Introduction
Hepatocellular carcinoma (HCC) is the most common
type of primary liver cancer in adults (1). Worldwide, the number of new HCC
diagnoses has increased annually, making HCC the second most common
cause of cancer-related mortality (2). The survival rate of patients with HCC
remains <30% (3). Viral
infection, alcoholism, obesity and diabetes are the major risk
factors of HCC (4), which result
in chronic inflammation, leading to the destruction and
regeneration of hepatocytes, as well as genetic mutations and
dysregulation of growth signals (5,6).
Since its etiology is so complex, monotherapy has been shown to be
insufficient to cure all types of HCC (7).
The ubiquitin proteasome system (UPS) plays an
important role in biological processes, especially in tumor cells
(8), and ~95 putative
deubiquitinating enzymes have been identified in humans (9). As the reverse process of
ubiquitination, deubiquitination also serves an important role
(10,11).
Ubiquitin-specific protease 12 (USP12) belongs to
the UPS family, and promotes tumor pathogenesis through
deubiquitination (12,13). The WD repeat-containing protein,
WDR48, in combination with USP12, suppresses AKT signaling by
deubiquitinating PH domain leucine-rich repeat-containing protein
phosphatase (PHLPP) in HCT116 cells. Overexpression of USP12
induces apoptosis and suppresses the proliferation of HCT116 cells
(14). Also, USP12 regulates
prostate cancer (PC) cells via the AKT/PHLPP pathway, and
USP12-knockdown reduces cell proliferation, increases apoptosis and
causes G1 arrest in PC cells (15,16).
The cell cycle consists of a series of events, where
cellular components are doubled and then accurately segregated into
daughter cells. In eukaryotes, DNA replication is confined to
discrete synthesis in the S-phase, and chromosomal segregation
takes place at mitosis (the M-phase). The S- and M-phases are
separated by two ‘gap’ phases, called G1 and
G2. During these periods, cells acquire mass, integrate
growth signals, arrange a replicated genome, and prepare for
chromosomal segregation (17).
Such ordered progression is tightly regulated by cell cycle
checkpoints, at which the cell actively pauses proliferation until
earlier processes, such as DNA replication or mitosis, are
completed (18). These mechanisms
play an indispensable role in maintaining genomic integrity in
response to endogenous and exogenous DNA damage (19).
Materials and methods
Tissue array and
immunohistochemistry
HCC tissue arrays were obtained from the National
Engineering Center for Biochips. Immunohistochemical staining was
conducted to examine USP12 expression in the tissues using a
specific antibody against USP12. Briefly, the tissues were fixed
with 4% paraformaldehyde overnight at room temperature, embedded
with paraffin and serially cut at 4 µm. The sections were
deparaffinized as follows: Xylene 10 min, xylene 10 min, absolute
ethyl alcohol 5 min, 95% ethanol 5 min, 90% ethanol 5 min, 80%
ethanol 5 min, 70% ethanol 5 min, water 3 min and treated by sodium
citrate, and then blocked with 3% H2O2.
Sections were incubated with the rabbit anti-USP12 primary antibody
(1:150, cat. no. ab89870, Abcam) at 4°C overnight. Sections were
then treated with peroxidase-conjugated secondary antibody
(1:2,000, cat. no. ab205718, Abcam) and DAB chromogen. Then the
samples were counterstained with hematoxylin, and observed under
light microscope. The expression level was reflected by the
staining intensity and the proportion of positively-stained cells,
Positive reactions were defined as the presence of brown staining
in the cell cytoplasm, nucleus and membrane. For USP12, a staining
index (values 0–12) was determined by multiplying the score for
staining intensity (0, no staining; 1, weak staining; 2, moderate
staining; and 3, strong staining) by the score for the positive
stained area (1, positive staining in 0–25% of tumor cells; 2,
positive staining in >25–50% of tumor cells; 3, positive
staining in 51–75% of tumor cells; 4, positive staining in
>75–100% of tumor cells). Finally, scores of 0–4 were considered
low expression (−), and scores of 5–12 were considered high
expression (+) (20,21).
Cell culture
HCC cell lines (Huh7 and Hep3b) and 293T cells were
purchased from the Shanghai Institutes of the Chinese Academy of
Sciences. All cells were maintained in DMEM high glucose (Gibco;
Thermo Fisher Scientific, Inc.) containing 10% fetal bovine serum
(Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml penicillin and
100 µg/ml streptomycin (complete medium) at 37°C in a humidified
atmosphere containing 5% CO2. SB202190 (MedChemExpress),
an inhibitor of the p38 pathway, was added into the cells at 20 µM
concentration and incubated for 24 h to determine the effect of
USP12 on the p38 pathway.
Cell proliferation analysis
Cell proliferation was determined by multiparametric
high content screening. HCC cells were infected with either
negative control (NC) or USP12 short hairpin RNA (shRNA,
5′-CCGGCCAGATGTCTTACTTGTGAAACTATTCTCGAGAATAGTTTCACAAGTAAGACATCTGGTTTTTG-3′)
lentiviruses (Shanghai GeneChem Co., Ltd.). Transfected cells were
seeded into 96-well plates at a density of 2×103
cells/well, followed by incubation in complete medium at 37°C for 5
days. The Cellomics assay was carried out on an ArrayScan High
Content Platform (cat. no. ASN00004F; Thermo Fisher Scientific,
Inc.). The infected cells were identified, and the intensity and
distribution of fluorescence in each cell were reported (>800
cells were examined). The acquired images and obtained data were
stored in a Microsoft SQL database (https://www.compuwork.ca/canada/bc/vancouver/database/expert/microsoft/SQL_Server.aspx).
USP12-knockdown in HCC cell lines
The lentiviral vector pGCSIL-GFP (Shanghai GeneChem
Co., Ltd.) was digested using the restriction enzymes AgeI
and EcoRI. Subsequently, small interfering RNA (siRNA)
targeting the USP12 sequence (knockdown,
5′-CCAGAUGUCUUACUUGUGAAACUAU-3′) and a non-silencing sequence
(5′-TTCTCCGAACGTGTCACGT-3′) were transformed into shRNA
(stem-loop-stem structure), followed by cloning into the
predigested lentiviral vector. The recombinant plasmid and two
viral plasmids (Shanghai GeneChem Co., Ltd.) were transfected into
the human 293T cells using Lipofectamine® 3000
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocols. Transfected cells were incubated for 3
days at 37°C, and lentiviral particles was collected from the
culture medium. For stable infection, 10,000 HCC cells were seeded
into 6-well plates, followed by infection with the USP12
shRNA-expressing (USP12-shRNA) or non-silencing shRNA-expressing
lentivirus (control) at a multiplicity of infection of 10.
