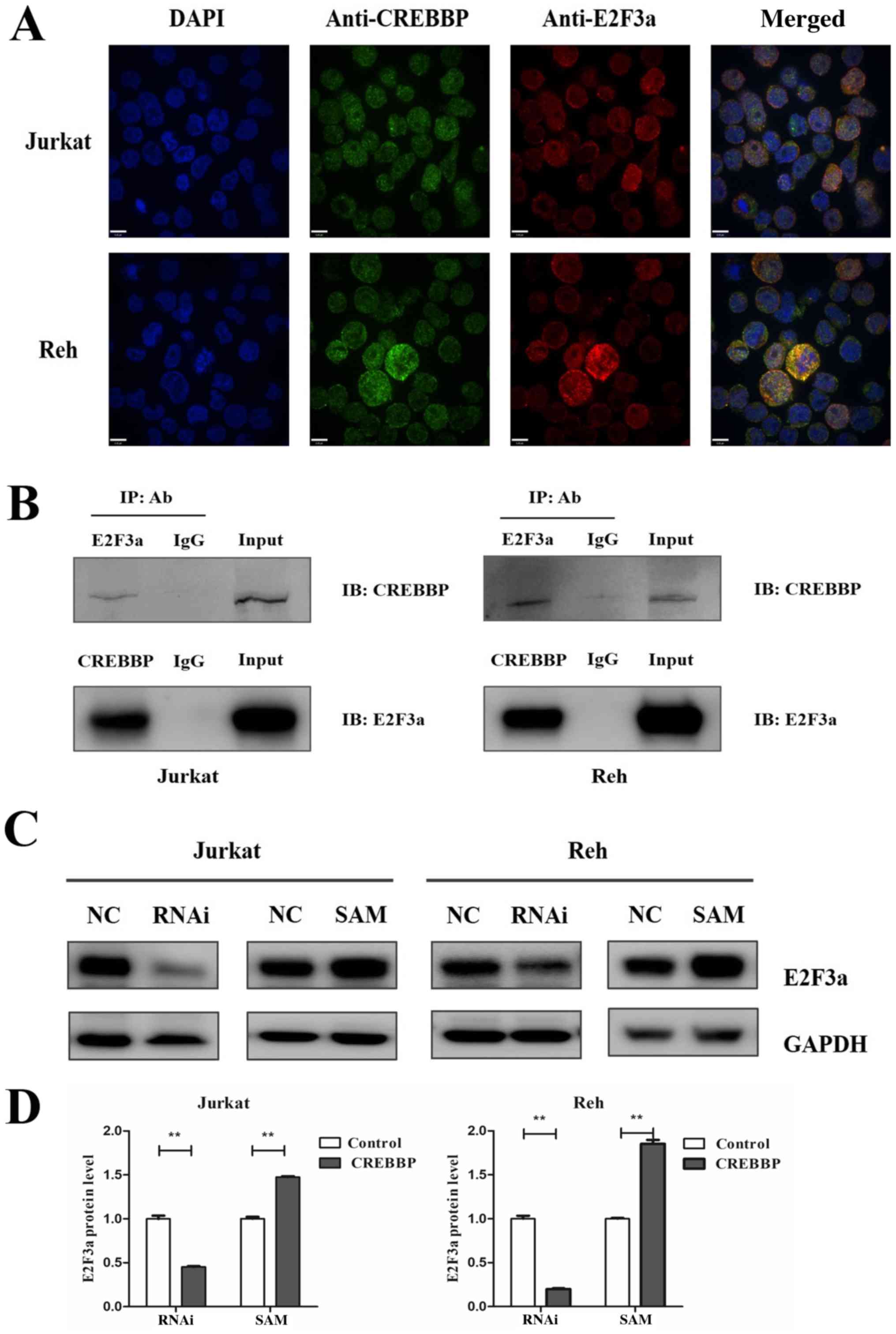 | Figure 5.Co-localization and interaction of
CREBBP and E2F3a in Jurkat and Reh cells. (A) CREBBP co-localized
with E2F3a in the nuclei of Jurkat and Reh cells. Primary
antibodies against CREBBP (green) and E2F3a (red) antibodies were
used, and DAPI (blue) was used to stain the nuclei (scale bar, 6
µm; magnification, ×40). (B) CREBBP interacts with E2F3a. Cell
lysates were IP with mouse anti-CREBBP, mouse anti-E2F3 or mouse
anti-IgG antibodies. IB was analyzed with rabbit anti-CREBBP or
rabbit anti-E2F3 antibodies. IgG was used as the negative control,
while the input (whole cell lysates) was used as the positive
control. (C) Western blotting analysis of E2F3a protein expression
in Jurkat and Reh cells following downregulation or overexpression
of CREBBP. (D) Relative E2F3a protein expression in Jurkat and Reh
cells following downregulation or overexpression of CREBBP. Data
are presented as the mean ± standard error of the mean (n=3).
