Introduction
Acute lymphoblastic leukemia (ALL) is a
heterogeneous group of diseases with multiple, prognostically
relevant genetic abnormalities and is characterized by over
production of abnormal immature white blood cells (1). Philadelphia chromosome-positive
(Ph+) ALL is characterized by the existence of a
constitutively active breakpoint cluster region-tyrosine-protein
kinase ABL1 (BCR-ABL) protein tyrosine kinase, which subverts the
transcriptional programs that normally control lymphoid development
(2). Ph+ ALL comprises
2–3% of pediatric patients and 20–30% of adult patients with ALL
worldwide (3), and this ALL
subgroup has been regarded as a prognostically unfavorable subgroup
as it has an increased risk of relapse/refractory disease (1,4).
Imatinib, a selective inhibitor of the BCR-ABL tyrosine kinase, is
effective in treating Ph+ chronic myelogenous leukemia
(CML) (5). However, Ph+
ALL is less sensitive to imatinib than CML (6). Thus, there is an urgent requirement
to develop novel strategies to treat Ph+ ALL.
CD9, which belongs to the tetraspanin membrane
proteins, is expressed in a variety of different types of blood
cells, including precursor B-lymphocytes (7). CD9 plays crucial roles in a wide
range of physiological processes, including cell motility and
fertilization (8). CD9 is also
expressed in numerous solid cancer types, including prostate
carcinoma, melanoma and glioblastoma, and serves a role in several
pathological processes, including cancer cell motility,
invasiveness and proliferation (9–15).
Previous studies have been conducted to investigate the association
between CD9 and ALL with respect to the biological and clinical
characteristics. For example, Nishida et al (16) and Yamazaki et al (17) identified that CD9+
B-cell ALL (B-ALL) cells exhibited an asymmetric cell division-like
proliferation with greater leukemogenic potential than
CD9− cells, while CD9+ B-ALL cells exhibited
drug-resistance. In addition, an anti-CD9 monoclonal antibody has
an anti-proliferative effect on B-ALL cells, and knockdown of CD9
expression suppresses the leukemogenic potential of the B-ALL cell
line (17). Thus, these findings
suggested that targeted therapies against CD9 may be a novel
therapeutic approach for B-ALL. Moreover, Arnaud et al
(18) identified that CD9 promoted
Ras-related C3 botulinum toxin substrate 1 (RAC1) activation and
enhanced C-X-C motif chemokine receptor 4-mediated B-ALL cell
migration and engraftment to the bone marrow or testis. Recently,
Liang et al (19) revealed
that patients with CD9+ ALL exhibited a higher positive
rate of the BCR-ABL fusion gene compared with patients who were
CD9−, and CD9 expression indicated an poor prognosis in
patients with ALL. However, the role of CD9 in the pathogenesis of
Ph+ ALL and the potential benefit of applying
loss-of-function strategies targeting CD9 for treatment of
Ph+ ALL require further examination.
Therefore, the aims of the present study were: i) To
determine the effects of CD9 on leukemic cell progression and the
efficacy of therapeutic agents in Ph+ ALL cells; and ii)
to assess the in vitro anti-leukemia activity of
CD9-targeted RNA interference in Ph+ ALL cells.
Materials and methods
Cell lines and culture conditions
The human Ph+ ALL cell line SUP-B15 and
human embryonic kidney cell line 293T were purchased from The
American Type Culture Collection. The SUP-B15 cell line has been
authenticated by short tandem repeat DNA profiling analysis by
Genetic Testing Biotechnology Corporation (Suzhou). SUP-B15 cells
were cultured in Iscove's modified Dulbecco's medium (IMDM;
Sigma-Aldrich; Merck KGaA) with 20% FBS (Gibco; Thermo Fisher
Scientific, Inc.), and 293T cells were cultured in DMEM
(Sigma-Aldrich; Merck KGaA) with 10% FBS. All cells were maintained
in a humidified incubator at 37°C in an atmosphere of 5%
CO2.
Lentiviral vector construction and
transduction
A total of three interference sequences, shCD9-1,
shCD9-2 and shCD9-3, that target human CD9 mRNA (NCBI reference
sequence NM_001330312.1; Gene ID 928) were designed with an online
small interfering RNA tool (http://bioinfo.clontech.com/rnaidesigner/frontpage.jsp;
Clontech Laboratories, Inc.). Primer Designer™ Tool (Invitrogen;
Thermo Fisher Scientific, Inc.) was used to design the short
hairpin RNA (shRNA) primers for targeting the gene interference
sequences of CD9. The primer sequences used in the present study
are presented in Table I. The
single-stranded oligonucleotides of the sequences were chemically
synthesized, annealed to form double-stranded DNA and then inserted
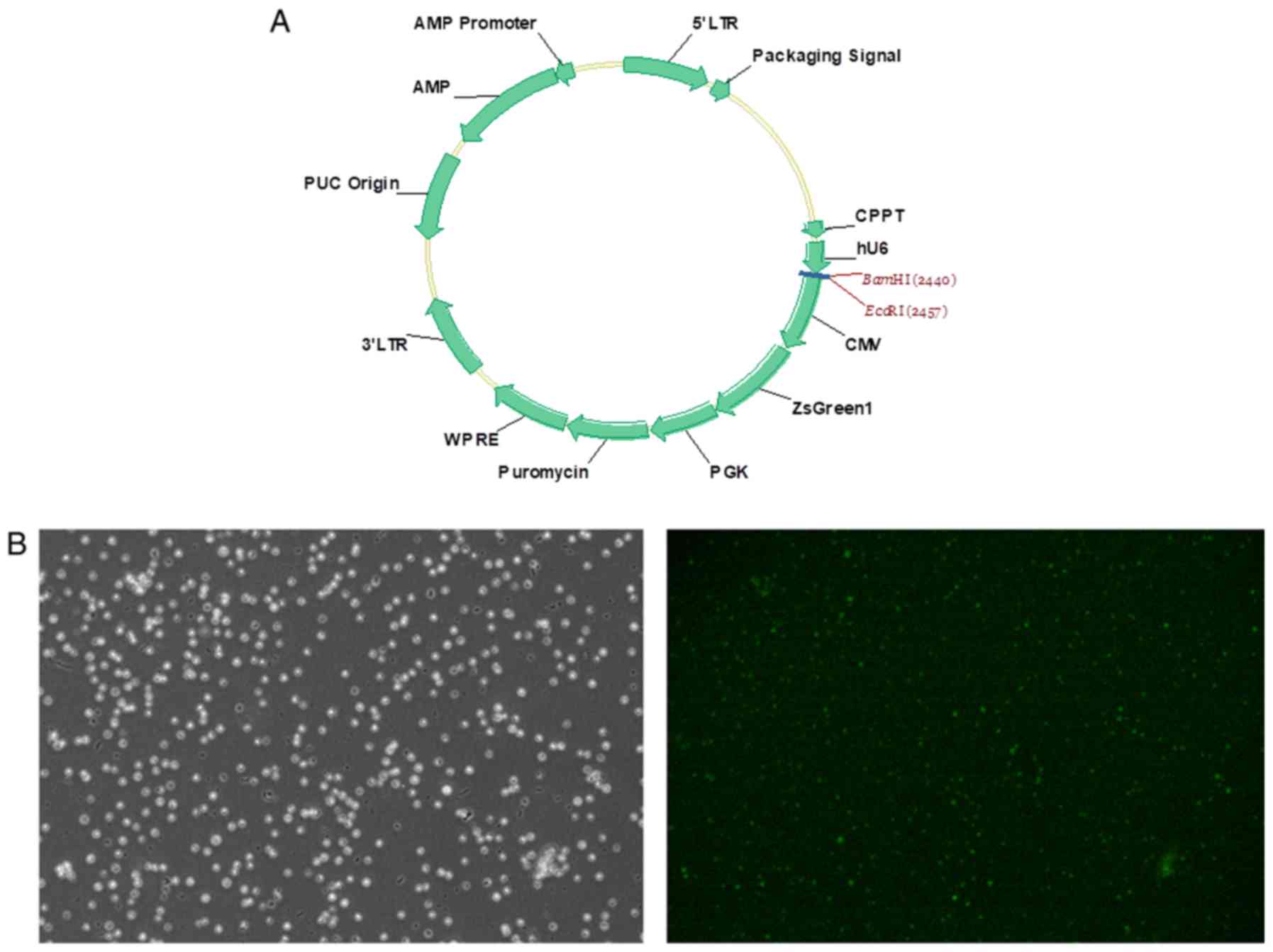
into the PHY-310 lentiviral vector [hU6-MCS-CMV-ZsGreen1-PGK-Puro;
Han Yin Biotechnology (Shanghai) Co., Ltd.; Fig. 1A], which expresses ZsGreen1
fluorescent protein as a cell-tracking marker, to produce the
recombinant shRNA-expressing lentivirus vectors: PHY-310/shCD9-1,
PHY-310/shCD9-2 and PHY-310/shCD9-3.