Tumor xenografts
A total of 10 female BALB/c nude mice (4–6 weeks
old, 18–20 g weight) were purchased from the Model Animal Research
Centre of Nanjing University. The animals were housed in a
purpose-built designated pathogen-free facility under standard
conditions maintained in standard conditions (temperature, 18–22°C;
humidity, 50–60%; 12-h light/dark cycle). The mice were provided
with ad libitum access to food and water. The animal-related
protocols were approved by the Institutional Animal Care and Use
Committee of Wannan Medical College. Briefly, cell suspensions
containing 2×106 tumor cells in 0.2 ml serum-free DMEM
(Gibco; Thermo Fisher Scientific, Inc.) were subcutaneously
injected into the flank of each mouse. Tumor cells containing
USP12-shRNA or control tumor cells were injected into the right
flank (five mice in each group). Tumor growth was recorded by
determining the length (L) and width (W) of each tumor with a
caliper, and the formula L × W2 × (π/6) was employed to
calculate the tumor size. If the diameter of the largest tumor was
>15 mm, the mice were deemed as ineffective, although this did
not occur in the present study. The total duration of the xenograft
experiment was 22 days, and the mice were anesthetized by the
intraperitoneal injection of pentobarbital sodium (60 mg/kg) and
then euthanized by carbon dioxide asphyxiation at the end of the
experiment. The flow rate of carbon dioxide used for euthanasia did
not displace >30% of the chamber volume/minute.
Flow cytometry analysis
Cells (1×106) undergoing lentiviral
transfection were harvested and washed twice with cold PBS,
followed by fixation in cold 70% ethanol overnight. Fixed cells
were resuspended in PBS, and the cell suspension was filtered
through a 400-mesh membrane. The cells were stained with propidium
iodide (PI) (eBioscience; Thermo Fisher Scientific, Inc.) for 1 h
at room temperature for cell cycle analysis, and then analyzed
using a BD FACSCalibur flow cytometer (BD Biosciences). Each
experiment was carried out at least in triplicate.
Western blotting
Tumor cells were lysed in RIPA buffer (Nanjing
KeyGen Biotech Co., Ltd., cat. no. KGP702) containing fresh 1 mM
protease inhibitor (Nanjing KeyGen Biotech Co., Ltd., cat. no.
KGP603) and 1 mM phosphatase inhibitor (Nanjing KeyGen Biotech Co.,
Ltd., cat. no. KGP602). The protein concentration was measured
using a bicinchoninic acid assay kit (Pierce; Thermo Fisher
Scientific, Inc.). Equal amounts of protein (20 µg) were subjected
to 10% SDS-PAGE and then electro-transferred onto a PVDF membrane.
The blot was incubated in 10 mM Tris-HCl (pH 7.4), consisting of 3%
BSA (Nanjing KeyGen Biotech Co., Ltd., cat. no. KGY00810) and 0.05%
Tween-20 at room temperature for 2 h, to block non-specific
bindings. Subsequently, the membrane was incubated with primary
antibody at 4°C for 12 h, followed by incubation with a
corresponding peroxidase-conjugated secondary antibody (Santa Cruz
Biotechnology) at room temperature for 2 h. The immunoreactive
bands were visualized using the SuperSignal West Pico
Chemiluminescent Substrate (Pierce; Thermo Fisher Scientific,
Inc.), and the band density was quantified by densitometric
analysis and using GAPDH as normalization control by a Versadoc
Imaging System Model 3000 (Bio-Rad Laboratories, Inc.) at room
temperature for 1 min. The details of primary and secondary
antibodies used in the experiment are shown in Table SI. The experiment was conducted ≥3
times.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the HCC cell lines
using TRIzol® (Invitrogen; Thermo Fisher Scientific,
Inc.) following the manufacturer's instructions. RT-qPCR was
performed as reported previously (7). Briefly, 1 µg of total RNA was reverse
transcribed using random primers and Primescript reverse
transcriptase (Vazyme). qPCR reactions for the indicated genes were
carried out using the SYBR-Green qPCR kit (Vazyme) on a fluorescent
temperature cycler (ABI 7500 Real-Time PCR system; Thermo Fisher
Scientific, Inc.). The following primers were used to detect the
expression of USP12 forward, 5′-AGACCTTTCTGTTGACGTGGA-3′ and
reverse, 5′-TGTTTGCTGCGACACTCTTC-3′; and GAPDH forward,
5′-TGACTTCAACAGCGACACCCA-3′ and reverse,
5′-CACCCTGTTGCTGTAGCCAAA-3′. Briefly, after an initial denaturation
step at 95°C for 5 min, 45 cycles of amplification were carried out
with 45 cycles at 95°C for 15 sec, and an annealing temperature of
60°C for 1 min. GAPDH was selected as the housekeeping gene, and
the relative gene expression of the target gene was determined by
the 2−ΔΔCq method (13). The experiment was repeated ≥3
times.