**P<0.01. CREBBP, CREB-binding protein; IP, immunoprecipitated;
Ab, antibody; IB, immunoblotting; RNAi, RNA interference; SAM,
synergistic activation mediator; NC, negative control. |
Identification of CASP8AP2 as a
downstream gene of CREBBP
Our previous study revealed that E2F3a activates the
transcription of CASP8AP2 (15).
Since CREBBP was indicated to interact with E2F3a, the role of
CREBBP in the regulation of CASP8AP2 expression in leukemia cells
was further investigated. Bioinformatics analysis revealed two
CREBBP putative binding sites (C1 and C2) within the CASP8AP2
promoter region (−120 bp and −70 bp), based on Homo sapiens
chromosome 6, GRCh38 (Fig. 6A).
ChIP assay identified CREBBP binding to CASP8AP2 promoters that
contained these two binding sites in Jurkat and Reh cells (Fig. 6A). Furthermore, CASP8AP2 mRNA
expression decreased following downregulation and increased
following overexpression of CREBBP in Jurkat and Reh cells
(Fig. 6B). Similarly, CASP8AP2
protein expression was also affected in relation to CREBBP
expression (Fig. 6C and D).
Overall, these results indicated that CREBBP and E2F3a regulate
CASP8AP2 expression.
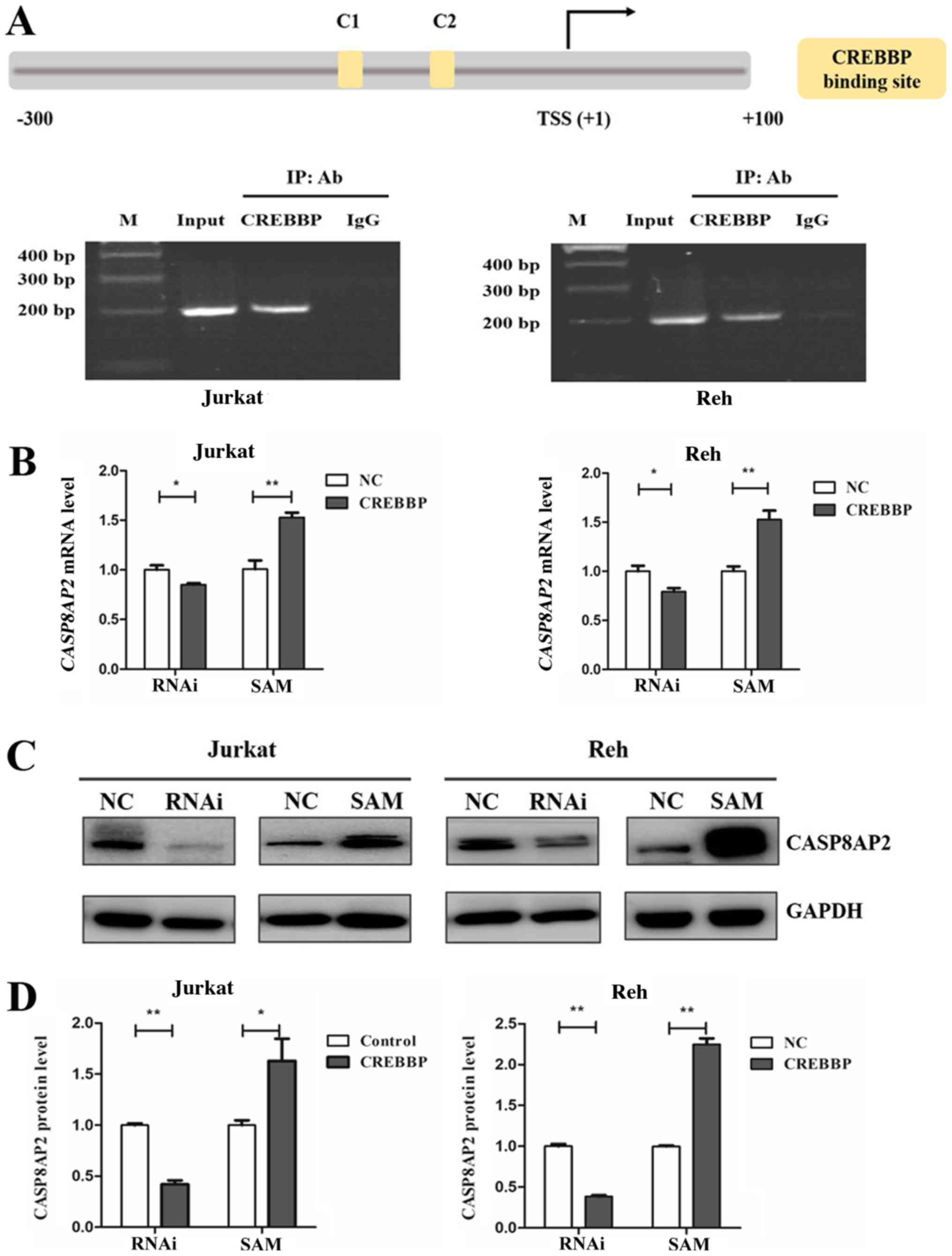 | Figure 6.CASP8AP2 is a downstream target of
CREBBP. (A) Schematic diagram of CASP8AP2 promoter fragments. The
putative CREBBP binding sites are presented as yellow boxes: C1
(−120 bp) and C2 (−70 bp). TSS is indicated with an arrow. The
numbers represent bp relative to TSS. (B) Chromatin
immunoprecipitation assay identified CREBBP-binding sites in the
CASP8AP2 promoter. The predicted binding sites were successfully
amplified, from both the input DNA and the chromatin
immunoprecipitated by anti-CREBBP antibody. Normal mouse IgG was
used as the negative control. (C) Relative CASP8AP2 mRNA expression
levels following downregulation or overexpression of CREBBP in
Jurkat and Reh cells. Gene transcription levels were normalized to
that of ABL and set to 1 in the control group. (D) Relative
CASP8AP2 protein expression in Jurkat and Reh cells following
downregulation or overexpression of CREBBP. Data are presented as
the mean ± standard error of the mean (n=3). *P<0.05;
