Introduction
Bronchial asthma is a heterogeneous disease
characterized by chronic airway inflammation (1). There are ~235 million individuals
suffering from asthma worldwide (2). Toluene diisocyanate (TDI), a chemical
intermediate used in the manufacture of several synthetic
materials, is considered as the most common causative agent of
occupational asthma and accounts for 9–15% of cases in adults
(3–6). The clinical manifestations and
pathological changes of TDI-induced asthma are similar to those of
allergic asthma; however, the pathogenesis of TDI-induced asthma
still remains obscure (7). In an
animal model of TDI-induced asthma, chronic airway inflammation and
bronchial hyperreactivity were observed, along with the
overexpression of inflammatory cytokines (8). These conditions were found to be
associated with the airway submucosal infiltration of neutrophils,
lymphocytes and eosinophils (8).
In addition, previous studies have suggested that animals with
asthma exhibit elevated concentrations of interleukin (IL)-4, −5,
and −13 in the bronchoalveolar lavage fluid (BALF), lung and serum
(9–11).
High mobility group box 1 (HMGB1) is a classic
inflammatory cytokine that is overexpressed in the lungs of
asthmatic animals (12). HMGB1 is
a highly conserved nuclear protein that is secreted by immune cell
lineages following the stimulation of inflammatory cytokines such
as tumor necrosis factor-α and IL-1 (13). IL-1 functions upstream of HMGB1
(14) however, there are no
relevant reports regarding the mechanisms through which IL-1 can
stimulate the secretion of HMGB1. In mammals, HMGB1 serves a
pivotal role in mediating inflammation, and is strongly associated
with the pathological processes of sepsis, pneumonia, arthritis and
other diseases (13,15).
Airway eosinophilia and related inflammatory
cytokines contribute substantially to the airway
hyperresponsiveness of asthma (1,16).
In this process, the typical pathological alterations in airway
remodeling include the hyperplasia of goblet cells and
myofibroblasts, and deposition of collagen, which is secondary to
airway inflammation in the pathology of asthma (17). As a marker for fibroblasts and a
factor involved in the differentiation of epithelial cells into
myofibroblasts, the expression levels of α-smooth muscle actin
(α-SMA) indicate the formation of subepithelial fibrosis (18). In past decades, α-SMA was reported
to be an important biomarker of airway remodeling in asthma
(19) by indicating the deposition
of collagen I. Furthermore, overexpression of fibroblastic collagen
I and α-SMA have been reported to further aggravate airway
remodeling (20).
MK2206 is an allosteric small-molecule inhibitor of
AKT and exhibits promising clinical potential in the treatment of
solid tumors (21,22). AKT is an archetypal family member
of the Ser/Thr protein kinases and downstream effector of the PI3K
signaling pathway (23). Our
previous study reported the importance of PI3K in asthma (24). The AKT signaling pathway is
activated by phosphorylation of AKT, which subsequently activates
downstream inflammatory cytokines to regulate airway smooth muscle
(ASM) and inflammatory cells (23). Studies have observed that the
structure and function of ASM change in patients with asthma and
animal models (25,26). However, the potential role of
MK2206 in airway inflammation or remodeling as a result of asthma
is still unclear.
Our previous study reported that the pathological
changes of asthma in a TDI-induced animal model closely resemble
the acute stage of human TDI asthma (27). Using this model, the effect of the
AKT inhibitor, MK2206, on airway inflammation, airway remodeling
and airway hyperresponsiveness (AHR) triggered by TDI-induced
asthma, and the possible mechanisms were investigated. The results
of the present study indicated the potential application of AKT
inhibitors in the treatment of occupational asthma.
Materials and methods
Animals and drugs
A total of 24 male BALB/C mice (6–8 weeks old, 20±2
g) were purchased from the Experimental Animal Center of Southern
Medical University. Mice were housed in a specific pathogen-free
environment at a constant temperature of 23±1°C and 55±5% humidity,
and provided with standard laboratory diet and drinking water ad
libitum in a 12-h dark/light cycle. All mice were fed
irradiated food and given access to sterile water. The study
protocol for animals was approved by Southern Medical University
Experimental Animal Ethics Committee (approval no. L2017177).
TDI (≥98.0%), methacholine and acetone (all
Sigma-Aldrich; Merck KGaA) were administered to mice to establish
TDI-induced asthma models. A 2:3 mixture of acetone and olive oil
(AOO) was used to dissolve TDI, and was also used as the vehicle
treatment for the study control group. For airway challenge, a 1:4
mixture of AOO was used.
Models and groups
All mice were separated into four groups randomly
(n=6/group): i) Acetone and olive oil (AOO); ii) TDI; iii) TDI +
MK2206; and iv) TDI + dexamethasone (DEX). Mice in the control AOO
group were treated using the same procedure as the TDI asthma model
group (except for the use of TDI). In this control group (AOO),
mice were sensitized with AOO on the dorsa of both ears dermally
(20 µl/ear applied topically) on day 1 and day 8. They received an
injection of saline 24 h prior to challenge. On days 15, 18 and 21,
the mice were raised in horizontal cylindrical niches individually
and challenged using air with AOO. The TDI group was established
according to our previously published studies (24,28).
On days 1 and 8, mice were sensitized using TDI (0.3%) on the dorsa
of both ears dermally (20 µl/ear). Then, the mice were managed in
horizontal cylindrical niches individually and challenged using air
with TDI (3%) that was dissolved in a 1:4 mixture of AOO with a
compressed air nebulizer (NE-C28; Omron) for 3 h on days 15, 18 and
21. Saline was injected intraperitoneally 24 h before the TDI
challenge, which acted as a vehicle control for the MK2206 and DEX
treatments. In the TDI + MK2206 group, mice were treated using the
same procedure as that for the TDI group; however, they were
treated with 100 mg/kg MK2206 in saline using oral gavage 24 h
before the air challenge. Mice in the TDI + DEX group were treated
using the same procedure as that for the TDI group; they were
injected with 200 mg/kg DEX in saline 24 h before the air
challenge. All animals were sacrificed using cervical dislocation
under anesthesia (60 mg/kg intraperitoneal pentobarbital sodium) at
the end of the study. The total duration of the study was less than
4 weeks and lung tissues were collected after animal sacrifice.
Immunohistochemistry
The lung tissues were fixed in 10% formalin at room
temperature for 24 h, and embedded in paraffin. Sections of the
samples were deparaffinized and submerged into citrate buffer for
antigen retrieval (pH 6.0). To block endogenous peroxidase
activity, each section (2.5-µm thick) was incubated with 0.3%
H2O2 at room temperature for 10 min. After
blocking in 5% bovine serum albumin (Beijing ZSGB-BIO Technology,
Ltd.) for 20 min at room temperature, the sections were incubated
with primary antibody either rabbit anti-α-SMA (1:100; cat. no.
23081-1-AP; Proteintech Group, Inc.) or rabbit
anti-phosphorylated-AKT (p-AKT; 1:100; cat. no. 66444-1-Ig;
Proteintech Group, Inc.) antibodies overnight at 4°C. The next day,
sections were washed three times using PBS, then incubated with
biotin-conjugated anti-rabbit IgG (1:100; cat. no. BM2004; Wuhan
Boster Biological Technology, Ltd.) secondary antibody for 20 min
at room temperature. Lastly, the sections were incubated with
HRP-streptavidin (1:1,000; cat. no. BIR701-3; Beijing Borsi
Technology Co., Ltd.) at room temperature for 10 min, then
visualized using a DAB peroxidase kit (1:20; cat. no. AR1000; Wuhan
Boster Biological Technology, Ltd.) at room temperature for 1 min,
then counterstained with hematoxylin at room temperature for 3 min.
Each stained sections were examined in at least five random visual
fields using an Olympus BX53 light microscope (Olympus Corporation;
magnification, ×200).
Western blotting (WB)
Total protein from each lung tissue was extracted
from the cells via lysis with RIPA lysis buffer (Beyotime Institute
of Biotechnology). The protein content of the lysate was determined
using a bicinchoninic acid assay kit (Beyotime Institute of
Biotechnology) according to the manufacturer's protocol. Protein
samples (40 ng protein/sample) were resolved using 10% SDS-PAGE;
the separated proteins were transferred onto polyvinylidene
difluoride (PVDF) membranes. Next, the blots were blocked with 5%
BSA in TBS-0.05% Tween-20 (TBST) at room temperature for 1 h, and
incubated with antibodies against AKT (1:1,000; cat. no.
10176-2-AP), p-AKT (1:1,000; cat. no. 66444-1-Ig), α-SMA (1:1,000;
cat. no. 23081-1-AP), HMGB1 (1:1,000; cat. no. 66525-1-Ig),
collagen I (1:1,000; cat. no. 66761-1-Ig) and GAPDH (1:1,000; cat.
no. 60004-1-Ig) all purchased from ProteinTech Group, Inc.
overnight at 4°C. The membranes were washed three times with TBST
and incubated with biotin-conjugated goat anti-mouse IgG secondary
antibody (1:1,000; cat. no. SA00004-1; ProteinTech Group, Inc.) or
biotin-conjugated donkey anti-rabbit IgG (1:1,000; cat. no.