Statistical analyses
All statistical analyses were performed using SPSS
18.0 software (SPSS, Inc.) and the data were expressed as mean ±
standard deviation. Differences among categorical variables were
analyzed using the χ2 test. The expression levels of
USP12 in xenografted tumors were analyzed by the independent sample
Student's t-test. The immunoreactive scores for USP12 for tissue
arrays were analyzed using the non-parametric Mann-Whitney U,
Kruskal-Wallis H and Wilcoxon tests followed by the Dunn's test
post hoc test. Multiple groups were analyzed with one-way ANOVA
followed by the Tukey post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
USP12 expression in human HCC
tissues
The array consisted of 90 HCC patient tissue
samples, including tumor tissues and paired-adjacent normal
tissues. Of these 90 patients, 53 were male and 37 female, with a
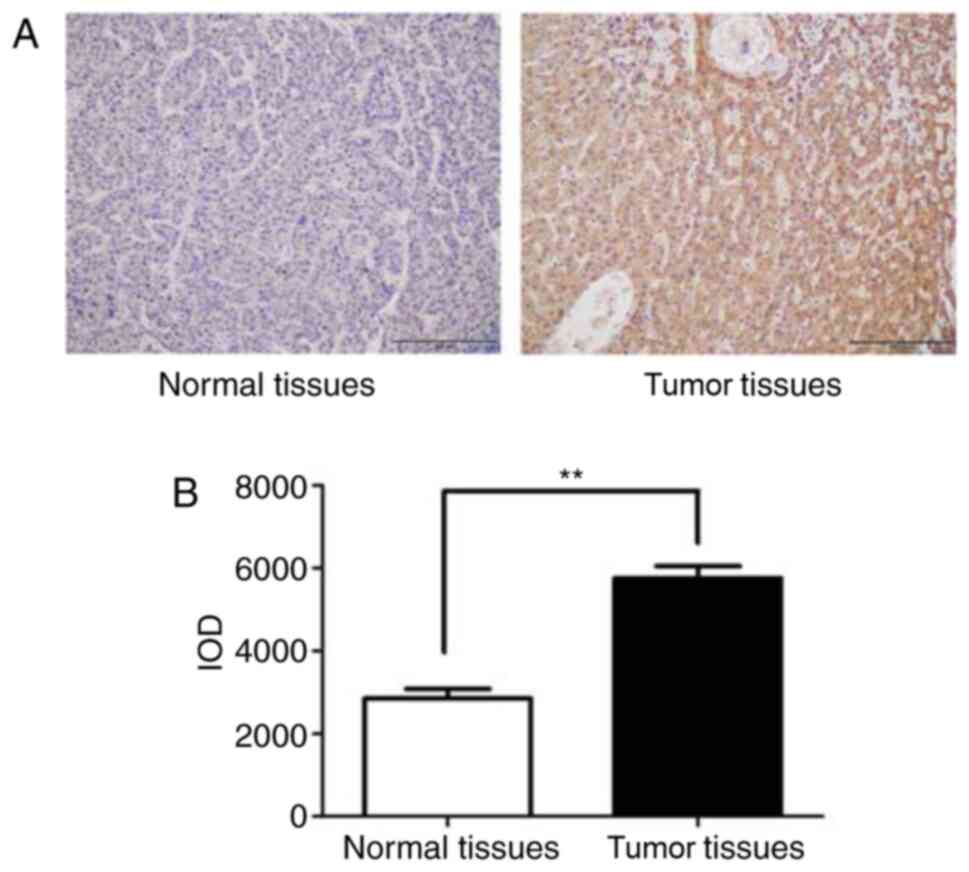
median age of 68 years (range, 25–87 years). Table I and Fig. 1A show that USP12 was highly
expressed in HCC tumor tissue samples By contrast, the USP12
expression in adjacent normal tissue samples was significantly
lower than that in the tumor tissues (Table I; P<0.001). Moreover, USP12 high
expression may indicated poor differential HCC, and the expression
level of USP12 was not associated with other factors. However,
integrated optical density analysis revealed that the USP12
expression was lower in tumor tissues compared with that in
corresponding normal tissues (Fig.
1B).
 | Table I.Expression of USP12 in human liver
cancer tissue array. |
Table I.
Expression of USP12 in human liver
cancer tissue array.
|
|
| USP12
expression |
|
|---|
|
|
|
|