 | Table I.Primers used for targeting the
interference sequences of the CD9 gene. |
Table I.
Primers used for targeting the
interference sequences of the CD9 gene.
| Primer | Primer sequence
(5′-3′) |
|---|
| shCD9-1 | F:
GATCCGCGGGAAACGCTGAAAGCCATTCAAGAGATGGCTTTCAGCGTTTCCCGTTTTTTGR:
AATTCAAAAAACGGGAAACGCTGAAAGCCATCTCTTGAATGGCTTTCAGCGTTTCCCGCG |
| shCD9-2 | F:
GATCCGCCACAAGGATGAGGTGATTAGAGAACTTAATCACCTCATCCTTGTGGTTTTTTG |
|
| R:
AATTCAAAAAACCACAAGGATGAGGTGATTAAGTTCTCTAATCACCTCATCCTTGTGGCG |
| shCD9-3 | F:
GATCCAGGAAGTCCAGGAGTTTTATTCAAGAGATAAAACTCCTGGACTTCCTTTTTTTG |
|
| R:
AATTCAAAAAAAGGAAGTCCAGGAGTTTTATCTCTTGAATAAAACTCCTGGACTTCCTG |
Transduction of SUP-B15 cells with the recombinant
lentivirus was performed as described previously (20). Briefly, the subconfluent 293T cells
(1.5×108/dish) in a 10-cm culture dish were
co-transfected with 12 µg lentiviral vector and 9 µg lentiviral
packaging vector LV-PV001 [Han Yin Biotechnology (Shanghai) Co.,
Ltd.] using polyethylenimine (Sigma-Aldrich; Merck KGaA). At 48 h
after transfection, lentiviral particles were harvested and
purified by ultra-centrifugation at 3,000 × g for 2.5 h at 4°C. The
viral titer was determined by hole-dilution method (21). SUP-B15 cells
(5×105/well) were seeded into 6-well plates. The
lentiviral particles produced from the transfected 293T cells were
used to transduce SUP-B15 cells in the presence of 8 µg/ml
polybrene (Sigma-Aldrich; Merck KGaA). In the shControl group,
SUP-B15 cells were transduced with a blank vector. After 72 h of
transduction, fluorescence microscopy (magnification, ×200) was
used to detect the transduction efficiency, and the efficiency of
CD9 knockdown was evaluated by reverse transcription-quantitative
PCR (RT-qPCR) and western blotting, as well as flow cytometry
analysis. After 96 h of transduction, the transduced SUP-B15 cells
were used for subsequent experiments.
RT-qPCR assay
Total RNA in the SUP-B15 cells was isolated using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). Total RNA was reverse transcribed into cDNA using the
ExScript RT reagent kit (Takara Bio, Inc.) at 37°C for 15 min. qPCR
was performed as previously described (20). PCR primers were designed for
amplifying DNA fragments that span intron-exon boundaries, and the
primer sequences were as follows: CD9 forward,
5′-TCGCCATTGAAATAGCTGCGGC-3′ and reverse,
5′-CGCATAGTGGATGGCTTTCAGC-3′; and ribosomal protein L13a forward,
5′-CGAGGTTGGCTGGAAGTACC-3′ and reverse 5′-CTTCTCGGCCTGTTTCCGTAG-3′.
qPCR was subsequently performed in the ABI 7500 real-time PCR
machine (Applied Biosystems; Thermo Fisher Scientific, Inc.) using
a SYBR-Green PCR Master Mix (Takara Bio, Inc.). The following
thermocycling conditions were used for the qPCR: Initial
denaturation at 95°C for 5 min; followed by 40 cycles of
denaturation at 95°C for 10 sec and combined annealing/extension at
60°C for 20 sec. Ribosomal protein L13a was used as an internal
control and the relative gene expression levels were calculated
using the 2−ΔΔCq method (22).
Western blotting
The methods of protein extraction and western blot
analysis were carried out according to a previous study (20). Briefly, cells were lysed with RIPA
lysis buffer (Beyotime Institute of Biotechnology) and the total
protein concentration was determined using a BCA protein assay kit
(Beyotime Institute of Biotechnology). Total protein (30 µg/lane)
was separated by 10% SDS-PAGE and then transferred to a PVDF
membrane. The membranes were blocked with 5% skim-milk diluted in
TBS-0.1% Tween-20 for 1 h at room temperature, and then the blots
were incubated overnight with primary antibodies at 4°C. The
primary antibodies used were anti-p53 (1:1,000, cat. no. 9282, Cell
Signaling Technology, Inc.), anti-p21 (1:1,000, cat. no. 2947, Cell
Signaling Technology, Inc.), anti-caspase 3 (1:1,000, cat. no.
9662, Cell Signaling Technology, Inc.), anti-cleaved caspase 3
(1:1,000, cat. no. 9579, Cell Signaling Technology, Inc.), anti-CD9
(1:1,000, cat. no. 20597-1-AP, ProteinTech Group, Inc.) and
anti-β-actin (1:3,000, cat. no. 4970, Cell Signaling Technology,
Inc.). Following incubation using horseradish peroxidase-conjugated
anti-rabbit antibody (1:10,000, cat. no. A6154, Sigma-Aldrich;
Merck KGaA) or anti-rat antibody (1:10,000, cat. no. A5795,
Sigma-Aldrich; Merck KGaA) at room temperature for 1 h, the protein
bands were analyzed with a freshly prepared enhanced
chemiluminescent solution (GE Healthcare Life Sciences). The
relative density of protein expression was quantified using ImageJ
software (version 1.49p; National Institutes of Health). Protein
levels were standardized against β-actin.
To investigate the effect of CD9 blockade on the
expression of p53 and p53-related proteins, SUP-B15 cells
(5×105) were untreated or treated with the IgG isotype
control (cat. no. 2729S, Cell Signaling Technology, Inc.) or
anti-CD9 antibody (at a concentration of 50 µg/ml; cat. no.
20597-1-AP; Protein Tech Group, Inc.) at 37°C for 24 h, and then
were lysed and subjected to western blot analysis.