SA00004-11; ProteinTech Group, Inc.). After incubating with
HRP-streptavidin (1:5,000; BIR701-3; Beijing Borsi Technology Co.,
Ltd.) at room temperature for 30 min, the blots were visualized
using ECL reagent (100 µl per membrane; cat. no. ab133406; Abcam)
with a Tanon 5200 Automatic chemiluminescence imaging analysis
system (Tanon Science and Technology Co., Ltd.). The gray value of
the blots were quantified by ImageJ software (1.48v; National
Institutes of Health).
Measurement of bronchoalveolar lavage
fluids
BALF was collected and centrifuged at 1,000 × g for
10 min at 4°C. The recovered lavage solution was centrifuged at
room temperature at 162 × g for 10 min The total cell number in the
BALF of each mouse was counted manually by direct microscopic
counting using an Olympus BX53 light microscope (Olympus
Corporation; magnification, ×400). The supernatant was collected
and stored at −80°C for multiplex immunoassay analyses of IL-4, −5,
−6 and −13 (cat. no. EK0405; EK0408; EK0411; EK0425; Wuhan Boster
Biological Technology, Ltd.) in accordance with the manufacturer's
protocols. The cell pellet was resuspended in 50 µl saline, and the
total number of cells was counted. The cells were then smeared
rapidly and uniformly, and allowed to dry naturally. The slides
were then fixed with 4% paraformaldehyde for 30 min, then stained
with hematoxylin and eosin (H&E; Beijing Solarbio Science &
Technology Co., Ltd.). The slides were examined using an Olympus
BX53 light microscope (Olympus Corporation; magnification,
×400).
Hematoxylin and eosin
The left lung was fixed with 4% paraformaldehyde for
24 h, then dehydrated with gradient alcohol. Tissues were then
incubated in with xylene for 15 min and embedded with paraffin.
Tissue was sectioned to a thickness of 4 µm, and individual
sections were allowed to adhere on slides and dried naturally. The
slides were then incubated at 60°C overnight, dewaxed, stained with
hematoxylin for 1 min, and rinsed with tap water for several
seconds to remove excess dye. The cells were acidified with 0.5%
hydrochloric acid alcohol (prepared with 70% ethanol) for 2–5 sec
until the nuclei were blue and the cytoplasm was almost colorless.
The slides were then stained with 1% eosin for 30 sec, then rinsed
with running water. The slides were then dehydrated with an
ascending ethanol gradient, treated with xylene and sealed with
neutral gum. The infiltration of inflammatory cells in bronchial
mucosa and perivascular were examined (magnification, ×200 for lung
tissue or ×400 for cells in the BALF) under an Olympus BX53 light
microscope (Olympus Corporation). The experiment was carried out at
room temperature, unless otherwise specified.
Serum IgE measurements
Blood samples from the retro-orbital venous sinus
were collected immediately after the mice were sacrificed by
decapitation following anesthesia with intraperitoneal injection of
50 mg/kg pentobarbital, and stored at room temperature for 1 h. The
supernatants were harvested and stored at −80°C after
centrifugation at 3,000 × g for 10 min at 37°C. Serum IgE levels
were measured using an IgE ELISA kit (cat. no. ASB-OKIA00100;
NeoBioscience Technology Co., Ltd.) according to the manufacturer's
protocol.
Assessment of AHR
On day 22 of model establishment, mice were placed
in a barometric plethysmographic chamber (Data Sciences
International; Harvard Bioscience, Inc.) and challenged by
increasing gradient concentrations of methacholine (3.12, 6.25,
12.5, 25 or 50 mg/ml) using ultrasonic nebulization. The aerosols
were prepared using an ultrasonic nebulizer (Data Sciences
International; Harvard Bioscience, Inc.) and nebulized into the
chamber for 3 min. After each atomization, the pressure
fluctuations caused due to the breathing of mice were monitored for
3 min and then quantified using the enhanced pause algorithm (Penh,
dimensionless parameter), which represented the precise resistance
index for airway recovery (29,30).
Histological measurements in
lungs
The left lungs of mice were harvested and fixed in
4% paraformaldehyde at room temperature for 24 h. After dehydration
and paraffin embedding, coated glass slides with 4-µm sections were
stained with H&E. Blinded histological assessment was performed
as previously described (31).
Briefly, peribronchial and perivascular inflammation was measured
by using the following scoring standard: 0=normal; 1=infrequent
inflammatory cells; 2=a ring of inflammatory cells 1 cell-layer
deep; 3=a ring of inflammatory cells 2–4 cells deep; and 4=a ring
of inflammatory cells >4 cells deep. The histological scores and
average epithelial reticular basement membrane thickness (measured
manually) were calculated using an Olympus BX53 light microscope
(Olympus Corporation; agnification, ×200) and at least 40 fields of
20 sections from each mouse were recorded.
To quantify goblet cell numbers in the epithelium,
the paraffin-embedded samples were cut into 5–6-µm sections and
stained with periodic acid-Schiff (PAS) at room temperature for 10
min. The results of PAS staining were assessed in at least five
random fields under an Olympus BX53 light microscope (Olympus
Corporation; magnification, ×200). The hyperplasia goblet cells
were quantified as previously described (32). The pathological changes were
classified using a modified five-point scoring system (grades 0–4)
based on the percentage of goblet cells in the epithelium: Grade 0
(no goblet cells); grade 1 (<25%); grade 2 (25–50%); grade 3
(51–75%); and grade 4 (>75%). The mean scores of the 6 mice in
each group were calculated.
Immunofluorescence measurements
The 16HBE cell line (American Type Culture
Collection) was cultured with RPMI-1640 containing 10% FBS (both
from Gibco; Thermo Fisher Scientific, Inc.) in a cell-culture
incubator at 37°C with 5% CO2. At 90% confluence, cells
were placed on a coverslip in six-well plates at a density of
2×105 cells/well. MK2206 (1 µg/ml) or DEX (1 µg/ml) was
added to the medium 1 h before TDI (60 µg/ml) exposure for 6 h. For
immunocytochemistry, cells were fixed in 4% formaldehyde-PBS at
room temperature for 10 min, permeabilized in 0.5% Triton X-100 at
room temperature for 5 min and blocked with 5% (w/v) BSA for 1 h at
room temperature, followed by overnight incubation with anti-α-SMA
(1:100; cat. no. 23081-1-AP; ProteinTech Group, Inc.) and
anti-HMGB1 (1:100; cat. no. 66525-1-Ig; ProteinTech Group, Inc.)
antibodies at 4°C. The next day, cells were washed in three times
PBS and incubated with Alexa Fluor® 488-labeled goat
anti-mouse IgG antibody (1:1,000; cat. no. SA00004-11; ProteinTech
Group, Inc.) at room temperature for 1 h in the dark. The nuclei
were stained with DAPI (Beyotime Institute of Biotechnology) at
room temperature for 10 min. The immunofluorescence of α-SMA and
HMGB1 was observed at ×200 magnification using a laser-scanning
confocal microscope (Olympus Corporation). The immunofluorescence
was quantified by ImageJ software (1.48v; National Institutes of
Health).
Statistical analysis
Data were presented as the mean ± SEM of three
experiments. One-way ANOVA followed by Bonferroni post hoc testing
was performed to compare differences between multiple groups for
continuous data; Kruskal-Wallis test followed by Dunn's multiple
comparison test was used for ordinal data. All statistical analyses
were conducted using SPSS 13.0 (SPSS, Inc.) or GraphPad Prism 8
(GraphPad Software, Inc.) for continuous or ordinal data,
respectively. P<0.05 was considered to indicate a statistically
significant difference.