**P<0.01. CREBBP, CREB-binding protein; bp, base pair; TSS,
transcription start site; M, DNA marker; IP, immunoprecipitated;
Ab, antibody; NC, negative control; RNAi, RNA interference; SAM,
synergistic activation mediator. |
Discussion
A previous study reported that patients with low
CREBBP expression are associated with high levels of MRD following
induction therapy, as well as inferior long-term outcomes in
pediatric patients with ALL (8).
However, the molecular mechanisms associated with low CREBBP on
prognosis have not yet been fully investigated. Furthermore, it has
been shown that the prognosis of patients with low CREBBP
expression who have received an intermediate risk regimen improves
compared with those taking standard risk regimen (8). Thus, it was hypothesized that CREBBP
expression may affect the sensitivity of leukemia cells to
chemotherapy. The present study assessed the IC50 values
of key chemotherapeutic drugs (DNR, VCR and L-ASP) used for
induction therapy in ALL cell lines (Jurkat and Reh), with varying
levels of CREBBP expression. The results demonstrated that
downregulation of CREBBP decreased the sensitivity of Jurkat and
Reh cells to DNR, while overexpression of CREBBP benefited DNR with
regards to leukemia cell death. However, CREBBP expression levels
did not affect sensitivity to VCR and L-ASP. VCR primarily exerts
its antitumor effect by blocking the cell cycle at the mitotic
phase, thus inhibiting the polymerization of tubulin and the
formation of spindle microtubules (20). L-ASP degrades and depletes
asparagine in the blood and starves leukemia cells (21). DNR is an anthracycline
chemotherapeutic drug that inhibits DNA and DNA-dependent RNA
synthesis (22), and has been
extensively used in the treatment of leukemia. Moreover, DNR is a
non-specific cell cycle agent, and thus exerts cytotoxic effects at
every phase of the cell cycle, including G0,
G1, S, G2 and M (23). The results of the present study
suggested that low CREBBP expression potentially resulted in DNR
chemoresistance via regulation of cell cycle progression.
It has been reported that tumors with low
proliferation rates are insensitive to chemotherapy as most of the
cells are non-proliferative (24).