Flow cytometry assay
To detect the membrane expression of CD9,
3×105 cells were harvested and treated with Fc Block
(1:100; BD Biosciences) for 15 min at 4°C. Thereafter, the cell
surface was stained with phycoerythrin (PE)-conjugated anti-CD9
(1:50; cat. no. 12-0098-41; eBioscience; Thermo Fisher Scientific,
Inc.) for 30 min at 4°C. PE-conjugated mouse IgG1 κ isotype control
(1:50; cat. no. 12-4714-82; eBioscience; Thermo Fisher Scientific,
Inc.) was used as a negative control. The membrane expression of
CD9 in SUP-B15 cells was then analyzed using a FACSCalibur flow
cytometer (BD Biosciences) and FlowJo version 10 software (FlowJo
LLC).
Cell proliferation assay
Cell Counting Kit-8 (CCK-8; Dojindo Molecular
Technologies, Inc.) was used for a cell proliferation assay,
according to the manufacturer's protocol. Cells were cultured in
96-well plates at a concentration of 3×103 cells/well.
After incubation at 37°C for 24, 48, 72 and 96 h, CCK-8 solution
(10 µl) was added to each well and cells were incubated for 2 h at
37°C. The absorbance was detected at 450 nm using a SpectraMax M5
microplate reader (Molecular Devices, LLC).
Apoptosis assay
Allophycocyanin-labeled Annexin V (BD Biosciences)
and DAPI (BD Biosciences) double staining (both stained at room
temperature for 15 min) was used to detect early and late
apoptosis. The cells were analyzed using a FACSCalibur flow
cytometer (BD Biosciences) and FlowJo version 10 software (FlowJo
LLC). Annexin V-positive cells were defined as apoptotic.
To investigate the effect of the caspase 3 inhibitor
in combination with CD9 knockdown on apoptosis, cells were
preincubated with 0.6 µmol/l Z-DEVD-FMK (MedChemExpress) at 37°C
for 24 h prior to the apoptosis assay.
Cell cycle assay
Cells were washed with PBS, treated with 0.1 mg/ml
RNAse A at 37°C for 30 min, stained with propidium iodide (0.05
mg/ml; Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C for 1 h,
and then analyzed using a FACSCalibur flow cytometer (BD
Biosciences). Multicycle version 4.0 software (Phoenix Flow
Systems, Inc.) was used for calculating the cell cycle phase
distribution from the resultant DNA histogram.
Drug sensitivity assay
In vitro drug sensitivity to therapeutic
agents was assessed with CCK-8 as previously described (20). Cells were exposed to different
concentration gradients of chemotherapeutic agents [including
vincristine (VCR; 10, 20, 30, 40 and 50 µg/l; Selleck Chemicals),
daunorubicin (DNR; 10, 20, 30, 40 and 50 µg/l; Selleck Chemicals),
cyclophosphamide (CPM; 50, 100, 200, 400, 600, 800 and 1,000 µg/ml;
Selleck Chemicals), dexamethasone (DXM; 0.5, 1, 1.5, 2 and 2.5
nmol/l; Selleck Chemicals) or imatinib (10, 20 and 30 µmol/l;
Sigma-Aldrich; Merch KGaA) at 37°C for 48 h, and CCK-8 was used to
determine the cell viability. Optical density (OD) values were
measured at 450 nm using a SpectraMax M5 microplate reader
(Molecular Devices, LLC). The cytotoxicity was calculated using the
following formula: Cytotoxicity (%)=(1-mean OD of treated/mean OD
of control) ×100.
Cell adhesion assay
An artificial basement membrane used for the cell
adhesion assay was prepared by adding 0.5 µg Superfibronectin
(Sigma-Aldrich; Merck KGaA) into each well of a 96-well plate and
incubating at 4°C overnight. Subsequently, 1×105 SUP-B15
cells/well were seeded into the 96-well plate and allowed to adhere
to the Superfibronectin at 37°C for 90 min in a humidified
incubator with an atmosphere of 5% CO2. Non-adherent
cells were then carefully removed by rinsing with PBS. Finally,
adherent cells were quantified by CCK-8 using the same method
described previously.
Cell migration and invasion
assays
In the cell migration assay, a total of
1×104 SUP-B15 cells suspended in 100 µl serum-free IMDM
(Sigma-Aldrich; Merck KGaA) was placed in the upper chamber of a
8-µm pore size Transwell plate (Corning, Inc.) and 800 µl IMDM
containing 10% FBS (Gibco; Thermo Fisher Scientific, Inc.) was
added to the bottom chamber as a chemoattractant. After incubation
for 72 h at 37°C and 5% CO2 atmosphere, cells that
migrated to the bottom chamber were fixed in 4% paraformaldehyde at