Results
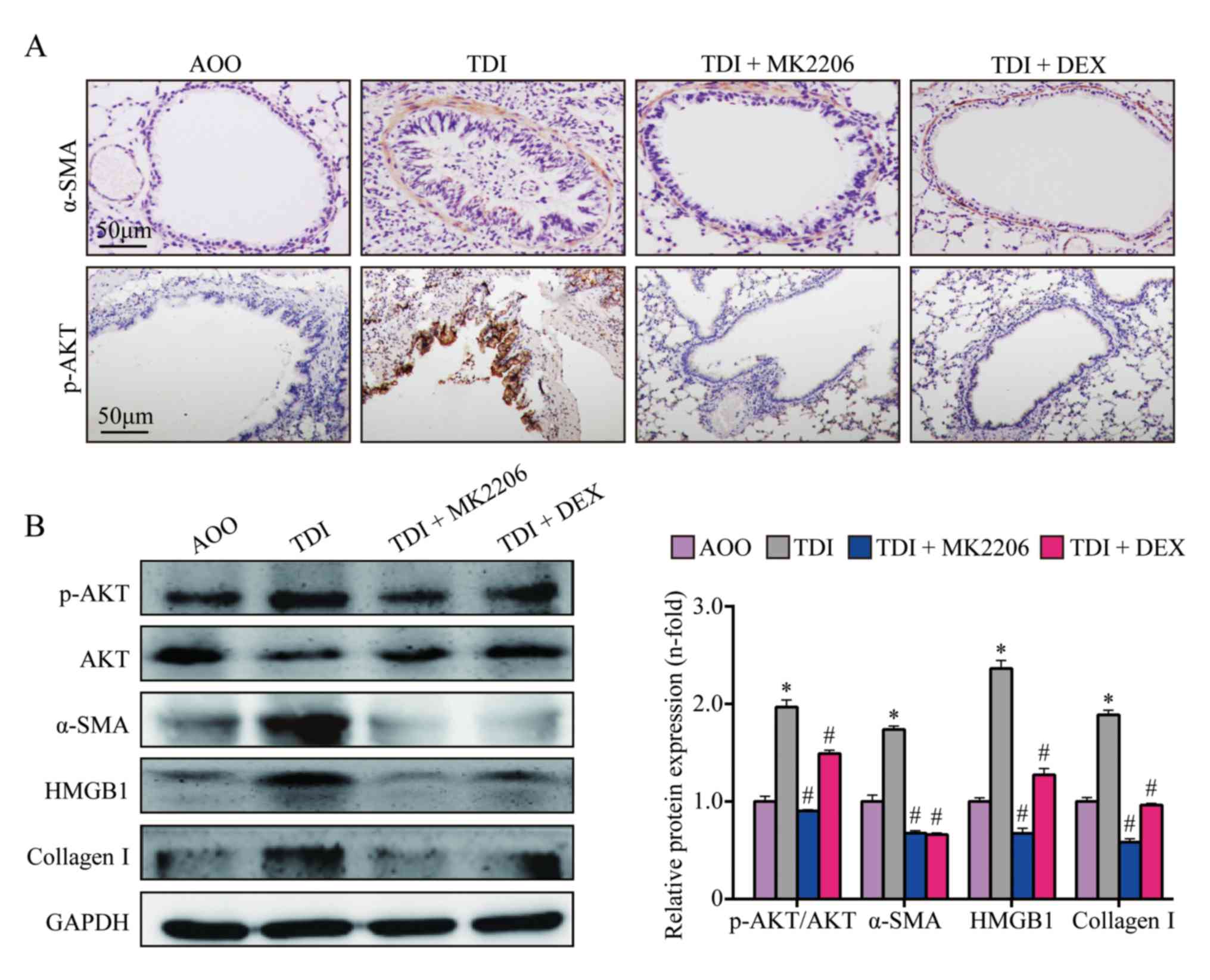
MK2206 inhibits AKT, p-AKT, collagen
I, α-SMA and HMGB1 in lung tissue
The expression levels of collagen I, α-SMA, HMGB1
and p-AKT were significantly upregulated in the TDI group compared
with those in the AOO group (P<0.05; Fig. 1A-B). In addition, compared with the
TDIgroup, the expression levels of collagen I, α-SMA, HMGB1 and
p-AKT in the TDI + MK2206 and TDI + DEX groups were significantly
decreased (P<0.05; Fig.
1A-B).
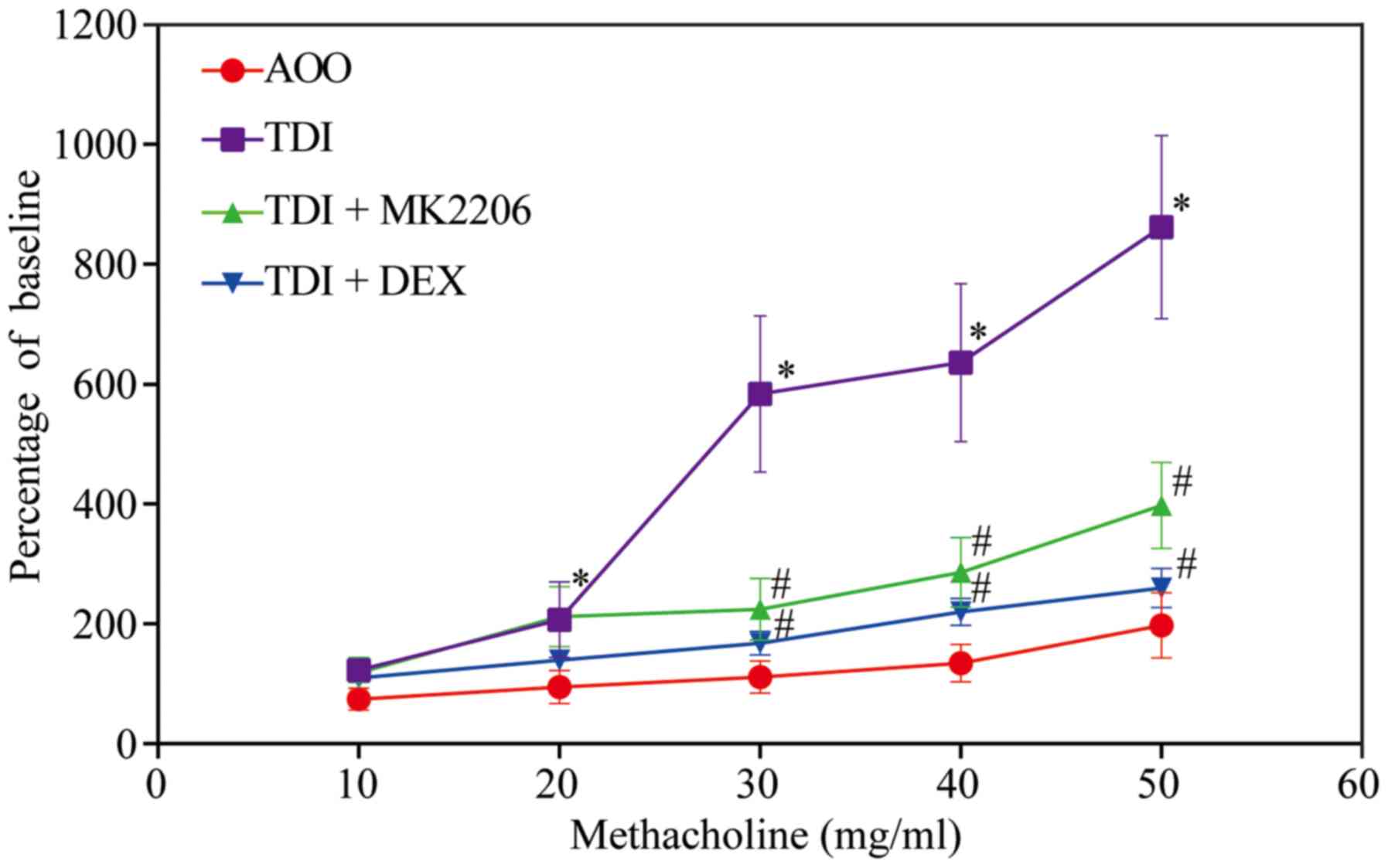
MK2206 reduces AHR
In the present study, airway reactivity was measured
at 24 h after an air challenge. With an increase in methacholine
concentration, the Penh value of airway reactivity demonstrated an
upward trend in all groups. The airway reactivity was significantly
increased in the TDI group compared with the AOO, TDI + MK2206 and
TDI + DEX groups (P<0.05; Fig.
2). In addition, no significant differences were observed
between the airway responsiveness of the AOO, TDI + MK2206 and TDI
+ DEX groups (P>0.05; Fig.
2).
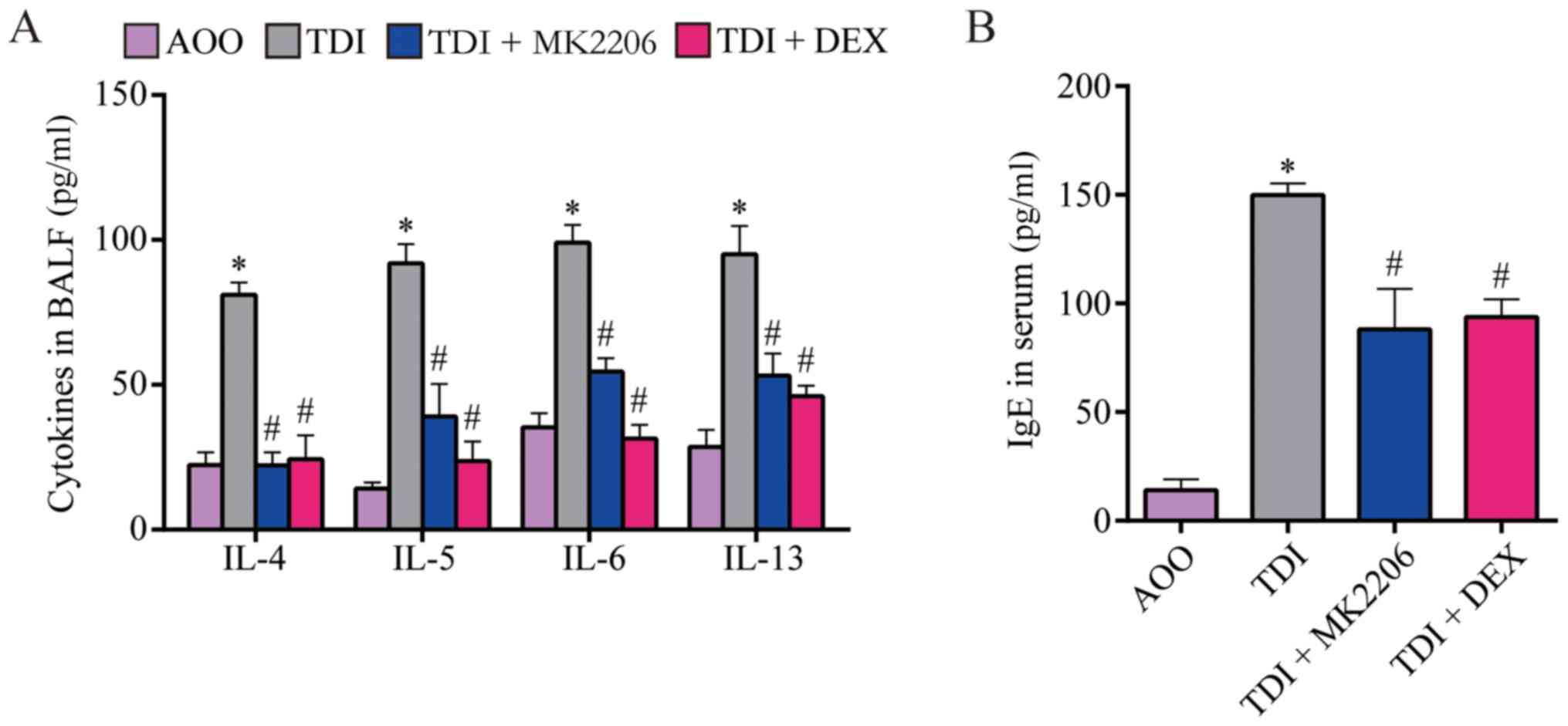
MK2206 reduces airway
inflammation
To assess the effect of MK2206 on TDI-induced
allergic airway inflammation, the levels of cytokines in BALF and
total serum IgE were determined. The results demonstrated that the
levels of IL-4, −5, −6 and −13, and serum IgE were significantly
increased in the TDI group compared with those of the AOO group
(P<0.05; Fig. 3A and B). In
addition, the TDI + MK2206 and TDI + DEX groups demonstrated
significantly lower levels of IL-4, −5, −6 and-13, and serum IgE
compared with those of the TDI group (P<0.05; Fig. 3A and B).