The present results demonstrated that downregulation of CREBBP
significantly inhibited the proliferation of Jurkat and Reh cells,
whereas overexpression of CREBBP significantly accelerated cell
proliferation. Furthermore, downregulation of CREBBP was
significantly associated with accumulation of Jurkat and Reh cells
at the G0/G1 phase. These results suggested
that CREBBP may affect drug sensitivity via proliferation and cell
cycle progression. Previous studies have revealed the key role of
CREBBP in promoting cell proliferation and regulating cell cycle
progression (25,26). For example, depletion of CREBBP has
been reported to limit S phase entry, a phenomenon that can be
reversed by overexpressing CREBBP (26), which is consistent with the results
of the present study.
CREBBP functions as a coactivator, which regulates
cell cycle progression partly through its influence on
transcription (27). The E2F
transcription factor family is involved in the regulation of gene
expression, which is central for G1/S transition
(28–30). CREBBP may interact and acetylate
E2F family proteins to activate target gene transcription (31,32).
Gene microarray analysis identified E2F3 mRNA expression in a
diagnostic sample from patients with ALL, while other members were
low or undetectable (13,14). There are two isoforms of E2F3,
E2F3a and E2F3b. E2F3a is only expressed at the G1/S
boundary (33), while E2F3b is
expressed throughout the cell cycle and is associated with
retinoblastoma protein (Rb) and Rb-associated proteins in a
growth-arrested G0 state (34). A previous study reported that low
E2F3a expression was associated with adverse outcomes in
pediatric patients with ALL, and overexpression of E2F3a may
enhance the sensitivity to DNR, which increased the proportion of
leukemia cells in the S and G2/M phases and accelerated
cell proliferation (15). These
findings are consistent with the biological manifestations of
altered CREBBP expression as demonstrated in the present study,
which suggested that CREBBP may influence E2F3a expression in
leukemia cells. The present results also indicated that CREBBP and
E2F3a co-localized in the nuclei and interacted with each other in
Jurkat and Reh cells. Furthermore, E2F3a expression was identified
to be consistent with CREBBP expression levels. Collectively, these
results suggested that CREBBP and E2F3a may play a synergistic role
in leukemia drug resistance.
It has been reported that CASP8AP2 is a target gene
of E2F3a, and low CASP8AP2 expression is associated with an
unfavorable prognosis of pediatric patients with ALL (15,35).
Furthermore, CASP8AP2 is a key protein involved in cell
proliferation and S phase progression (36,37).
The present study demonstrated that CREBBP may bind to the promoter
of CASP8AP2 and regulate gene transcription and protein expression.
In addition, the present results indicated that CREBBP may serve as
a scaffold to interpret molecular signals via its interaction with
E2F3a in leukemia cells. Thus, CREBBP and E2F3a may synergistically
control leukemia cells crossing the G1 phase and the
regulation of target gene transcription, which plays a key role in
S phase progression.
In conclusion, the results of the present study
suggested that downregulation of CREBBP inhibited leukemia cell
proliferation and caused cell cycle arrest at the
G0/G1 phase. These results expand the current
understanding of the molecular mechanisms associated with CREBBP
insufficiency on chemotherapy resistance to DNR, and the
association with adverse outcomes. Furthermore, CREBBP may interact
with and regulate E2F3a to promote cell cycle progression, and
serve as a scaffold to bind to the CASP8AP2 promoter as part of
transcription regulation. It is still necessary to investigate the
other regulatory pathways of CREBBP in ALL. In addition, further
study targeting CREBBP in clinical intervention may lead to
improved prognosis of ALL.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank the Dr Hui Chen at
the Beijing Key Laboratory of Pediatric Hematology Oncology,
Hematology and Oncology Center, Beijing Children's Hospital,
Capital Medical University (Beijing, China) for her assistance with
the flow cytometric analysis.
Funding
The present study was funded by the National Natural
Science Foundation of China (grant nos. 81300432 and 81300434), the
Beijing Natural Science Foundation (grant no. 7192066) and the
Major National Science and Technology Projects (grant no.
2017ZX09304029).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding authors upon reasonable
request.
Authors' contributions
CG was the principal investigator who designed the
study, performed the majority of the experiments and drafted the
initial manuscript. SGL, WTL, ZXY, XXZ, TYX and ZPC performed some
experiments. HYZ and ZGL designed the study and analyzed the data.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zhang J, Ding L, Holmfeldt L, Wu G,
Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, et
al: The genetic basis of early T-cell precursor acute lymphoblastic
leukaemia. Nature. 481:2905–163. 2012. View Article : Google Scholar
|
|
2
|
Holmfeldt L, Wei L, Diaz-Flores E, Walsh
M, Zhang J, Ding L, Payne-Turner D, Churchman M, Andersson A, Chen
SC, et al: The genomic landscape of hypodiploid acute lymphoblastic
leukemia. Nat Genet. 45:242–52. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Paulsson K, Lilljebjörn H, Biloglav A,
Olsson L, Rissler M, Castor A, Barbany G, Fogelstrand L, Nordgren
A, Sjögren H, et al: The genomic landscape of high hyperdiploid
childhood acute lymphoblastic leukemia. Nat Genet. 47:672–676.