room temperature for 20 min, stained with 1% crystal violet at room
temperature for 30 min, and then counted using a hemocytometer
under a light microscope (magnification, ×100). The invasion assay
was performed by the same procedure except that the upper chamber
was coated with 50 µl Matrigel (1 mg/ml; BD Biosciences) at 37°C
for 5 h.
In addition, in order to assess the effect of CD9
blockade on adhesion, migration and invasion of Ph+ ALL
cells, SUP-B15 cells (2.5×104) were untreated or treated
with the IgG isotype control or anti-CD9 antibody (10 µg/ml) at
37°C for 4 h before cell adhesion, migration and invasion assays
were performed.
Statistical analysis
Data are presented as the mean ± standard deviation
from three individual experiments. The statistical significance of
differences among groups was assessed with one-way ANOVA followed
by Tukey's post-hoc test for multiple comparisons. P<0.05 was
considered to indicate a statistically significant difference. All
statistical analyses were performed with Stata software (version
12.0; StataCorp LP).
Results
Downregulation of CD9 mRNA and protein
expression in SUP-B15 cells using a lentivirus-based approach
In order to establish an efficient method of
permanent knockdown of CD9 in SUP-B15 cells, a lentiviral-mediated
shRNA approach was used. To demonstrate the delivery efficiency of
shRNA, the SUP-B15 cells were transduced with PHY-310,
PHY-310/shCD9-1, PHY-310/shCD9-2 and PHY-310/shCD9-3 carrying the
ZsGreen1 gene. Successful lentiviral transduction was demonstrated
by fluorescence microscopy after transduction for 72 h (Fig. 1B). Lentiviral delivery of shRNA
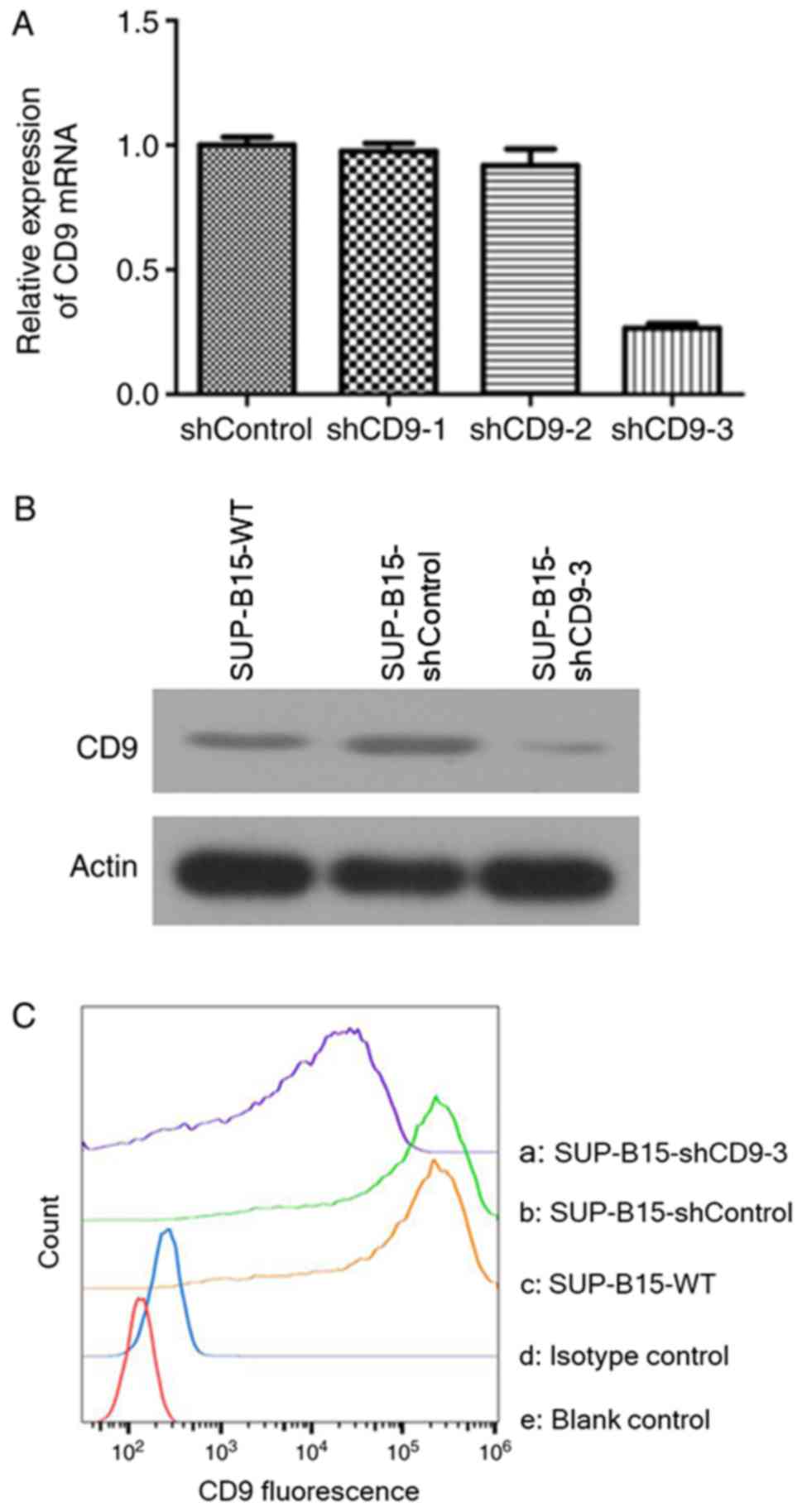
targeted against CD9 led to a downregulation of CD9 mRNA in SUP-B15
cells, to 2.51±3.00% in the PHY-310/shCD9-1 group, 8.83±1.41% in
the PHY-310/shCD9-2 group and 73.51±3.22% in the PHY-310/shCD9-3
group, compared with the shControl group, as measured by RT-qPCR
(Fig. 2A). Based on maximal mRNA
inhibition of CD9 in the PHY-310/shCD9-3 group, PHY-310/shCD9-3 was
utilized for the subsequent experimental paradigms. The protein
expression of CD9 in the stably transduced SUP-B15 cells after
PHY-310/shCD9-3 transduction is presented in Fig. 2B. The results suggested that
PHY-310/shCD9-3 induced a marked downregulation of the CD9 protein
expression compared with the SUP-B15-wild-type (WT) and shControl
groups.
The membrane expression of CD9 was measured by flow
cytometry. Transduction with PHY-310/shCD9-3 resulted in notable
reduction of the mean fluorescence intensity of the CD9 molecule in
SUP-B15 cells compared with the SUP-B15-WT (89.9% reduction) and
shControl groups (90.2% reduction; Fig. 2C). These results suggested that
lentiviral-mediated delivery of shRNA targeting the CD9 gene was
able to markedly downregulate the expression of CD9 mRNA and
protein in SUP-B15 cells.
Silenced CD9 inhibits cell
proliferation, promotes apoptosis and arrests the cell cycle in
SUP-B15 cells
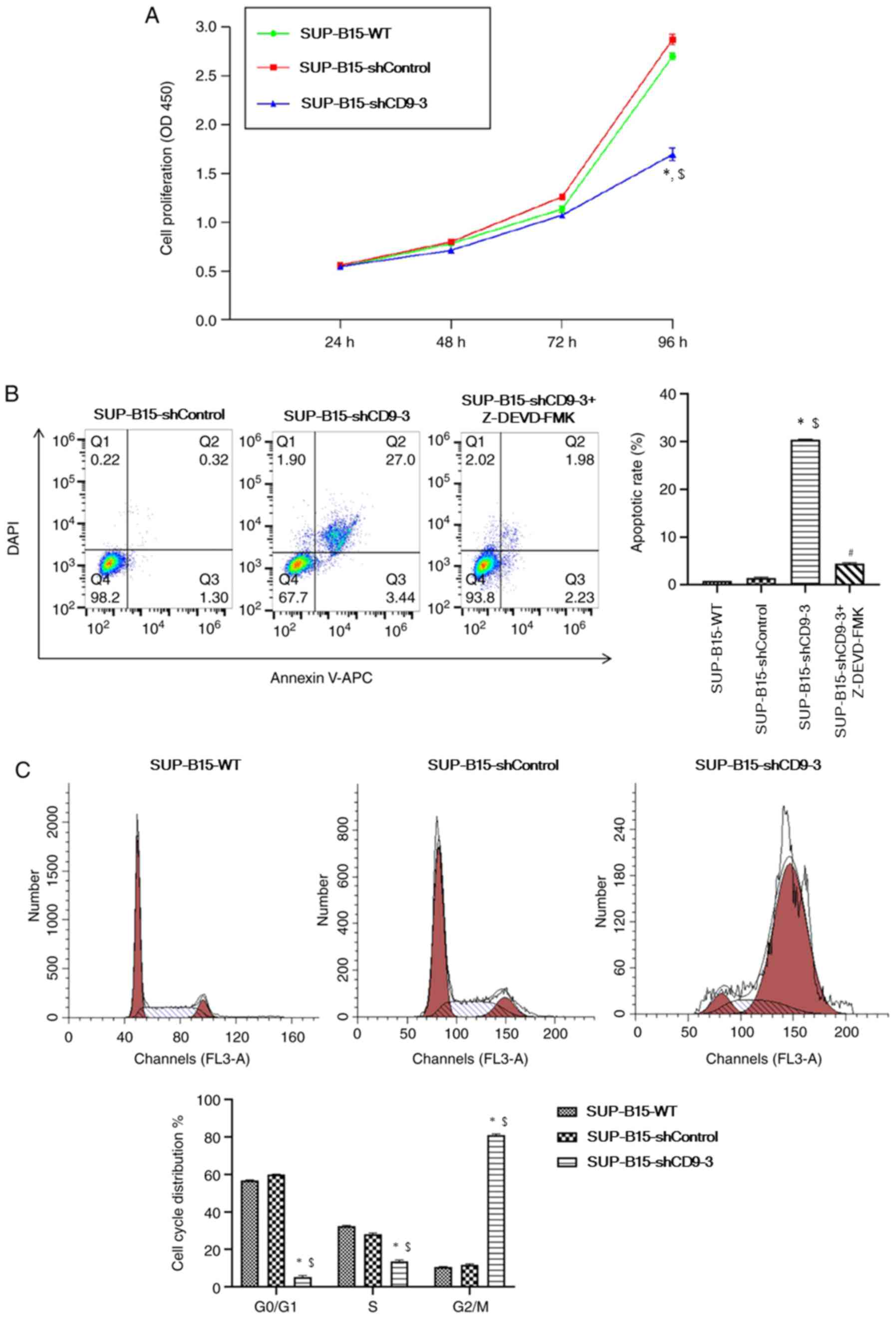
In order to evaluate the impact of silenced CD9 on
SUP-B15 cell survival, the CCK-8 assay and flow cytometry were used
to analyze cell proliferation and apoptosis, respectively. From the
CCK-8 assay, it was identified that the proliferation of cells
transduced with CD9-shRNA was significantly reduced after 96 h of
incubation compared with the SUP-B15-WT and shControl groups
(Fig. 3A). The study examined
whether the attenuated proliferation of SUP-B15 cells with CD9
knockdown was associated with an increase in apoptosis. SUP-B15
apoptosis was evaluated by flow cytometry analysis with CD9
knockdown cells, together with treatment with caspase 3 inhibitor
Z-DEVD-FMK (MedChem Express). Flow cytometry results demonstrated
that knockdown of CD9 significantly increased apoptosis compared
with the SUP-B15-WT and shControl groups (Fig. 3B). However, pre-treatment with 0.6
µmol/l Z-DEVD-FMK for 24 h prior to apoptosis assay ameliorated CD9
knockdown-induced apoptosis of SUP-B15 cells. These findings
suggested that apoptosis in CD9-knockdown cells may be regulated
via caspase-dependent pathways.