|
|---|
| Characteristic | Patients
(n=90) | High | Low | P-value |
|---|
| Gender |
|
|
| 0.890 |
|
Male | 53 | 28 | 25 |
|
|
Female | 37 | 19 | 18 |
|
| Age |
|
|
| 0.203 |
|
≤65 | 40 | 23 | 17 |
|
|
>65 | 50 | 22 | 28 |
|
| Tumor Size |
|
|
| 0.209 |
| ≤5
cm | 44 | 20 | 24 |
|
| >5
cm | 46 | 27 | 19 |
|
|
Differentiation |
|
|
| 0.034a |
|
I/II | 32 | 13 | 19 |
|
|
III | 58 | 39 | 19 |
|
| TNM Stage |
|
|
| 0.249 |
|
I/II | 38 | 18 | 20 |
|
|
III/IV | 52 | 31 | 21 |
|
| Location |
|
|
|
<0.0001b |
| Tumor
tissue | 90 | 67 | 23 |
|
|
Adjacent tissue | 90 | 31 | 59 |
|
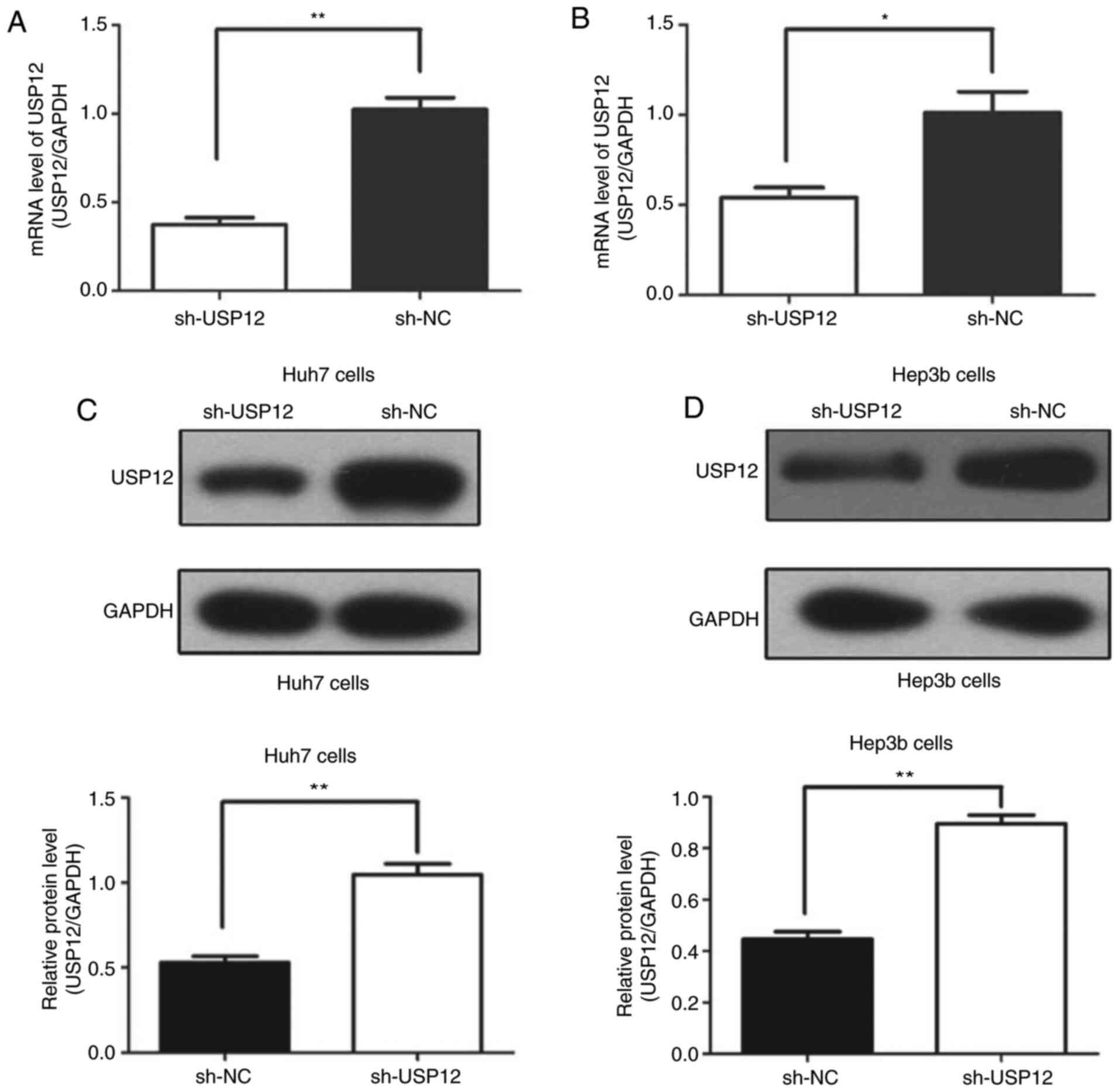
Efficiency of USP12-knockdownin HCC
cell lines in vitro
In the present study, USP12 expression in HCC cell
lines was knocked down by shRNA in order to assess its role in the
tumorigenesis of HCC. Fig. 2A and
C show that the USP12 expression at both the mRNA and protein
levels were significantly decreased in the target cells compared
with those in the control cells after transfection in Huh7 cells; a
similar effect was observed in Hep3b cells (Fig. 2B and D).
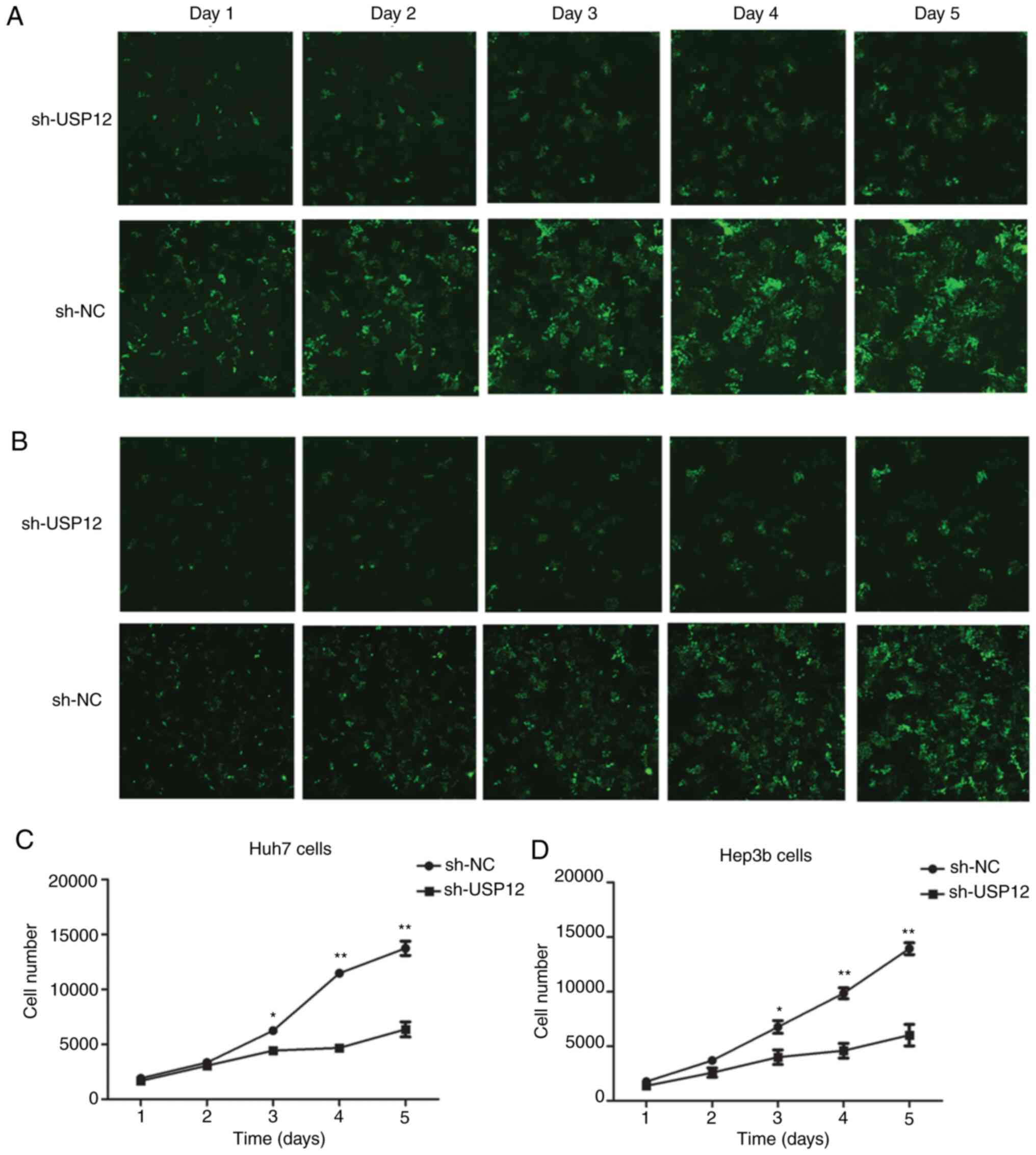
USP12-knockdown inhibits the
proliferation of HCC cell lines in vitro
To investigate the effect of USP12 on cell
proliferation, HCC cells transfected with either the USP12-shRNA or
the NC lentiviral vector were examined by Cellomics assay. The
proliferation of HCC cells containing NC or USP12-shRNA was
determined each day for 5 days (Fig.
3A and B). The results revealed that proliferation was
inhibited following USP12-knockdown (Fig. 3C and D). Therefore, these findings
suggest that USP12-knockdown suppressed the proliferation of HCC
cells.