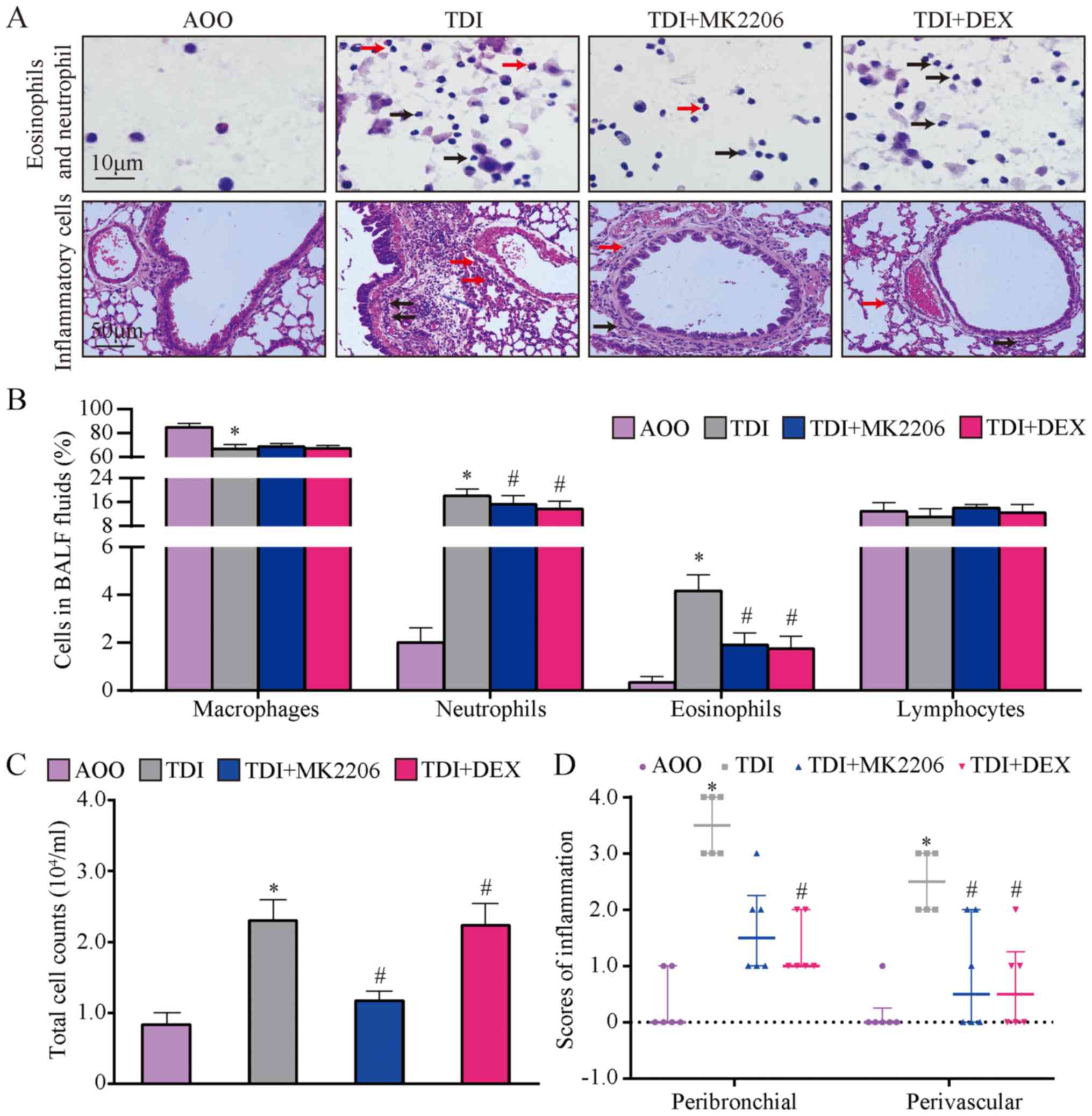
Total cell counts and percentages of different
immune cells were assessed in the BALF. The results demonstrated
that the number of neutrophils and eosinophils were significantly
higher in the TDI group compared with those of the AOO group
(P<0.05; Fig. 4A and B). This
finding suggested that TDI triggered inflammatory responses.
Conversely, compared with the AOO group, the number of macrophages
in the TDI group was significantly reduced (P<0.05; Fig. 4B). In addition, the number of
neutrophils and eosinophils in the TDI + MK2206 and TDI + DEX
groups were significantly lower compared with those of the TDI
group (P<0.05; Fig. 4A and B),
but no significant differences were observed in the lymphocyte and
macrophage count (Fig. 4A and B).
Additionally, the total cell count in the BALF was significantly
increased in the TDI group compared to that of AOO group
(P<0.05; Fig. 4C), whereas that
of the TDI + MK2206 was significantly lower compared to that of the
TDI group (P<0.05; Fig. 4C).
These results suggested that MK2206 and DEX served protective roles
in airway inflammation.
In the AOO group, bronchial, alveolar and vascular
structures appeared normal and intact; the infiltration of
inflammatory cells around the bronchial wall was not observed and
the bronchial epithelial cells were not proliferated or shed
(Fig. 4A). In contrast, in the TDI
group, the airway epithelium started to shed, epithelial cell
proliferation and mucous secretion increased, and numerous
neutrophils, eosinophils and lymphocytes infiltrated the tube wall
(Fig. 4A). In addition, compared
with the TDI group, the inflammation scores of the TDI + MK2206 and
TDI + DEX groups were significantly decreased (P<0.05; Fig. 4D), manifestation of inflammation
appeared to be slightly attenuated and airway epithelial
hyperplastic differentiation was weakened (Fig. 4B-D).