To determine whether CD9 knockdown has an effect on
the cell cycle, the DNA content of SUP-B15 cells was analyzed by
flow cytometry. Cell cycle analysis demonstrated that silencing of
CD9 significantly increased the percentage of cells in the
G2/M phase, but decreased cells in
G0/G1 and S phases (Fig. 3C). Therefore, it was speculated
that CD9 knockdown was able to arrest cell cycle progression in
SUP-B15 cells.
Silenced CD9 increases the
cytotoxicity of therapeutic agents in SUP-B15 cells
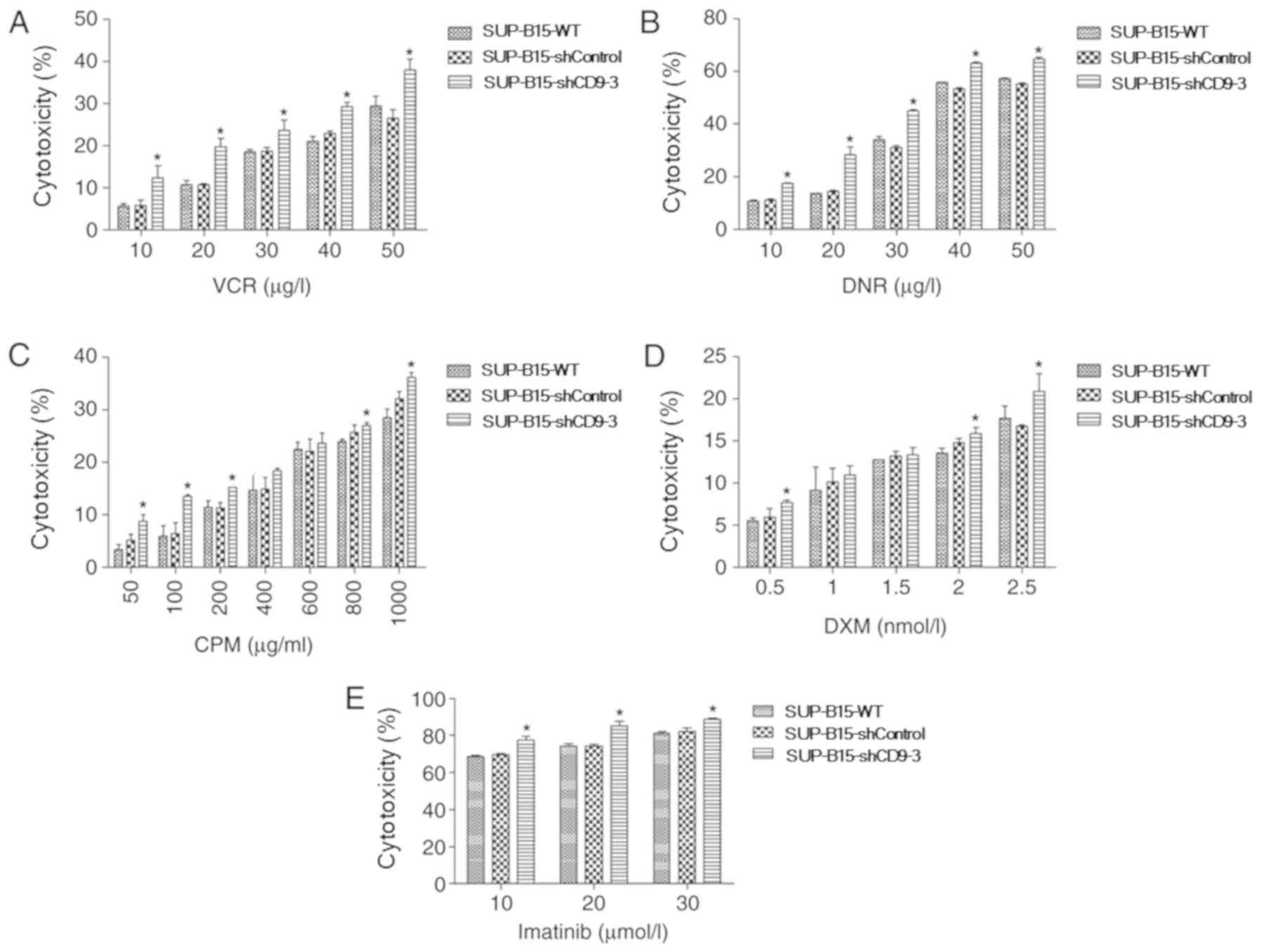
To assess the potential functional significance of
CD9 in chemotherapeutic resistance of Ph+ ALL cells, the
impact of CD9 knockdown in SUP-B15 cells on their susceptibility to
chemotherapeutic agent-induced cytotoxicity was investigated.
Different concentration gradients of chemotherapeutic drugs (such
as VCR, DNR, CPM and DXM) were incubated with SUP-B15 cells for 48
h, and then, the CCK-8 assay was used to determine the
cytotoxicity. Silencing of CD9 significantly increased the
susceptibility of SUP-B15 cells to the cytotoxicity of VCR
(Fig. 4A), DNR (Fig. 4B), CPM (Fig. 4C) and DXM (Fig. 4D). In addition, whether CD9
knockdown in SUP-B15 cells increased the cytotoxicity of the
BCR-ABL tyrosine kinase inhibitor imatinib was assessed. A
concentration gradient of imatinib was used to treat SUP-B15 cells.
The results demonstrated that silencing of CD9 also significantly
increased the cytotoxicity of imatinib in SUP-B15 cells (Fig. 4E).