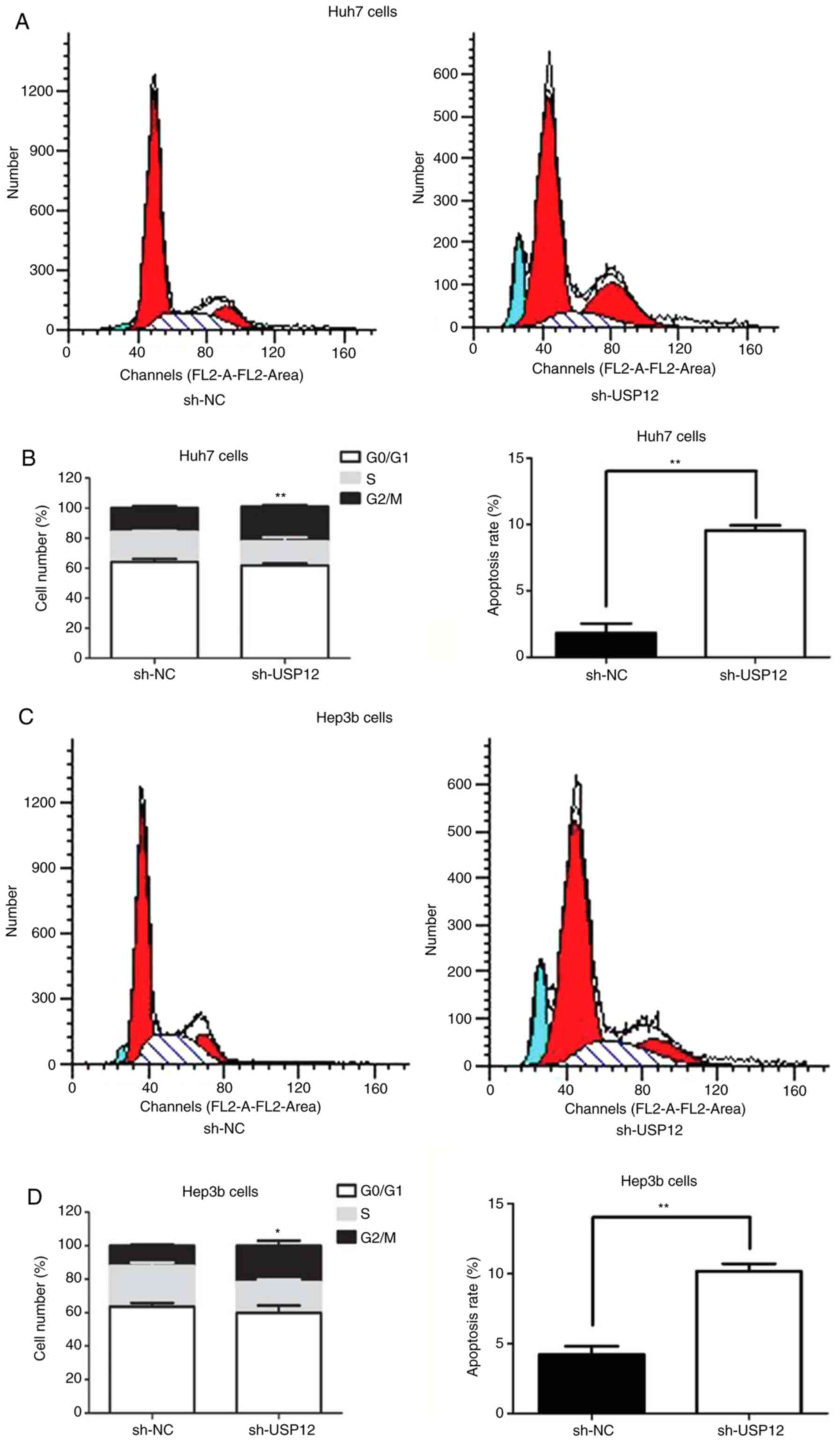
The regulatory role of USP12 in the proliferation of
HCC cells was also assessed by flow cytometry. The results
indicated that USP12-knockdown resulted in a reduced proportion of
cells in the G0/G1 phase and an increased
proportion in the G2/M phase, and that apoptosis was
increased after USP12-knockdown (Fig.
4A and D). These results suggest that USP12 induces apoptosis
and G2/M arrest in HCC cells.
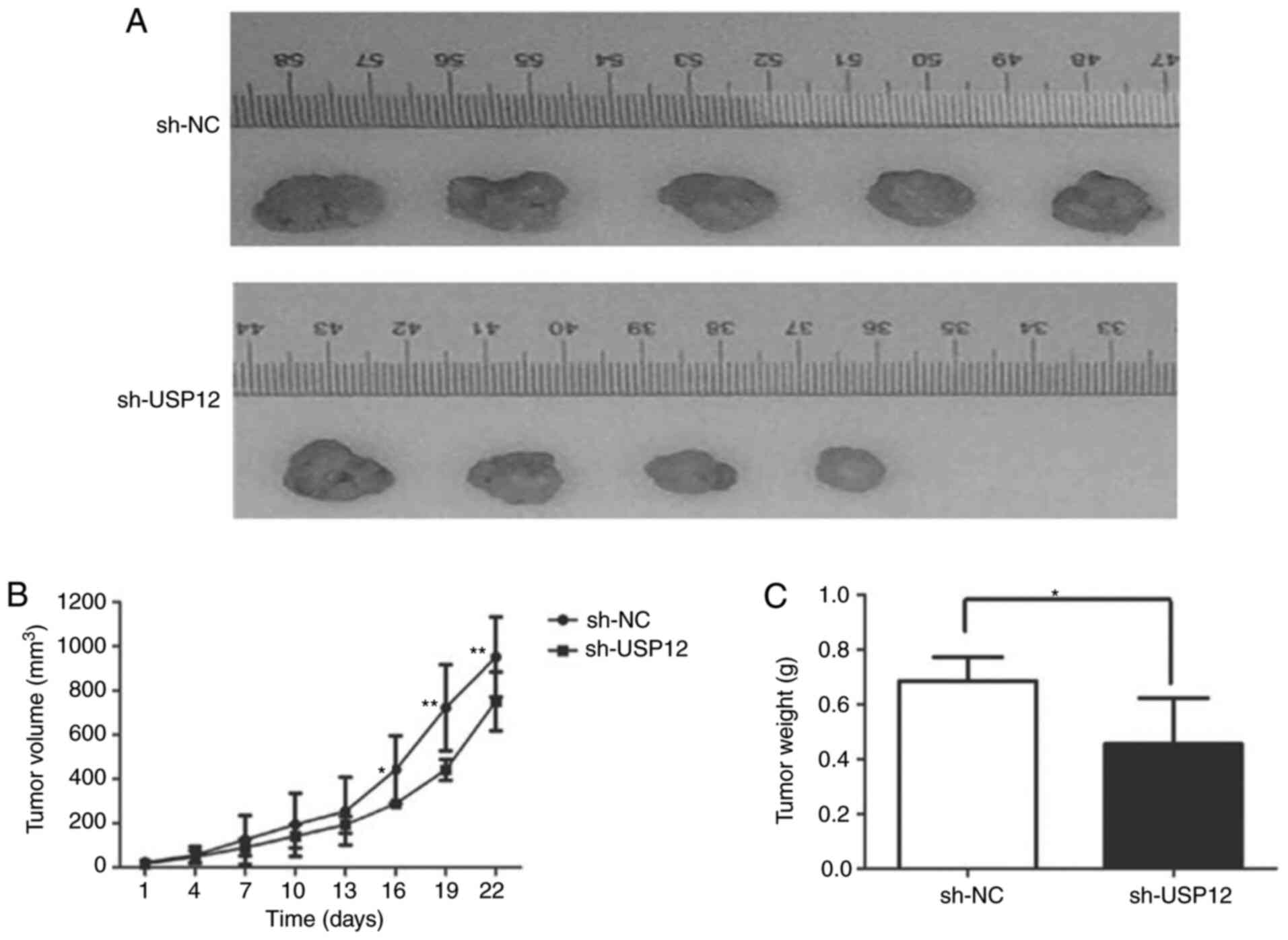
USP12-knockdown inhibits the growth of
HCC cells in vivo
To explore the mechanisms underlying the regulatory
effect of USP12 on cell proliferation in vivo, Huh7 cells
containing USP12 shRNA or non-silencing RNA were injected into nude
mice. The weight and volume of tumors were significantly decreased
after USP12-knockdown, which suggests that USP12-knockdown is
associated with decreased tumor growth (Fig. 5).
USP12 induces apoptosis via the
mitogen-activated protein kinase (MAPK) pathway, and inhibits the
proliferation of HCC cells, via G2/M arrest
Since the apoptotic rate of HCC cells was
significantly increased after USP12-knockdown, it was hypothesized
that USP12 regulated cell growth via the p38/MAPK pathway.
Phosphorylated p38 (p-p38) and phosphorylated JNK (p-JNK) were
activated after USP12-knockdown, and inhibition of the MAPK pathway
could eliminate such activation. Furthermore, the apoptosis
markers, cleaved-caspase 9 and cleaved-caspase 3, were upregulated
following USP12-knockdown (Fig.
6A). Therefore, it was concluded that USP12 regulated the
proliferation of HCC cells, at least partly, through the p38/MAPK
pathway. The MAPK pathway is an important pathway in the cell
cycle, and it can induce a wide variety of downstream events,
including apoptosis and G2/M arrest (22). The molecular mechanisms underlying
the regulatory effect of USP12 on cell proliferation were further
investigated and the expression of cell cycle proteins was
determined. Fig. 6B displays that
the expression of M-phase inducer phosphatase 3 (Cdc25c), Cdk1 and
cyclin B1 were all decreased after USP12-knockdown. Fig. 6B shows that inhibition of the
p38/MAPK pathway rescued the activation of the p38/MAPK pathway
induced by the downregulation of USP12.