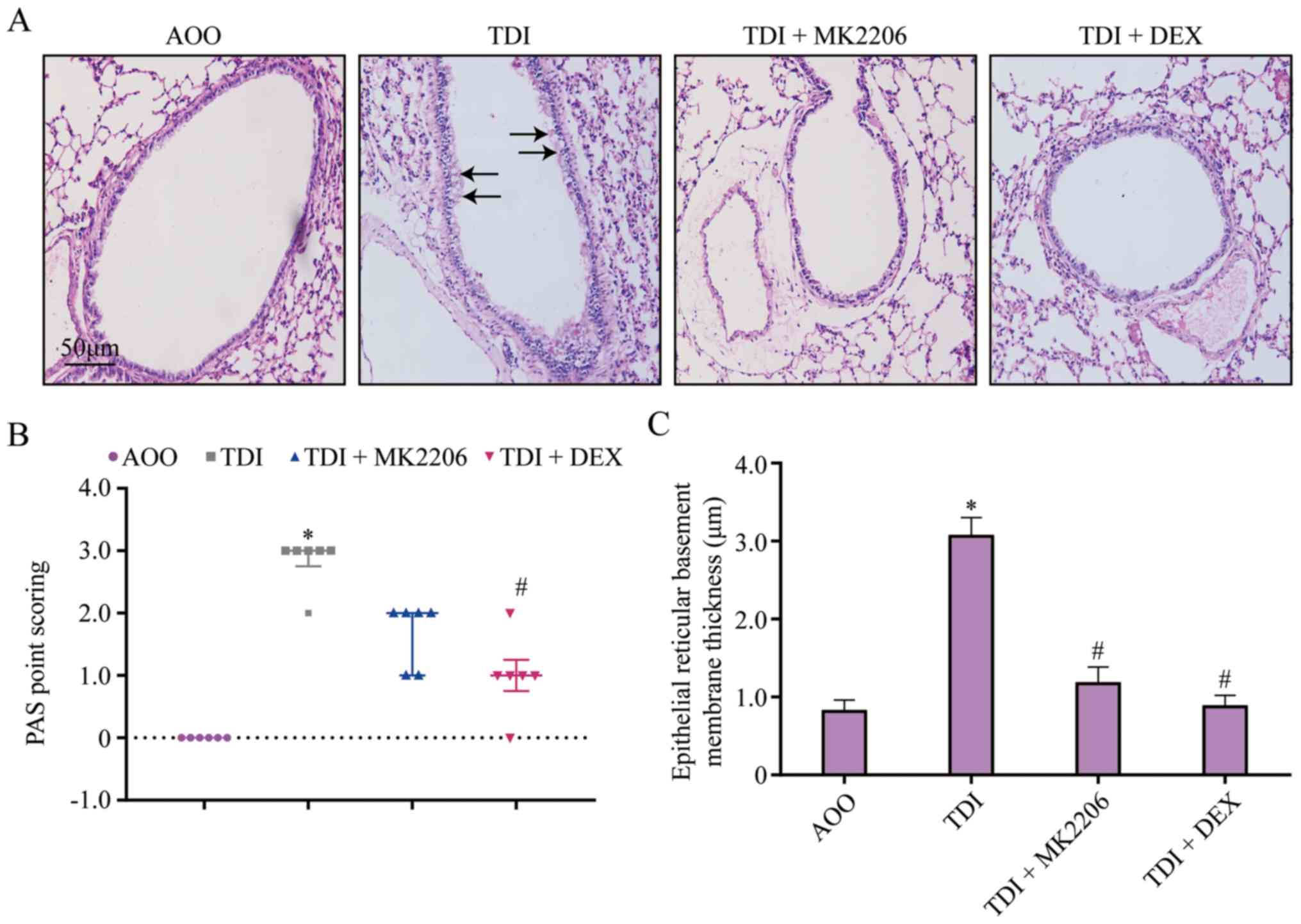
MK2206 inhibits airway remodeling
PAS staining demonstrated that goblet cells occupied
the largest proportion of cells in the TDI group, compared with the
AOO group. The proportion of goblet cells was reduced following
MK2206 and DEX treatment (Fig.
5A). In addition, the PAS score and basement membrane thickness
in the TDI group were significantly increased compared with the AOO
group (P<0.05; Fig. 5B and C).
Conversely, the PAS scores and basement membrane thickness in the
TDI + MK2206 and TDI + DEX groups were significantly decreased
compared with those of the TDI group (P<0.05; Fig. 5B and C).
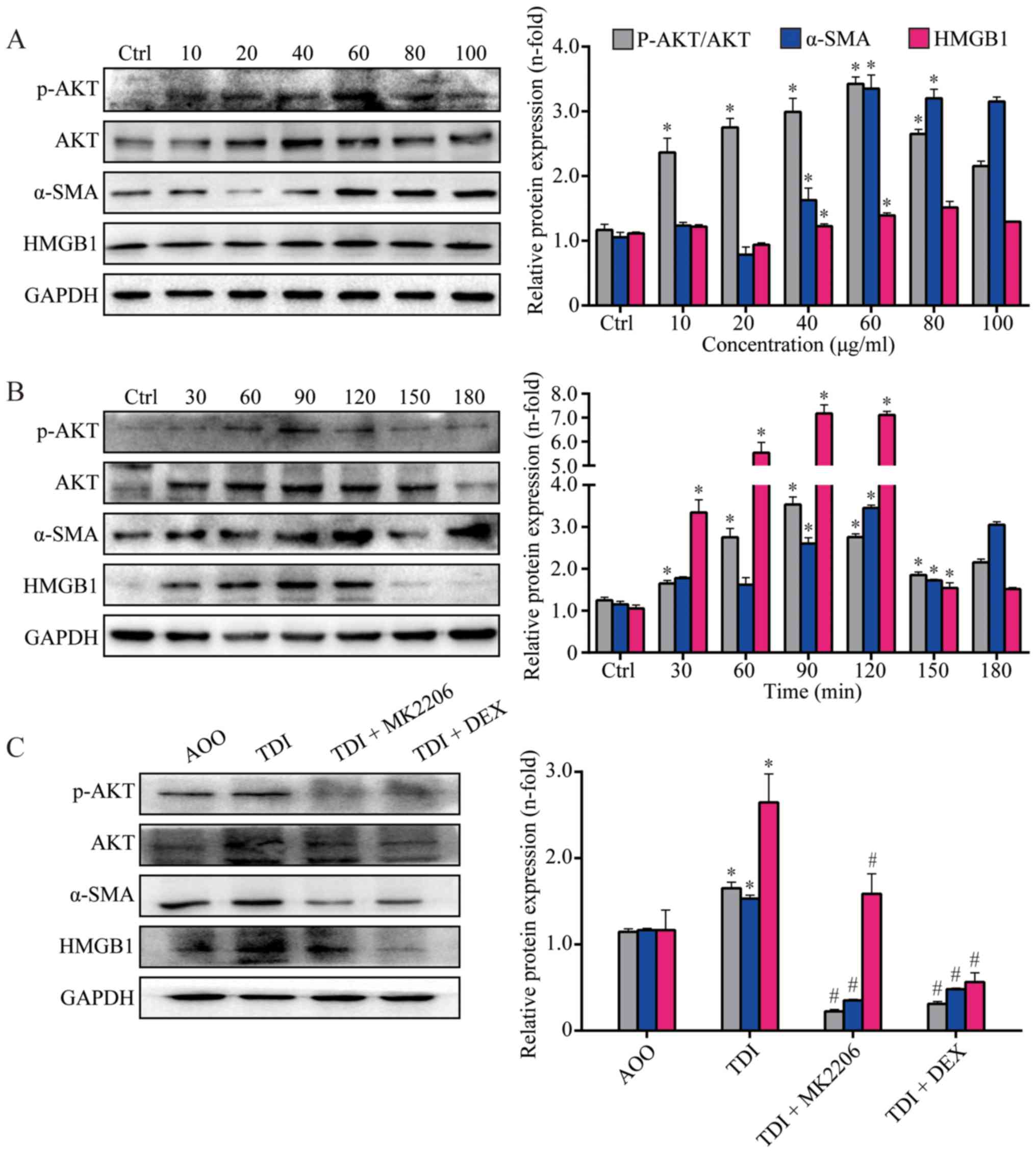
MK2206 suppresses AKT activation in
TDI-treated 16HBE cells
Results from in vitro studies demonstrated
that AKT was activated, and the expression of α-SMA and HMGB1 was
upregulated in 16HBE cells after TDI stimulation (P<0.05;
Fig. 6A and B). In addition, the
results from WB demonstrated that the activation of AKT was
inhibited, and the expression of α-SMA and HMGB1 was downregulated
by MK2206 (P<0.05; Fig. 6C). In
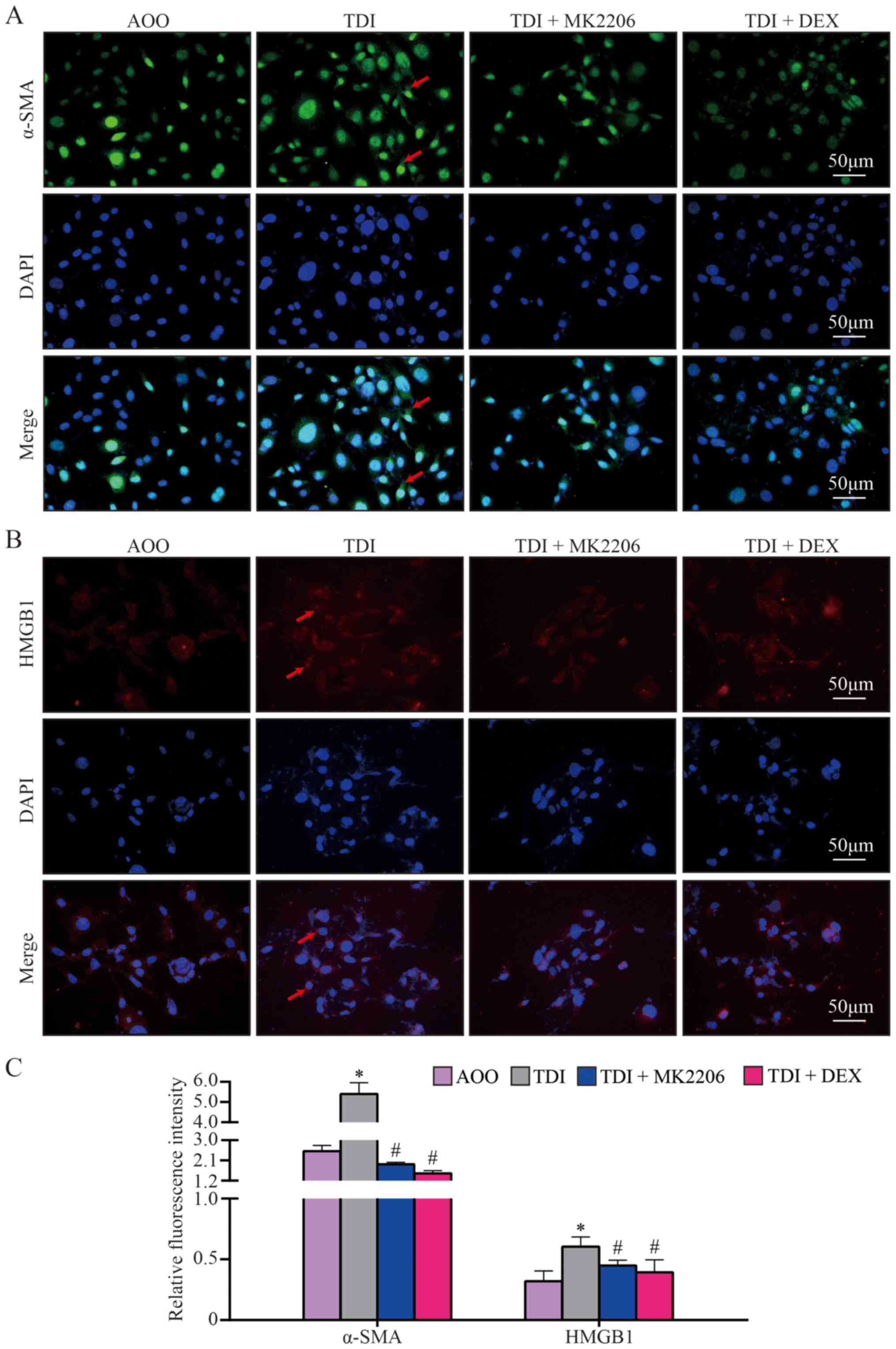
order to further elucidate the redistribution of α-SMA and HMGB1 in
16HBE cells, the distribution of α-SMA and HMGB1 were examined and
quantified. Confocal microscopy demonstrated different
distributions of α-SMA and HMGB1 between the TDI group and the
other groups, and increased α-SMA and HMGB1 translocation into the
cytosol in the TDI group, compared with all other groups
(P<0.05; Fig. 7A-C). These data
suggested that AKT phosphorylation inhibition may serve a crucial
role in TDI-induced redistribution of α-SMA and HMGB1.