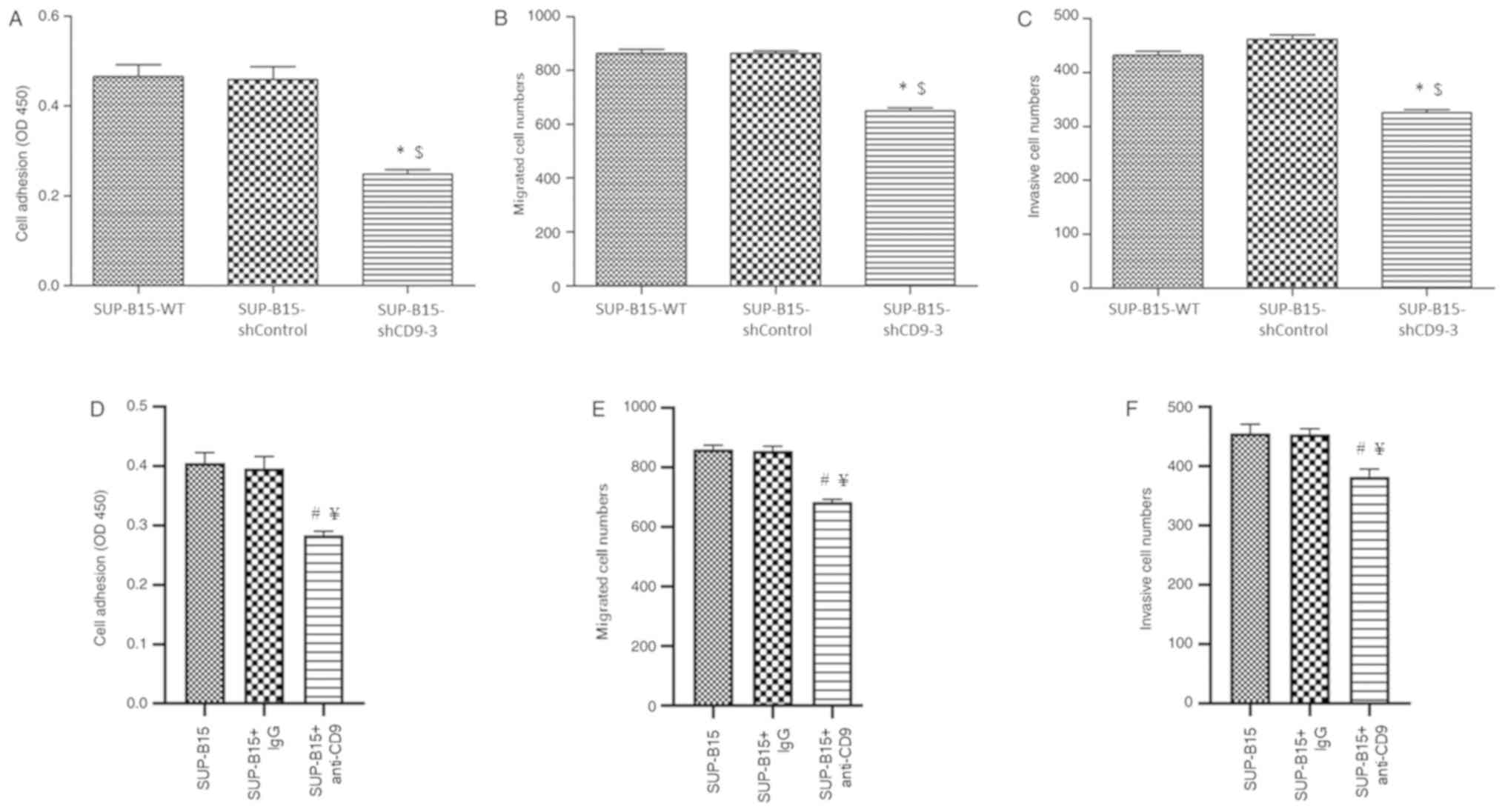
Silenced CD9 inhibits adhesion,
migration and invasion of SUP-B15 cells
Invasion and metastasis are key clinicopathological
features of acute leukemia (23).
Thus, whether CD9 knockdown affected cell adhesion, migration and
invasion in SUP-B15 cells was analyzed. The results indicated that
CD9-silenced SUP-B15 cells exhibited less potential of adhesion
(Fig. 5A), migration (Fig. 5B) and invasion (Fig. 5C) compared with the SUP-B15-WT and
shControl cells. In addition, in order to verify the effect of CD9
knockdown on adhesion, migration and invasion of Ph+ ALL
cells, SUP-B15 cells were incubated with anti-CD9 antibody before
cell adhesion, migration and invasion assays were performed. It was
found that anti-CD9 antibody-treated SUP-B15 cells exhibited
significantly less potential of adhesion (Fig. 5D), migration (Fig. 5E) and invasion (Fig. 5F) compared with the untreated cells
and IgG isotype-treated control cells.
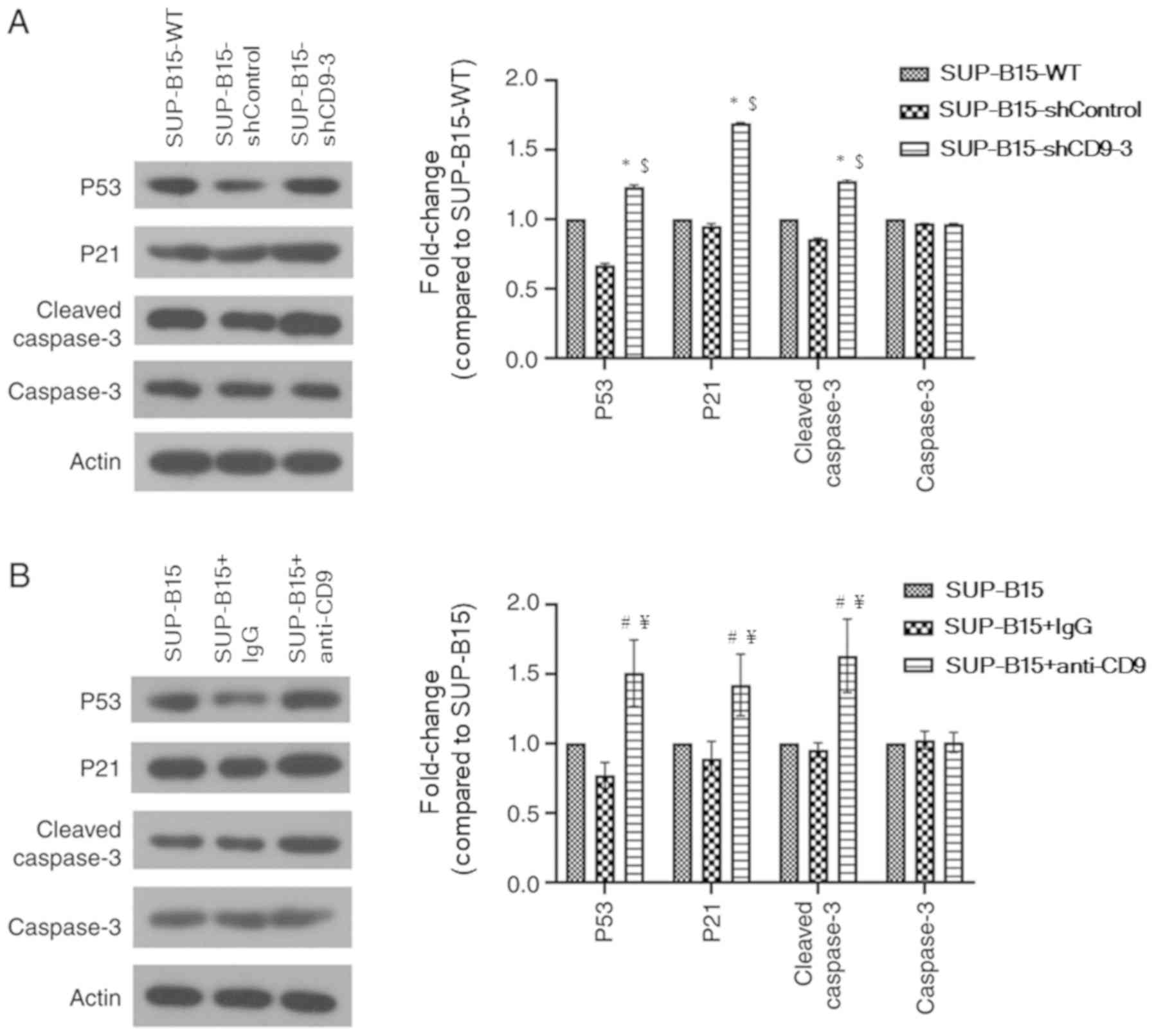
Silenced CD9 inhibits SUP-B15 cell
proliferation and promotes apoptosis via a p53-dependent
pathway
To identify the potential mechanism via which
knockdown of CD9 suppresses proliferation and induces apoptosis in
SUP-B15 cells, the expression levels of proliferation- and
apoptotic-related proteins (such as p53, p21 and cleaved caspase 3)
were detected using western blotting (Fig. 6A). Compared with the SUP-B15-WT and
shControl groups, p53 and p21 protein expression levels were
significantly increased in the CD9 shRNA group. In addition,
silenced CD9 promoted cleaved caspase 3 protein expression. These
findings suggested that silenced CD9 inhibited SUP-B15 cell
proliferation and promoted apoptosis via a p53-dependent
pathway.