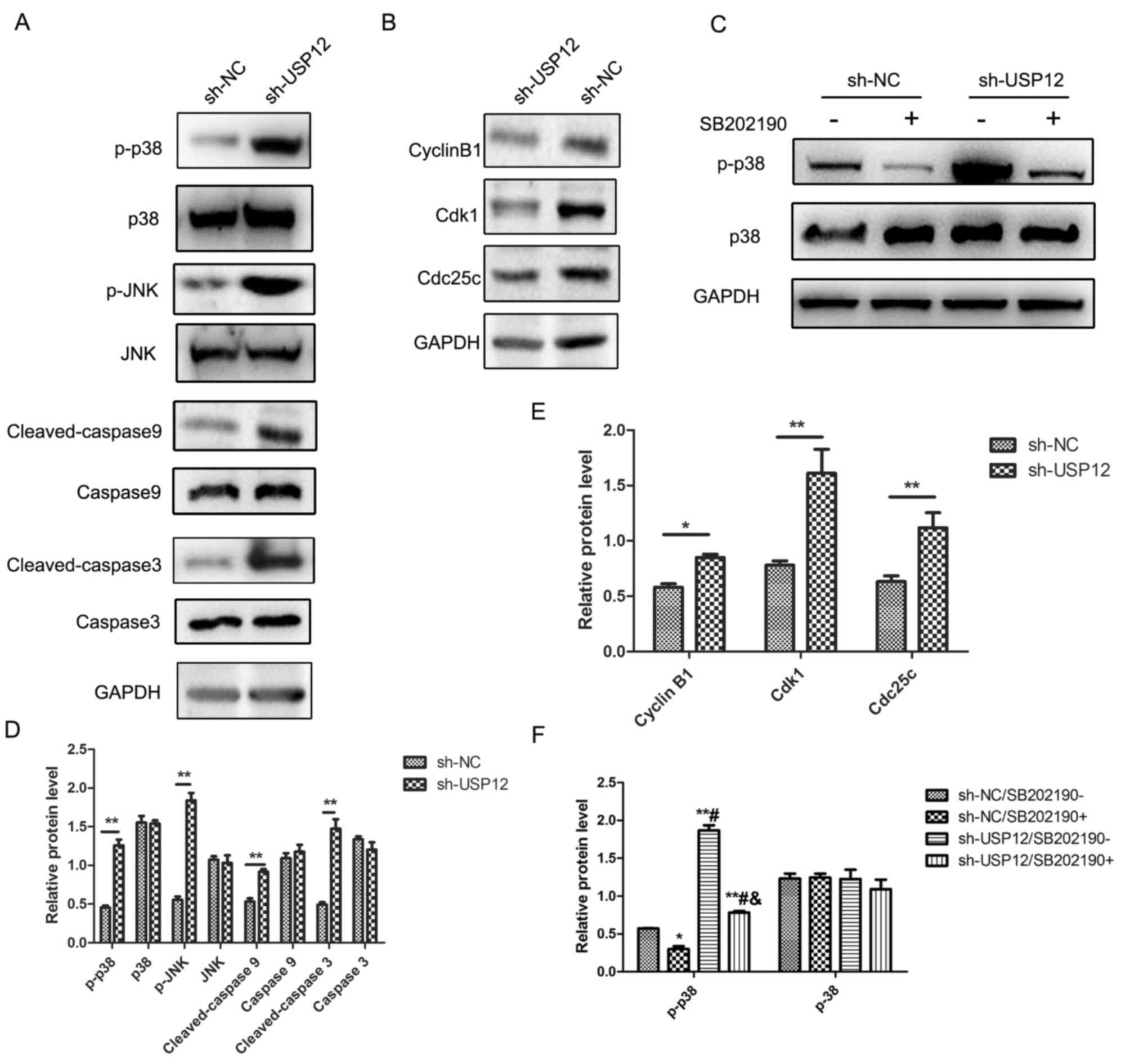 | Figure 6.Mechanisms of USP12 regulation of
tumor growth and apoptosis. Western blot analysis was carried out
to detect the expression of cell cycle checkpoint proteins and
apoptosis markers in Huh7 cells. (A) The level of p-p38 and p-JNK
were upregulated while the expression levels of p38 and JNK were
not changed after USP12-knockdown. The levels of cleaved-caspase 9
and cleaved-caspase 3 were upregulated after USP12-knockdown, while
caspase 9 and caspase 3 were not changed after USP12-knockdown. (B)
The level of cyclin B1, Cdk1 and Cdc25c was decreased after
USP12-knockdown. GAPDH was used as an internal control. (C) The
level of p-p38 was downregulated after addition of SB202190 (20
µM), an inhibitor of the p38/MAPK pathway. SB202190 decreased the
upregulation of p-p38 after USP12-knockdown. (D) Statistical
analysis of (A), **P<0.01 vs. the corresponding group. (E) The
statistical analysis of (B), *P<0.05 vs. the corresponding
group, **P<0.01 vs. the corresponding group. (F) The statistical
analysis of (C), *P<0.05 vs. sh-NC/SB202190-, **P<0.01 vs.
sh-NC/SB202190-. #P<0.01 vs. sh-NC/SB202190+ and
&P<0.01 vs. sh-USP12/SB202190-. USP12,
ubiquitin-specific protease 12; p-, phosphorylated; Cdc25c, M-phase
inducer phosphatase 3; MAPK, mitogen-activated protein kinase; sh,
short hairpin; NC, negative control. |
Discussion
As one of the most commonly detected tumors
worldwide, HCC has a high mortality rate, particularly in China
(23). Identification of novel
target genes for further research is in progress. The present study
found that USP12 expression was higher in human HCC tissues
compared with the adjacent normal tissues. In vitro and
in vivo experiments showed that USP12-knockdown inhibited
the proliferation of HCC cells. Moreover, the molecular mechanisms
underlying the regulatory role of USP12 were also investigated.
Taken together, these findings provide evidence that USP12 may be
used as a candidate for therapeutic intervention during HCC.
The correct timing of cell cycle events is regulated
by cell-cycle checkpoints (24).
Consequently, checkpoint blockade results in cell-cycle arrest and
alters cellular proliferation. The results of the present study