Discussion
Although asthma can be currently managed by
attenuating the symptoms in most cases, 5–10% of patients still
suffer from severe asthma and cannot be treated effectively
(33). Severe asthma displays a
high rate of morbidity and mortality, and has been reported to be
responsible for 50% of all asthma-related treatment costs (34,35).
Activation of AKT can accelerate tumorigenesis and suppress tumor
invasion in transgenic mouse models (36,37).
Clinical studies mainly focus on the inhibition of AKT by MK2206 in
cancer; for example, a phase II study revealed that MK2206 +
erlotinib is an effective combination in advanced non-small cell
lung cancer (38). However, the
effect of MK2206 in asthma remains unclear. The present study aimed
to find alternative treatments with fewer side effects in the
management of asthma. The results of the present study suggested
that MK2206 may attenuate airway inflammation and remodeling
induced by TDI treatment by inhibiting AKT phosphorylation. These
findings suggested that MK2206 may be a potential drug candidate
for the treatment of asthma.
In the present study, an airway challenge with TDI
significantly elevated the levels of p-AKT and induced airway
inflammation. This inflammation was indicated by the upregulation
of inflammatory cytokines in the BALF, increased serum IgE levels
and elevated numbers of inflammatory cells. DEX was used as a
positive control to evaluate the role of MK2206 in the TDI mouse
model of asthma. Glucocorticoids, as the cornerstone of the
treatment of asthma disease, serve an important role in inhibiting
airway inflammation in asthma (35). In the TDI asthma model,
glucocorticoids can not only inhibit airway inflammation, but also
protect airway barrier function (39). However, glucocorticoids may
increase the risks of severe systemic diseases such as
osteoporosis, diabetes and obesity (8,40).
Notably, the results of the present study demonstrated that TDI
treatment resulted in airway remodeling of the lung as indicated by
goblet cell hyperplasia; it also increased the production of α-SMA
and collagen I. However, the levels of airway inflammation and
remodeling were significantly inhibited after MK2206
administration. MK2206 displayed similar inhibitory effects on AKT
phosphorylation, airway inflammation and airway remodeling as DEX,
a well-known drug recommended for the treatment of severe asthma
(39).
TDI exposure resulted in the infiltration of airway
inflammatory cells. Enhanced production of the inflammatory factors
IL-4, −5 and −13 was observed in asthma and also found to be
expressed abnormally in the TDI-induced asthma mouse model
(21). The activation of T cells
is mediated by IL-4, and IL-13 is associated with increased airway
mucous secretion and bronchial hyperresponsiveness (41,42).
IL-13 expression enhances smooth muscle strength, and results in
airway hyperresponsiveness (25)
independent of T and B cells (26). Additionally, IL-6, as a pivotal
regulator of inflammation, is elevated in the airways consistently
in children and adults with asthma. Additionally, IL-6 levels are
considered to be an indicator of forced expiratory volume in 1 sec
and forced vital capacity (24,27).
Consistent with previous studies (4,23,28),
the results of the present study indicated that p-AKT was found to
be significantly elevated in the TDI group, which demonstrated its
involvement in airway inflammation.
MK2206 inhibits the autophosphorylation of AKT and
phosphorylation of downstream signals that mediate airway
inflammation by regulating further granulocyte recruitment
(28). Thus, the inhibition of AKT
phosphorylation is one of the important steps in the signaling
pathway involved in the immune response, which regulates the
transcription activity of pro-inflammatory cytokine genes such as
IL-4 and IL-13 (43). Tang et
al (44) reported that AKT
activation increases IL-6 expression in human lung epithelial
cells, and AKT and AKT phosphorylation are both decreased after
small interfering RNA treatment. Consistent with previous reports,
the results of the present study demonstrated that MK2206 inhibited
the phosphorylation of AKT and production of cytokines (IL-4, −5,
−6 and −13) in a mouse model of TDI-induced asthma. In addition,
the present study also revealed that MK2206 inhibited the
phosphorylation of AKT in vitro. These results indicated a
potential role for MK2206 in a clinical setting for the management
of TDI-induced asthma.
AHR is reported in individuals with bronchial
remodeling and asthma (45);
reduced AHR is an important indicator of therapeutic benefit. The
present study indicated that MK2206 decreased AHR by controlling
bronchial remodeling. In a classic inflammatory microenvironment,
activated fibroblasts differentiate into myofibroblasts, and
release pro-inflammatory factors and extracellular matrix proteins
to increase airway remodeling (46,47).
The production of α-SMA is indicative of myofibroblast formation
and collagen I deposition (48).
Myofibroblasts are proposed to be the primary effectors of lung
fibrotic responses, which are characterized by the expression of
α-SMA stress fibers (48). The
results of the present study demonstrated that the number of goblet
cells and expression levels of α-SMA were greater in the asthma
group compared with those in the AOO and TDI + MK2206 groups. These
findings provided new experimental evidence supporting the
potential use of MK2206 as a new therapeutic drug for the treatment
of TDI-induced asthma.
Clinical studies and experiments using animal models
have demonstrated an important role of HMGB1 and its receptors in
airway inflammation and asthma (12,49).
MK2206 is a known antitumor agent that exerts its effect by
inhibiting p-AKT activation (50).
The present study demonstrated that MK2206 inhibited the expression
of HMGB1 in vitro and in vivo. Tumor growth factor-β
increases AKT phosphorylation (22), and MK2206 may have some effects on
this signaling pathway while reducing airway inflammation.
The present study demonstrated that the inhibition
of AKT activation using MK2206 attenuated AHR, airway inflammation,
and airway remodeling in a TDI-induced mouse model of asthma
(Fig. 8). These findings enriched
current knowledge regarding MK2206 and provided the basis for
future investigation. However, the current study has several
limitations; for example, the role of AKT signaling in asthma, and
the specific mechanism of HMGB1 in TDI-induced asthma and AKT
activation were not fully investigated.