Whether CD9 blockade may affect the expression of
p53 and p53-related proteins was also investigated. The results
suggested that p53, p21 and cleaved caspase 3 protein expression
levels were significantly increased in the anti-CD9-treated SUP-B15
cells compared with the untreated cells and IgG isotype-treated
cells (Fig. 6B).
Discussion
It is well known that the expression of CD9
fluctuates during B cell development. For instance, precursor B
cells have high CD9 expression, mature B cells display
downregulation of CD9 and plasma cells re-express CD9 (24). Previous studies conducted by
Nishida et al (16) and
Yamazaki et al (17)
reported that CD9 may be a marker for the identification of
leukemic stem cells in B-ALL, and that CD9 may regulate cancer stem
cell function. These authors also suggested that targeted therapies
against CD9 and its downstream signals may be novel strategies for
treating B-ALL. In addition, it has been revealed that CD9 is able
to increase the ability of B-ALL cells to disseminate via RAC1
activation (18). In ALL, CD9
expression is correlated with the expression of the BCR-ABL fusion
gene and indicates poor prognosis (19). Therefore, the present study
investigated the effects of CD9 on the biological features of
Ph+ ALL cells. In the present study, a loss-of-function
approach was used to analyze the biological effect of CD9 in
Ph+ ALL cells. A lentiviral shRNA expression vector
targeting CD9 gene was constructed and used to transduce the
Ph+ ALL cell line SUP-B15. Silenced CD9 significantly
inhibited SUP-B15 cell proliferation in vitro. From the flow
cytometry analysis, it was demonstrated that the SUP-B15 apoptotic
rate was significantly increased when the CD9 gene was knocked
down. Cell cycle analysis demonstrated that CD9 knockdown caused
G2/M phase cell cycle to arrest in SUP-B15 cells. Taken
together, these results from the in vitro experiments
suggested that the therapeutic suppression of CD9 expression may
lead to inhibition of the progression of Ph+ ALL.
However, in order to further evaluate the therapeutic effects of
CD9 knockdown on Ph+ ALL in vivo, it is necessary
to establish xenograft models of Ph+ ALL.
As a tumor suppressor protein, p53 has a key effect
on balancing between cell proliferation and death by suppressing
cell cycle progression via p21 upregulation and promoting apoptosis
via caspase 3 activation (25,26).
In the present study, it was identified that silenced CD9
upregulated protein expression levels of p53 and p21, which may
result in the suppression of proliferation in SUP-B15 cells. In
addition to attenuating proliferation, silenced CD9 also exhibited
pro-apoptotic properties. However, the relatively high apoptosis in
CD9-silenced SUP-B15 cells could be decreased by caspase 3 blocker
(Z-DEVD-FMK). Thus, it was hypothesized that caspase 3, the
executioner of cell death (27),
may be involved in apoptosis induced by silenced CD9. Western
blotting was used to analyze the expression of cleaved caspase 3,
and the results demonstrated that the expression of cleaved caspase
3 was upregulated in the SUP-B15 cells transduced with
CD9-shRNA.
Intensive multi-agent chemotherapy has produced a
high complete remission rate in pediatric ALL (28). However, Ph+ ALL remains
largely resistant to the currently available chemotherapy
strategies (29). The
CD9+ population of human precursor-B ALL cell lines has
been identified to be relatively more chemotherapy-resistant than
the CD9− population (17). In the present study, it was found
that silenced CD9 was able to increase VCR, DNR, CPM and
DXM-induced cytotoxicity in SUP-B15 cells. These findings suggested
that chemotherapy combined with knockdown of CD9 may be a novel
potential approach for treating Ph+ ALL. It is possible
that the combination of different anti-cancer strategies that
disrupt different cell signaling pathways leads to enhanced
destruction of cancer cells. The potential mechanisms of how CD9
knockdown may promote the efficacy of chemotherapeutic agents
include, but are not limited to, the shared intracellular signaling
pathways involved in cell cycle control, apoptosis or drug
metabolism and efflux, which should be further investigated.
In patients with Ph+ ALL, the response to
the selective BCR-ABL tyrosine kinase inhibitor imatinib is not as
effective as in Ph+ CML (6), and the underlying biological
mechanism remains largely unknown. The present study demonstrated
that silenced CD9 promoted sensitivity to imatinib in SUP-B15
cells. These findings suggested that imatinib combined with
knockdown of CD9 may be a promising strategy for treating
Ph+ ALL.
Invasion and metastasis are key clinicopathological
features of acute leukemia (23);
therefore, identifying the mechanism involved in the dissemination
of Ph+ ALL may lead to the development of novel and more
effective treatment approaches. In the present study, silenced CD9
markedly suppressed cell adhesion, migration and invasion in
SUP-B15 cells. These results suggested that CD9 may serve a key
role involved in leukemic cell invasion and metastasis in
Ph+ ALL. The present results are in line with other
previous studies reporting that CD9 enhances metastatic medullary
invasion of precursor-B leukemic cells in vivo (16,17),
and that CD9 promotes the adhesion of leukemic B lymphocytes to
fibronectin and the migration of these leukemic cells in response
to C-X-C motif chemokine ligand 12 (18). These results suggested that
therapeutic strategies involving CD9 gene silencing may inhibit the
invasion and migration of Ph+ ALL cells, and lead to
improved clinical outcome; however, further research is needed to
determine the potential mechanisms of the CD9 molecule on cell
motility in Ph+ ALL cells.
A limitation of the present study is that the
experiments were only performed in the Ph+ ALL cell line
SUP-B15. In contrast to cell lines, patient-derived primary
leukemia cells reflect the heterogeneous nature of cancer biology,
as they exist in patients. Thus, the potential anti-leukemia effect
of gene silencing of CD9 using an shRNA expressing lentivirus
vector should be further evaluated in primary Ph+ ALL
cells. Moreover, despite high transduction efficiency in SUP-B15
cells obtained in the shRNA approach, only one of the three tested
shRNA sequences significantly decreased the CD9 mRNA expression to
~26% compared with the shControl cells. RNA interference-mediated
gene silencing technologies can suffer from pervasive off-target
effects and non-specific toxicity (30–32).
Therefore, further experiments using additional loss-of-function
strategies with high on-target efficiencies and low off-target
effects (such as CRISPR/Cas9-mediated gene knockout) will be
required to elucidate the role of the CD9 gene in leukemogenesis in
Ph+ ALL.
In conclusion, the present study demonstrated that
the silencing of the CD9 gene induced apoptosis and G2/M
cell cycle arrest via a p53-dependent pathway in Ph+ ALL
SUP-B15 cells. Additionally, the downregulation of CD9 expression
inhibited the cell proliferation, adhesion, migration and invasion
of SUP-B15 cells, and increased the efficacy of chemotherapeutic
drugs and imatinib. Therefore, it was hypothesized that the genetic
silencing of CD9 using an shRNA-expressing lentivirus vector may
provide a promising treatment for Ph+ ALL.
Acknowledgements
Not applicable.