2015. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Qian M, Zhang H, Kham SK, Liu S, Jiang C,
Zhao X, Lu Y, Goodings C, Lin TN, Zhang R, et al:
Whole-transcriptome sequencing identifies a distinct subtype of
acute lymphoblastic leukemia with predominant genomic abnormalities
of EP300 and CREBBP. Genome Res. 27:185–195. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mullighan CG, Zhang J, Kasper LH, Lerach
S, Payne-Turner D, Phillips LA, Heatley SL, Holmfeldt L,
Collins-Underwood JR, Ma J, et al: CREBBP mutations in relapsed
acute lymphoblastic leukaemia. Nature. 471:235–239. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Malinowska-Ozdowy K, Frech C, Schönegger
A, Eckert C, Cazzaniga G, Stanulla M, zur Stadt U, Mecklenbräuker
A, Schuster M, Kneidinger D, et al: KRAS and CREBBP mutations: A
relapse-linked malicious liaison in childhood high hyperdiploid
acute lymphoblastic leukemia. Leukemia. 29:1656–1667. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Horton SJ, Giotopoulos G, Yun H, Vohra S,
Sheppard O, Bashford-Rogers R, Rashid M, Clipson A, Chan WI, Sasca
D, et al: Early loss of Crebbp confers malignant stem cell
properties on lymphoid progenitors. Nat Cell Biol. 19:1093–1104.
2017. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gao C, Zhang RD, Liu SG, Zhao XX, Cui L,
Yue ZX, Li WJ, Chen ZP, Li ZG, Rao Q, et al: Low CREBBP expression
is associated with adverse long-term outcomes in paediatric acute
lymphoblastic leukaemia. Eur J Haematol. 99:150–159. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chan HM and La Thangue NB: p300/CBP
proteins: HATs for transcriptional bridges and scaffolds. J Cell
Sci. 114:2363–2373. 2001.PubMed/NCBI
|
|
10
|
Yang XJ, Ogryzko VV, Nishikawa J, Howard
BH and Nakatani Y: A p300/CBP-associated factor that competes with
the adenoviral oncoprotein E1A. Nature. 382:319–324. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gorgoulis VG, Zacharatos P, Mariatos G,
Kotsinas A, Bouda M, Kletsas D, Asimacopoulos PJ, Agnantis N,
Kittas C and Papavassiliou AG: Transcription factor E2F-1 acts as a
growth-promoting factor and is associated with adverse prognosis in
non-small cell lung carcinomas. J Pathol. 198:142–156. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
DeGregori J: The genetics of the E2F
family of transcription factors: Shared functions and unique roles.
Biochim Biophys Acta. 1602:131–150. 2002.PubMed/NCBI
|
|
13
|
Ross ME, Zhou X, Song G, Shurtleff SA,
Girtman K, Williams WK, Liu HC, Mahfouz R, Raimondi SC, Lenny N, et
al: Classification of pediatric acute lymphoblastic leukemia by
gene expression profiling. Blood. 102:2951–2959. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li Z, Zhang W, Wu M, Zhu S, Gao C, Sun L,
Zhang R, Qiao N, Xue H, Hu Y, et al: Gene expression-based
classification and regulatory networks of pediatric acute
lymphoblastic leukemia. Blood. 114:4486–4493. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu FF, Wang KL, Deng LP, Liu X, Wu MY,
Wang TY, Cui L and Li ZG: Transcription factor E2F3a regulates
CASP8AP2 transcription and enhances sensitivity to chemotherapeutic
drugs in acute lymphoblastic leukemia. Cancer Cell Int. 18:402018.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Konermann S, Brigham MD, Trevino AE, Joung
J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS,
Nishimasu H, et al: Genome-scale transcriptional activation by an
engineered CRISPR-Cas9 complex. Nature. 517:583–588. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang H, Zou X, Zhong L, Hou Y, Zhou J,
Zhang Z, Xing X and Sun J: CRISPR/dCas9-mediated activation of
multiple endogenous target genes directly converts human foreskin
fibroblasts into Leydig-like cells. J Cell Mol Med. 23:6072–6084.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mei Y, Gao C, Wang K, Cui L, Li W, Zhao X,
Liu F, Wu M, Deng G, Ding W, et al: Effect of microRNA-210 on
prognosis and response to chemotherapeutic drugs in pediatric acute
lymphoblastic leukemia. Cancer Sci. 105:463–472. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Toso C and Lindley C: Vinorelbine: A novel
vinca alkaloid. Am J Health Syst Pharm. 52:1287–1304. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fung MKL and Chan GC: Drug-induced amino
acid deprivation as strategy for cancer therapy. J Hematol Oncol.