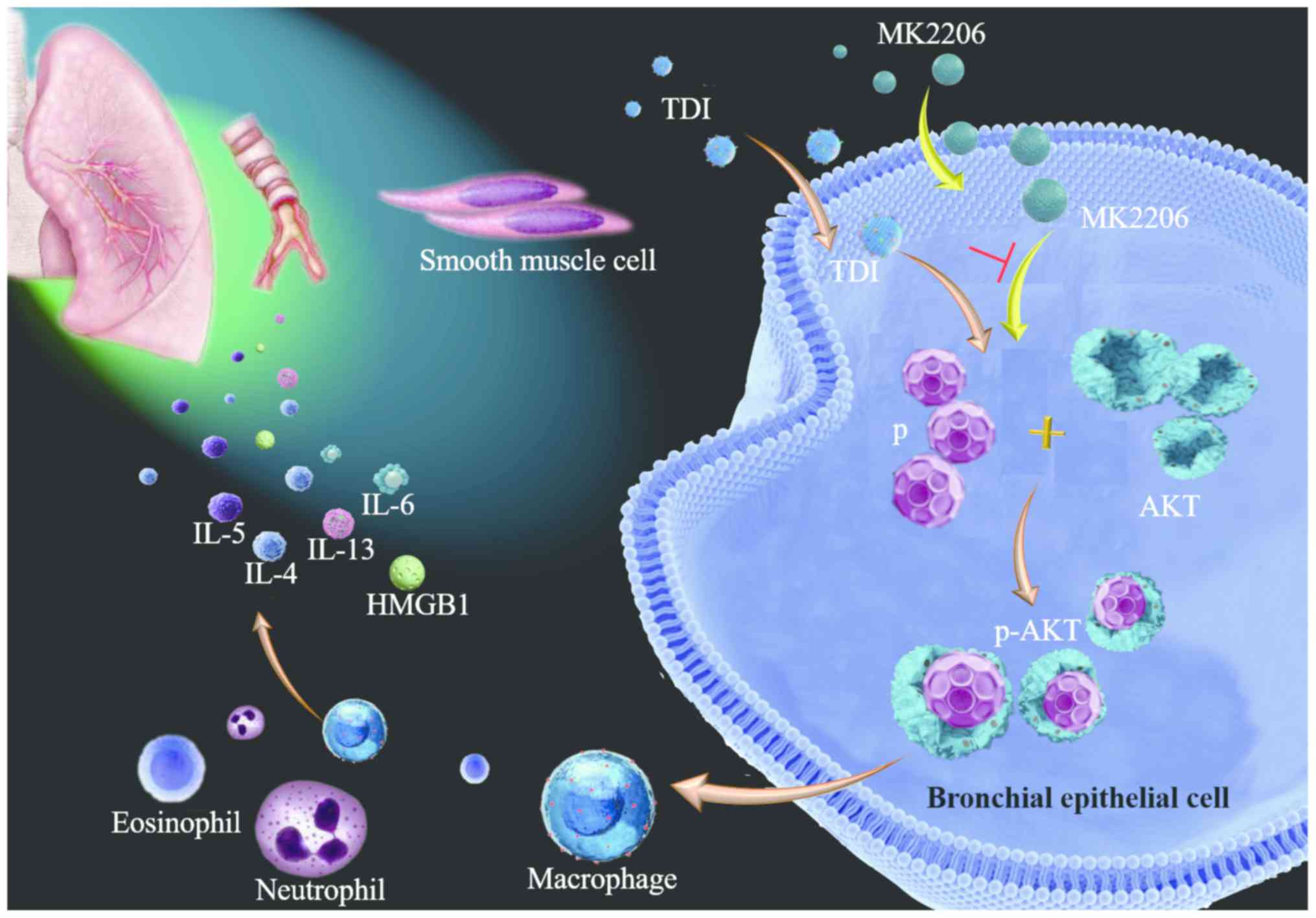 | Figure 8.Mechanism of MK2206 inhibiting
activation of AKT in TDI-induced asthma. In bronchial epithelial
cells, MK2206 suppresses the activation of AKT, which results in
the inhibition of neutrophils, eosinophil infiltration, the
expression of inflammatory mediators (IL-4, −5, −6 and −13, HMGB1),
and the production of α-smooth muscle actin and collagen I. TDI,
toluene diisocyanate; HMGB1, high mobility group box 1; p-AKT,
phosphorylated AKT; p, phosphate; IL, interleukin. |
In summary, the present study demonstrated that
MK2206 may serve a role in reversing airway inflammation and airway
remodeling in chemical-induced asthma. These findings may therefore
provide the basis for future development of MK2206 in the treatment
of occupational asthma.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural
Science Foundation of China (grant no. 81770033), a National Health
and Medical Research Council grant (grant no. 1158402), the
Scientific and Technological Project of Guangdong Province (grant
no. 2017B020226006), the National Key Research and Development Plan
of China (grant no. 2016YFC0905800), and the Science and Technology
Program of Guangzhou (grant no. 201804010069).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SC and JD conceived and designed the experiments.
HC, YC, YH, WC, WZ and HZ performed the experiments. HC, YC, LW,
FHZ and JD analyzed and interpreted the data. HC, LW, JD, SC and
FHZ drafted this manuscript. HC, SC, LW, FHZ and JD revised the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Southern Medical
University Experimental Animal Ethics Committee (approval no.
L2017177).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
α-SMA
|
α-smooth muscle actin
|
|
AHR
|
airway hyperresponsiveness
|
|
ASM
|
airway smooth muscle
|
|
PAS
|
periodic acid-Schiff
|
|
BALF
|
bronchoalveolar lavage fluid
|
|
HMGB1
|
high mobility group box 1
|
|
IL
|
interleukin
|
|
p-AKT
|
phosphorylated AKT
|
|
TDI
|
toluene diisocyanate
|
|
WB
|
western blotting
|
|
DEX
|
dexamethasone
|
References
|
1
|
Berend N, Salome CM and King GG:
Mechanisms of airway hyperresponsiveness in asthma. Respirology.
13:624–631. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chapman KR, Albers FC, Chipps B, Muñoz X,
Devouassoux G, Bergna M, Galkin D, Azmi J, Mouneimne D, Price RG
and Liu MC: The clinical benefit of mepolizumab replacing
omalizumab in uncontrolled severe eosinophilic asthma. Allergy.
74:1716–1726. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Balmes J, Becklake M, Blanc P, Henneberger
P, Kreiss K, Mapp C, Milton D, Schwartz D, Toren K, Viegi G, et al:
American Thoracic Society Statement: Occupational contribution to
the burden of airway disease. Am J Respir Crit Care Med.
167:787–797. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Padoan M, Pozzato V, Simoni M, Zedda L,
Milan G, Bononi I, Piola C, Maestrelli P, Boschetto P and Mapp CE:
Long-term follow-up of toluene diisocyanate-induced asthma. Eur
Respir J. 21:637–640. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Maestrelli P, Boschetto P, Fabbri LM and
Mapp CE: Mechanisms of occupational asthma. J Allergy Clin Immunol.
123:531–544. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Collins JJ, Anteau S, Conner PR, Cassidy
LD, Doney B, Wang ML, Kurth L, Carson M, Molenaar D, Redlich CA and
Storey E: Incidence of occupational asthma and exposure to toluene
diisocyanate in the United States Toluene Diisocyanate Production
Industry. J Occup Environ Med. 59 (Suppl 12):S22–S27. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mapp CE, Balboni A, Baricordi R and Fabbri
LM: Human leukocyte antigen associations in occupational asthma
induced by isocyanates. Am J Respir Crit Care Med. 156
(Suppl):S139–S143. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Song J, Zhao H, Dong H, Zhang D, Zou M,
Tang H, Liu L, Liang Z, Lv Y, Zou F and Cai S: Mechanism of
E-cadherin redistribution in bronchial airway epithelial cells in a
TDI-induced asthma model. Toxicol Lett. 220:8–14. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yao L, Zhao H, Tang H, Song J, Dong H, Zou
F and Cai S: Phosphatidylinositol 3-Kinase Mediates β-catenin
dysfunction of airway epithelium in a toluene diisocyanate-induced
murine asthma model. Toxicol Sci. 147:168–177. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kwak YG, Song CH, Yi HK, Hwang PH, Kim JS,
Lee KS and Lee YC: Involvement of PTEN in airway
hyperresponsiveness and inflammation in bronchial asthma. J Clin
Invest. 111:1083–1092. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Maltby S, Tay HL, Yang M and Foster PS:
Mouse models of severe asthma: Understanding the mechanisms of
steroid resistance, tissue remodelling and disease exacerbation.
Respirology. 22:874–885. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hou C, Kong J, Liang Y, Huang H, Wen H,
Zheng X, Wu L and Chen Y: HMGB1 contributes to allergen-induced
airway remodeling in a murine model of chronic asthma by modulating
airway inflammation and activating lung fibroblasts. Cell Mol
Immunol. 12:409–423. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Musumeci D, Roviello GN and Montesarchio
D: An overview on HMGB1 inhibitors as potential therapeutic agents
in HMGB1-related pathologies. Pharmacol Ther. 141:347–357. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ullah MA, Loh Z, Gan WJ, Zhang V, Yang H,
Li JH, Yamamoto Y, Schmidt AM, Armour CL and Hughes JM: Receptor
for advanced glycation end products and its ligand high-mobility
group box-1 mediate allergic airway sensitization and airway
inflammation. J Allergy Clin Immunol. 134:440–450. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang Q, Liu X, Yao Z, Mao S, Wei Q and
Chang Y: Penehyclidine hydrochloride inhibits the release of
high-mobility group box 1 in lipopolysaccharide-activated RAW264.7
cells and cecal ligation and puncture-induced septic mice. J Surg
Res. 186:310–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cockcroft DW and Davis BE: Mechanisms of
airway hyperresponsiveness. J Allergy Clin Immunol. 118:551–561.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Warner SM and Knight DA: Airway modeling
and remodeling in the pathogenesis of asthma. Curr Opin Allergy
Clin Immunol. 8:44–48. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Valatas V, Filidou E, Drygiannakis I and
Kolios G: Stromal and immune cells in gut fibrosis: The
myofibroblast and the scarface. Ann Gastroenterol. 30:393–404.