showed that USP12-knockdowninhibited cellular proliferation through
cell cycle arrest. A previous study showed that USP12-knockdown may
inactivate G0/G1 arrest in HeLa cells
(25). A similar effect has also
been observed in PC, and USP12 deficiency leads to decreased
proliferative ability, as well as increased apoptosis and
G1 arrest (15). The
present study showed that USP12-knockdown induced G2/M
arrest in HCC cell lines. In addition, USP12-knockdownincreased the
levels of apoptosis in HCC cell lines. Furthermore, the effect of
USP12 on the growth of HCC cells was also investigated in
vivo. The results showed that tumor growth was significantly
inhibited by USP12-knockdown.
Since the data from the present study showed that
USP12-knockdown induced G2/M arrest and apoptosis, the
expression of cell cycle checkpoint proteins was determined by
western blotting. The results were consistent with cell cycle
analysis; Cdc25c, Cdk1 and cyclin B1 were all significantly
downregulated after USP12-knockdown. Previous studies have
indicated that Cdc25c can be inactivated by p38, leading to
initiated G2/M cell cycle transition (26,27),
and that p38 activation can induce the apoptosis cascade (28–30).
Furthermore, the present study also assessed the status of the
p38/MAPK pathway, and found that it was activated by
USP12-knockdown. It was also found that activation of the p38/MAPK
pathway was reversed by SB202190. These results reveal that USP12
regulates the proliferation of HCC cells primarily via the p38/MAPK
pathway.
Collectively, by using in vitro and in
vivo approaches, the present study provided evidence that
USP12-knockdowninhibited tumor growth in human HCC by inducing
G2/M arrest and apoptosis, at least partly, via the
p38/MAPK pathway. In addition, USP12 expression was associated with
the pathological tumor stage of HCC, suggesting that USP12 could be
considered as a promising therapeutic target for HCC.
Supplementary Material
Supporting Data
Acknowledgements
We thank Dr Nanlin Jiao (Department of Pathology,
Yijishan Hospital of Wannan Medical College) for the kind
assistance in immunohistochemistry statistical analysis.
Funding
The present study was supported by the Key Project
of Anhui University Natural Science Research (grant no.