Funding
This study was supported by the Natural Science
Foundation of Zhejiang Province (grant nos. LQ14H080002 and
LQ19H080002), and the Public Welfare Science and Technology Project
of Wenzhou (grant no. Y20160099).
Availability of data and materials
The datasets generated or analyzed during this study
are not publicly available due to confidentiality of another study
from our group but are available from the corresponding authors
upon reasonable request.
Authors' contributions
JF and SG conceived of the project. CX, WX, YS, BZ,
DW and BL conducted the experiments and collected the data. CX, WX,
YS, YZ, SG and JF analyzed the data. CX, WX and YS wrote the paper.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Inaba H, Greaves M and Mullighan CG: Acute
lymphoblastic leukaemia. Lancet. 381:2791–1955. 2013. View Article : Google Scholar
|
|
2
|
Bernt KM and Hunger SP: Current concepts
in pediatric Philadelphia chromosome-positive acute lymphoblastic
leukemia. Front Oncol. 4:542014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fielding AK: How I treat Philadelphia
chromosome-positive acute lymphoblastic leukemia. Blood.
116:3409–3417. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pui CH, Sandlund JT, Pei D, Campana D,
Rivera GK, Ribeiro RC, Rubnitz JE, Razzouk BI, Howard SC, Hudson
MM, et al: Improved outcome for children with acute lymphoblastic
leukemia: Results of total therapy study XIIIB at St Jude
children's research hospital. Blood. 104:2690–2696. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Druker BJ, Guilhot F, O'Brien SG, Gathmann
I, Kantarjian H, Gattermann N, Deininger MW, Silver RT, Goldman JM,
Stone RM, et al: Five-year follow-up of patients receiving imatinib
for chronic myeloid leukemia. N Engl J Med. 355:2408–2417. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee HJ, Thompson JE, Wang ES and Wetzler
M: Philadelphia chromosome-positive acute lymphoblastic leukemia:
Current treatment and future perspectives. Cancer. 117:1583–1594.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Maecker HT, Todd SC and Levy S: The
tetraspanin superfamily: Molecular facilitators. FASEB J.
11:428–442. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hemler ME: Tetraspanin functions and
associated microdomains. Nat Rev Mol Cell Biol. 6:801–811. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang JC, Bégin LR, Bérubé NG, Chevalier S,
Aprikian AG, Gourdeau H and Chevrette M: Down-regulation of CD9
expression during prostate carcinoma progression is associated with
CD9 mRNA modifications. Clin Cancer Res. 13:2354–2361. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zöller M: Tetraspanins: Push and pull in
suppressing and promoting metastasis. Nat Rev Cancer. 9:40–55.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Peñas P, García-López M and del Río
Barreiro O: Inhibition of the motility of melanoma cells using
interference RNA against CD9. Actas Dermosifiliogr. 96:30–36.
2005.(In Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Herr MJ, Mabry SE, Jameson JF and Jennings
LK: Pro-MMP-9 upregulation in HT1080 cells expressing CD9 is
regulated by epidermal growth factor receptor. Biochem Biophys Res
Commun. 442:99–104. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang GP and Han XF: CD9 modulates
proliferation of human glioblastoma cells via epidermal growth
factor receptor signaling. Mol Med Rep. 12:1381–1386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rappa G, Green TM, Karbanová J, Corbeil D
and Lorico A: Tetraspanin CD9 determines invasiveness and
tumorigenicity of human breast cancer cells. Oncotarget.
6:7970–7991. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tang M, Yin G, Wang F, Liu H, Zhou S, Ni
J, Chen C, Zhou Y and Zhao Y: Downregulation of CD9 promotes
pancreatic cancer growth and metastasis through upregulation of
epidermal growth factor on the cell surface. Oncol Rep. 34:350–358.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nishida H, Yamazaki H, Yamada T, Iwata S,
Dang NH, Inukai T, Sugita K, Ikeda Y and Morimoto C: CD9 correlates
with cancer stem cell potentials in human B-acute lymphoblastic
leukemia cells. Biochem Biophys Res Commun. 382:57–62. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yamazaki H, Xu CW, Naito M, Nishida H,
Okamoto T, Ghani FI, Iwata S, Inukai T, Sugita K and Morimoto C:
Regulation of cancer stem cell properties by CD9 in human B-acute
lymphoblastic leukemia. Biochem Biophys Res Commun. 409:14–21.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Arnaud MP, Vallée A, Robert G, Bonneau J,
Leroy C, Varin-Blank N, Rio AG, Troadec MB, Galibert MD and
Gandemer V: CD9, a key actor in the dissemination of lymphoblastic
leukemia, modulating CXCR4-mediated migration via RAC1 signaling.
Blood. 126:1802–1812. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liang P, Miao M, Liu Z, Wang H, Jiang W,
Ma S, Li C and Hu R: CD9 expression indicates a poor outcome in
acute lymphoblastic leukemia. Cancer Biomark. 21:781–786. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu J, Liang B, Qian Y, Tang L, Xing C,
Zhuang Q, Shen Z, Jiang S, Yu K and Feng J: Down-regulation of CD19
expression inhibits proliferation, adhesion, migration and invasion
and promotes apoptosis and the efficacy of chemotherapeutic agents
and imatinib in SUP-B15 cells. Cell Biol Int. 42:1228–1239. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sanber KS, Knight SB, Stephen SL, Bailey
R, Escors D, Minshull J, Santilli G, Thrasher AJ, Collins MK and
Takeuchi Y: Construction of stable packaging cell lines for
clinical lentiviral vector production. Sci Rep. 5:90212015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Klein G, Vellenga E, Fraaije MW, Kamps WA
and de Bont ES: The possible role of matrix metalloproteinase
(MMP)-2 and MMP-9 in cancer, eg acute leukemia. Crit Rev Oncol
Hematol. 50:87–100. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Barrena S, Almeida J, Yunta M, López A,
Fernández-Mosteirín N, Giralt M, Romero M, Perdiguer L, Delgado M,
Orfao A and Lazo PA: Aberrant expression of tetraspanin molecules
in B-cell chronic lymphoproliferative disorders and its correlation
with normal B-cell maturation. Leukemia. 19:1376–1383. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vogelstein B, Lane D and Levine AJ:
Surfing the p53 network. Nature. 408:307–310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shen Y and White E: p53-dependent
apoptosis pathways. Adv Cancer Res. 82:55–84. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Slee EA, Adrain C and Martin SJ: Serial
killers: Ordering caspase activation events in apoptosis. Cell
Death Differ. 6:1067–1074. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kato M and Manabe A: Treatment and biology
of pediatric acute lymphoblastic leukemia. Pediatr Int. 60:4–12.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gleissner B, Gökbuget N, Bartram CR,
Janssen B, Rieder H, Janssen JWG, Fonatsch C, Heyll A, Voliotis D,
Beck J, et al: Leading prognostic relevance of the BCR-ABL
translocation in adult acute B-lineage lymphoblastic leukemia: A
prospective study of the german multicenter trial group and
confirmed polymerase chain reaction analysis. Blood. 99:1536–1543.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Barrangou R, Birmingham A, Wiemann S,
Beijersbergen RL, Hornung V and van Brabant Smith A: Advances in
CRISPR-Cas9 genome engineering: Lessons learned from RNA
interference. Nucleic Acids Res. 43:3407–3419. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Grimm D, Streetz KL, Jopling CL, Storm TA,
Pandey K, Davis CR, Marion P, Salazar F and Kay MA: Fatality in
mice due to oversaturation of cellular microRNA/short hairpin RNA
pathways. Nature. 441:537–541. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kaelin WG Jr: Use and abuse of RNAi to
study mammalian gene function. Science. 337:421–422. 2012.
View Article : Google Scholar : PubMed/NCBI
|