2017.PubMed/NCBI
|
|
19
|
Davies DE, Wicks J, Powell RM, Puddicombe
SM and Holgate ST: Airway remodeling in asthma: New insights. J
Allergy Clin Immunol. 111:215–226. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cao L, Liu F, Liu Y, Liu T, Wu J, Zhao J,
Wang J, Li S, Xu J and Dong L: TSLP promotes asthmatic airway
remodeling via p38-STAT3 signaling pathway in human lung
fibroblast. Exp Lung Res. 44:288–301. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Somnay Y, Simon K, Harrison AD,
Kunnimalaiyaan S, Chen H and Kunnimalaiyaan M: Neuroendocrine
phenotype alteration and growth suppression through apoptosis by
MK-2206, an allosteric inhibitor of AKT, in carcinoid cell lines in
vitro. Anticancer Drugs. 24:66–72. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yahiro Y, Maeda S, Shinohara N, Jokoji G,
Sakuma D, Setoguchi T, Ishidou Y, Nagano S, Komiya S and Taniguchi
N: PEG10 counteracts signaling pathways of TGF-β and BMP to
regulate growth, motility and invasion of SW1353 chondrosarcoma
cells. J Bone Miner Metab. 37:441–454. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang N, Zhang H, Cai X and Shang Y:
Epigallocatechin-3-gallate inhibits inflammation and
epithelial-mesenchymal transition through the PI3K/AKT pathway via
upregulation of PTEN in asthma. Int J Med. 41:818–828. 2018.
|
|
24
|
Liang J, Zhao H, Yao L, Tang H, Dong H, Wu
Y, Liu L, Zou F and Cai S: Phosphatidylinositol 3-kinases pathway
mediates lung caspase-1 activation and high mobility group box 1
production in a toluene-diisocyanate induced murine asthma model.
Toxicol Lett. 236:25–33. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lezmi G, Gosset P, Deschildre A, Abou-Taam
R, Mahut B, Beydon N and de Blic J: Airway remodeling in preschool
children with severe recurrent wheeze. Am J Respir Crit Care Med.
192:164–171. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wen X, Yan J, Han XR, Zheng GH, Tang R,
Liu LF, Wu DM, Lu J and Zheng YL: PTEN gene silencing contributes
to airway remodeling and induces airway smooth muscle cell
proliferation in mice with allergic asthma. J Thoracic Dis.
10:202–211. 2018. View Article : Google Scholar
|
|
27
|
Xiong J, Zhao W, Lin Y, Yao L, Huang G, Yu
C, Dong H, Xiao G, Zhao H and Cai S: Phosphorylation of low density
lipoprotein receptor-related protein 6 is involved in receptor for
advanced glycation end product-mediated β-catenin stabilization in
a toluene diisocyanate-induced asthma model. Int Immunopharmacol.
59:187–196. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yao L, Zhao H, Tang H, Liang J, Liu L,
Dong H, Zou F and Cai S: The receptor for advanced glycation end
products is required for beta-catenin stabilization in a
chemical-induced asthma model. Br J Pharmacol. 173:2600–2613. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lundblad LK, Irvin CG, Adler A and Bates
JH: A reevaluation of the validity of unrestrained plethysmography
in mice. J Appl Physiol. 93:1198–1207. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ye Z, Huang Y, Liu D, Chen X, Wang D,
Huang D, Zhao L and Xiao X: Obesity induced by neonatal overfeeding
worsens airway hyperresponsiveness and inflammation. PLoS One.
7:e470132012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tang H, Zhao H, Song J, Dong H, Yao L,
Liang Z, LV Y, Zou F and Cai S: Ethyl pyruvate decreases airway
neutrophil infiltration partly through a high mobility group box
1-dependent mechanism in a chemical-induced murine asthma model.
Int Immunopharmacol. 21:163–170. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Padrid P, Snook S, Finucane T, Shiue P,
Cozzi P, Solway J and Leff AR: Persistent airway
hyperresponsiveness and histologic alterations after chronic
antigen challenge in cats. Am J Respir Crit Care Med. 151:184–193.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chung KF, Wenzel SE, Brozek JL, Bush A,
Castro M, Sterk PJ, Adcock IM, Bateman ED, Bel EH, Bleecker ER, et
al: International ERS/ATS guidelines on definition, evaluation and
treatment of severe asthma. Eur Respir J. 43:343–373. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Soriano JB, Abajobir AA, Abate KH, Abera
SF, Agrawal A, Ahmed MB, Aichour AN, Aichour I, Aichour MTE, Alam
K, et al: Global, regional, and national deaths, prevalence,
disability-adjusted life years, and years lived with disability for
chronic obstructive pulmonary disease and asthma, 1990–2015: A
systematic analysis for the Global Burden of Disease Study 2015.
Lancet Respir Med. 5:691–706. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Al Efraij K and FitzGerald JM: Current and
emerging treatments for severe asthma. J Thorac Dis. 7:E522–E525.
2015.PubMed/NCBI
|
|
36
|
Hutchinson JN, Jin J, Cardiff RD, Woodgett
JR and Muller WJ: Activation of Akt-1 (PKB-alpha) can accelerate
ErbB-2-mediated mammary tumorigenesis but suppresses tumor
invasion. Cancer Res. 64:3171–3178. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chin YR and Toker A: Akt isoform-specific
signaling in breast cancer: Uncovering an anti-migratory role for
palladin. Cell Adh Migr. 5:211–214. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lara PN Jr, Longmate J, Mack PC, Kelly K,
Socinski MA, Salgia R, Gitlitz B, Li T, Koczywas M, Reckamp KL and
Gandara DR: Phase II study of the AKT Inhibitor MK-2206 plus
Erlotinib in patients with advanced non-small cell lung cancer who
previously progressed on erlotinib. Clin Cancer Res. 21:4321–4326.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Brooks SM, Werk EE, Ackerman SJ, Sullivan
I and Thrasher K: Adverse effects of phenobarbital on
corticosteroid metabolism in patients with bronchial asthma. N Engl
J Med. 286:1125–1128. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mylka V, Deckers J, Ratman D, De Cauwer L,
Thommis J, De Rycke R, Impens F, Libert C, Tavernier J, Vanden
Berghe W, et al: The autophagy receptor SQSTM1/p62 mediates
anti-inflammatory actions of the selective NR3C1/glucocorticoid
receptor modulator compound A (CpdA) in macrophages. Autophagy.
14:2049–2064. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Takeda M, Ito W, Tanabe M, Ueki S, Kihara
J, Kato H, Tanigai T, Kayaba H, Sasaki T and Chihara J: The
pathophysiological roles of PI3Ks and therapeutic potential of
selective inhibitors in allergic inflammation. Int Arch Allergy
Immunol. 152 (Suppl 1):S90–S95. 2010. View Article : Google Scholar
|
|
42
|
Kampe M, Lampinen M, Stolt I, Janson C,
Stalenheim G and Carlson M: PI3-kinase regulates eosinophil and
neutrophil degranulation in patients with allergic rhinitis and
allergic asthma irrespective of allergen challenge model.
Inflammation. 35:230–239. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chung MJ, Sohng JK, Choi DJ and Park YI:
Inhibitory effect of phloretin and biochanin A on IgE-mediated
allergic responses in rat basophilic leukemia RBL-2H3 cells. Life
Sci. 93:401–408. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tang L, Chen Q, Meng Z, Sun L, Zhu L, Liu
J, Hu J, Ni Z and Wang X: Suppression of Sirtuin-1 increases IL-6
expression by activation of the akt pathway during allergic asthma.
Cell Physiol Biochem. 43:1950–1960. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Le Cras TD, Acciani TH, Mushaben EM,
Kramer EL, Pastura PA, Hardie WD, Korfhagen TR, Sivaprasad U,
Ericksen M, Gibson AM, et al: Epithelial EGF receptor signaling
mediates airway hyperreactivity and remodeling in a mouse model of
chronic asthma. Am J Physiol Lung Cell Mol Physiol. 300:L414–L421.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Postma DS and Timens W: Remodeling in
asthma and chronic obstructive pulmonary disease. Proc Am Thoracic
Soc. 3:434–439. 2006. View Article : Google Scholar
|
|
47
|
Halwani R, Al-Muhsen S and Hamid Q: Airway
remodeling in asthma. Curr Opin Pharmacol. 10:236–245. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wang N, Yan D, Liu Y, Liu Y, Gu X, Sun J,
Long F and Jiang S: A HuR/TGF-β1 feedback circuit regulates airway
remodeling in airway smooth muscle cells. Respir Res. 17:1172016.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hou C, Zhao H, Liu L, Li W, Zhou X, Lv Y,
Shen X, Liang Z, Cai S and Zou F: High mobility group protein B1
(HMGB1) in Asthma: Comparison of patients with chronic obstructive
pulmonary disease and healthy controls. Mol Med. 17:807–815. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Winder A, Unno K, Yu Y, Lurain J and Kim
JJ: The allosteric AKT inhibitor, MK2206, decreases tumor growth
and invasion in patient derived xenografts of endometrial cancer.
Cancer Biol Ther. 18:958–964. 2017. View Article : Google Scholar : PubMed/NCBI
|