KJ2017A260).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CL, XL and MC performed the experiments. GF, FL and
GZ analyzed the data. YL and CL designed the experiments and wrote
the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The human samples used in the present study were
purchased from Shanghai Outdo Biotech (Shanghai, China). The
samples of this array were obtained from Taizhou Hospital of
Zhejiang Province, and the use of this array was approved by the
ethics committee of Taizhou Hospital of Zhejiang Province.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bellissimo F, Pinzone MR, Cacopardo B and
Nunnari G: Diagnostic and therapeutic management of hepatocellular
carcinoma. World J Gastroenterol. 21:12003–12021. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Goh GB, Chang PE and Tan CK: Changing
epidemiology of hepatocellular carcinoma in Asia. Best Pract Res
Clin Gastroenterol. 29:919–928. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dutta R and Mahato RI: Recent advances in
hepatocellular carcinoma therapy. Pharmacol Ther. 173:106–117.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu Z, Tu K and Liu Q: Effects of
microRNA-30a on migration, invasion and prognosis of hepatocellular
carcinoma. FEBS Lett. 588:3089–3097. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li C, Chen J, Zhang K, Feng B, Wang R and
Chen L: Progress and prospects of long noncoding RNAs (lncRNAs) in
hepatocellular carcinoma. Cell Physiol Biochem. 36:423–434. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Makita Y, Murata S, Katou Y, Kikuchi K,
Uejima H, Teratani M, Hoashi Y, Kenjo E, Matsumoto S, Nogami M, et
al: Anti-tumor activity of KNTC2 siRNA in orthotopic tumor model
mice of hepatocellular carcinoma. Biochem Biophys Res Commun.
493:800–806. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ciechanover A: The ubiquitin-proteasome
proteolytic pathway. Cell. 79:13–21. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Eletr ZM and Wilkinson KD: Regulation of
proteolysis by human deubiquitinating enzymes. Biochim Biophys
Acta. 1843:114–128. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sato Y, Yamagata A, Goto-Ito S, Kubota K,
Miyamoto R, Nakada S and Fukai S: Molecular basis of Lys-63-linked
polyubiquitination inhibition by the interaction between human
deubiquitinating enzyme OTUB1 and ubiquitin-conjugating enzyme
UBC13. J Biol Chem. 287:25860–25868. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Reyes-Turcu FE and Wilkinson KD:
Polyubiquitin binding and disassembly by deubiquitinating enzymes.
Chem Rev. 109:1495–1508. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Youle RJ and van der Bliek AM:
Mitochondrial fission, fusion, and stress. Science. 337:1062–1065.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Komander D, Clague MJ and Urbé S: Breaking
the chains: Structure and function of the deubiquitinases. Nat Rev
Mol Cell Biol. 10:550–563. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
McClurg UL, Summerscales EE, Harle VJ,
Gaughan L and Robson CN: Deubiquitinating enzyme Usp12 regulates
the interaction between the androgen receptor and the Akt pathway.
Oncotarget. 5:7081–7092. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Burska UL, Harle VJ, Coffey K, Darby S,
Ramsey H, O'Neill D, Logan IR, Gaughan L and Robson CN:
Deubiquitinating enzyme Usp12 is a novel co-activator of the
androgen receptor. J Biol Chem. 288:32641–32650. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wei R, Liu X, Yu W, Yang T, Cai W, Liu J,
Huang X, Xu GT, Zhao S, Yang J and Liu S: Deubiquitinases in
cancer. Oncotarget. 6:12872–12889. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stewart ZA and Pietenpol JA: Cell cycle
checkpoints as therapeutic targets. J Mammary Gland Biol Neoplasia.
4:389–400. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Medema RH and Macůrek L: Checkpoint
control and cancer. Oncogene. 31:2601–2613. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li F, Huang J, Ji D, Meng Q, Wang C, Chen
S, Wang X, Zhu Z, Jiang C, Shi Y, et al: Utility of urinary
circulating tumor DNA for EGFR mutation detection in different
stages of non-small cell lung cancer patients. Clin Transl Oncol.
19:1283–1291. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu Q, Li L, Han C, Wei L, Kong L and Lin
F: Sigma-1 receptor (σ1R) is downregulated in hepatic malignant
tumors and regulates HepG2 cell proliferation, migration and
apoptosis. Oncol Rep. 39:1405–1413. 2018.PubMed/NCBI
|
|
21
|
Yuan X, Sun X, Shi X, Jiang C, Yu D, Zhang
W, Guan W, Zhou J, Wu Y, Qiu Y and Ding Y: USP39 promotes the
growth of human hepatocellular carcinoma in vitro and in
vivo. Oncol Rep. 34:823–832. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nagappan A, Lee WS, Yun JW, Lu JN, Chang
SH, Jeong JH, Kim GS, Jung JM and Hong SC: Tetraarsenic hexoxide
induces G2/M arrest, apoptosis, and autophagy via PI3K/Akt
suppression and p38 MAPK activation in SW620 human colon cancer
cells. PLoS One. 12:e01745912017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sun XT, Yuan XW, Zhu HT, Deng ZM, Yu DC,
Zhou X and Ding YT: Endothelial precursor cells promote
angiogenesis in hepatocellular carcinoma. World J Gastroenterol.
18:4925–4933. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hegele A, Kamburov A, Grossmann A, Sourlis
C, Wowro S, Weimann M, Will CL, Pena V, Lührmann R and Stelzl U:
Dynamic protein-protein interaction wiring of the human
spliceosome. Mol Cell. 45:567–580. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tang LJ, Li Y, Liu YL, Wang JM, Liu DW and
Tian QB: USP12 regulates cell cycle progression by involving c-Myc,
cyclin D2 and BMI-1. Gene. 578:92–99. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bulavin DV, Higashimoto Y, Popoff IJ,
Gaarde WA, Basrur V, Potapova O, Appella E and Fornace AJ Jr:
Initiation of a G2/M checkpoint after ultraviolet radiation
requires p38 kinase. Nature. 411:102–107. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tarn C, Zou L, Hullinger RL and Andrisani
OM: Hepatitis B virus X protein activates the p38 mitogen-activated
protein kinase pathway in dedifferentiated hepatocytes. J Virol.
76:9763–9772. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huang Q, Liu X, Wu Y, Liao Y, Huang Y, Wei
X and Ma M: P38 MAPK pathway mediates cognitive damage in
pentylenetetrazole-induced epilepsy via apoptosis cascade. Epilepsy
Res. 133:89–92. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhu G, Qiu W, Li Y, Zhao C, He F, Zhou M,
Wang L, Zhao D, Lu Y, Zhang J, et al: Sublytic C5b-9 induces
glomerular mesangial cell apoptosis through the cascade pathway of
MEKK2-p38 MAPK-IRF-1-TRADD-caspase 8 in rat Thy-1 nephritis. J
Immunol. 198:1104–1118. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huang HL, Hsieh MJ, Chien MH, Chen HY,
Yang SF and Hsiao PC: Glabridin mediate caspases activation and
induces apoptosis through JNK1/2 and p38 MAPK pathway in human
promyelocytic leukemia cells. PLoS One. 9:e989432014. View Article : Google Scholar : PubMed/NCBI
|