10:1442017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Aubel-Sadron G and Londos-Gagliardi D:
Daunorubicin and doxorubicin, anthracycline antibiotics, a
physicochemical and biological review. Biochimie. 66:333–352. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Al-Aamri HM, Ku H, Irving HR, Tucci J,
Meehan-Andrews T and Bradley C: Time dependent response of
daunorubicin on cytotoxicity, cell cycle and DNA repair in acute
lymphoblastic leukaemia. BMC Cancer. 19:1792019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Frei E III and Sallan SE: Acute
lymphoblastic leukemia: Treatment. Cancer. 42 (2 Suppl):S828–S838.
1978. View Article : Google Scholar
|
|
25
|
Garcia-Carpizo V, Ruiz-Llorente S,
Sarmentero J, Graña-Castro O, Pisano DG and Barrero MJ:
CREBBP/EP300 bromodomains are critical to sustain the GATA1/MYC
regulatory axis in proliferation. Epigenetics Chromatin. 11:302018.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ait-Si-Ali S, Polesskaya A, Filleur S,
Ferreira R, Duquet A, Robin P, Vervish A, Trouche D, Cabon F and
Harel-Bellan A: CBP/p300 histone acetyl-transferase activity is
important for the G1/S transition. Oncogene. 19:2430–2437. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dutta R, Tiu B and Sakamoto KM: CBP/p300
acetyltransferase activity in hematologic malignancies. Mol Genet
Metab. 119:37–43. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Trouche D and Kouzarides T: E2F1 and
E1A(12S) have a homologous activation domain regulated by RB and
CBP. Proc Natl Acad Sci USA. 93:1439–1442. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Trouche D, Cook A and Kouzarides T: The
CBP co-activator stimulates E2F1/DP1 activity. Nucleic Acids Res.
24:4139–4145. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang H, Larris B, Peiris TH, Zhang L, Le
Lay J, Gao Y and Greenbaum LE: C/EBPbeta activates E2F-regulated
genes in vivo via recruitment of the coactivator CREB-binding
protein/P300. J Biol Chem. 282:24679–24688. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Martínez-Balbás MA, Bauer UM, Nielsen SJ,
Brehm A and Kouzarides T: Regulation of E2F1 activity by
acetylation. EMBO J. 19:662–671. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Marzio G, Wagener C, Gutierrez MI,
Cartwright P, Helin K and Giacca M: E2F family members are
differentially regulated by reversible acetylation. J Biol Chem.
275:10887–10892. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Leone G, DeGregori J, Yan Z, Jakoi L,
Ishida S, Williams RS and Nevins JR: E2F3 activity is regulated
during the cell cycle and is required for the induction of S phase.
Genes Dev. 12:2120–2130. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Leone G, Nuckolls F, Ishida S, Adams M,
Sears R, Jakoi L, Miron A and Nevins JR: Identification of a novel
E2F3 product suggests a mechanism for determining specificity of
repression by Rb proteins. Mol Cell Biol. 20:3626–3632. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jiao Y, Cui L, Gao C, Li W, Zhao X, Liu S,
Wu M, Deng G and Li Z: CASP8AP2 is a promising prognostic indicator
in pediatric acute lymphoblastic leukemia. Leuk Res. 36:67–71.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Barcaroli D, Bongiorno-Borbone L,
Terrinoni A, Hofmann TG, Rossi M, Knight RA, Matera AG, Melino G
and De Laurenzi V: FLASH is required for histone transcription and
S-phase progression. Proc Natl Acad Sci USA. 103:14808–14812. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
De Cola A, Bongiorno-Borbone L, Bianchi E,
Barcaroli D, Carletti E, Knight RA, Di Ilio C, Melino G, Sette C
and De Laurenzi V: FLASH is essential during early embryogenesis
and cooperates with p73 to regulate histone gene transcription.
Oncogene. 31:573–582. 2012. View Article : Google Scholar : PubMed/NCBI
|