Introduction
Primary ciliary dyskinesia (PCD) is a rare,
genetically heterogeneous disorder inherited in an autosomal
recessive manner in most cases (1). In half of affected individuals, PCD
occurs with situs inversus, which is characterized by the
mirror-image reversal of visceral organs, such as the heart, liver
and spleen, but without apparent physiological consequences. This
condition is referred to as Kartagener syndrome, which is the
combination of situs inversus, bronchiectasis, and
nasosinusitis (2). The aberrant
structure and/or function of motile cilia in the airways, paranasal
sinus, inner ear and other organs can prevent motility and may
affect mucus clearance in these organs, thus leading to conditions
like bronchiectasis, nasosinusitis and otitis. The flagella of
sperm are also a type of cilia, therefore flagellar dysfunction
decreases the motility of sperm and leads to infertility. The
absence of normal nodal ciliary function during embryogenesis can
lead to randomized organ placement, which explains why
approximately half of affected individuals have situs
inversus (3). The estimated
prevalence of PCD ranges from 1:2,200 to 1:40,000 in Western
countries (1). There is no
prevalence data for the Chinese population; however, PCD appears to
have a higher incidence compared with cystic fibrosis, which is
also a main cause of bronchiectasis, in this population (4). Thus far, 45 genes have been found to
be associated to the pathogenesis of PCD (5). These genes encode proteins that
contribute to the synthesis or assembly of axonemes, dyneins,
radial spokes or other similar structures. Dynein axonemal assembly
factor 1 (DNAAF1) primarily affects the preassembly of the inner
(IDA) and outer (ODA) dynein arms, which are multisubunit ATPase
complexes (6). Mutations in
DNAAF1 have been shown to cause PCD (6,7);
however, the incidence of DNAAF1 mutations is quite low
(7). The present study analyzed
clinical and genetic data from seven Chinese patients with PCD.
Among these patients, two were found to carry novel DNAAF1
mutations.
Materials and methods
Patients
Seven Chinese Han patients with Kartagener syndrome
(four males and three females; age, 8–41 years; median age, 32
years). All of the enrolled patients had chronic cough, excessive
sputum production and shortness of breath. The study was approved
by the Ethics Committee of Zhongshan Hospital, Qingpu Branch
(Shanghai, China) and all participants or their guardians provided
written informed consent.
Clinical examination
All patients underwent paranasal and chest computed
tomography (CT) and pulmonary function tests. The bronchoscope
examination was conducted in one patient and a lung biopsy specimen
was acquired. Five of the seven subjects agreed to transmission
electron microscopy (TEM) analysis of nasal mucosal cilia or sperm
flagella to identify cilial defects. TEM analyses were performed
after specimen.preparation. In brief, nasal biopsy specimens
obtained from the infraturbinal region and sperm specimens were
centrifuged at 2,000 × g for 5 min at 4°C. All specimens were fixed
in 2.5% glutaric dialdehyde at room temperature for 2–3 h. After
dehydration with alcohol series, samples were embedded in acetonum
overnight at room temperature. The solidified specimens were then
sliced into 50–60 nm sections and then examined using TEM after
double-staining at room temperature with uranyl acetate for 8 min
and lead citrate for 5 min.
Whole-exome sequencing (WES)
WES analysis was conducted by Gemple Biotech, and
was performed as previously described (8). Genomic DNA was isolated from blood or
buccal swab samples for WES or Sanger sequencing. Blood samples
were obtained from the peripheral vein. The concentration and
integrity of the genomic DNA samples were determined using a Qubit
dsDNA HS Assay kit (Thermo Fisher Scientific, Inc.) and by 2%
agarose gel electrophoresis, and visualized with ethidium bromide.
Exome sequencing libraries were prepared using the KAPA Hyper Prep
kit (Kapa Biosystems; Roche Diagnostics) and enriched using the
SeqCapEZ Exome version 3.0 kit (NimbleGen; Roche Diagnostics).
Captured libraries were sequenced on an HiSeq platform (Illumina,
Inc.), using 150 bp paired-end sequencing.
Variant filtering and bioinformatics
analysis
Adapter sequences were removed from the raw data and
low-quality reads, containing an excessive number of Ns or having
low base quality, were discarded. Sequencing reads were then
aligned to the hg19 human reference genome using the
Burrows-Wheeler Aligner (version 0.7.15). After further processing
in Samtools (version 1.3.1; github.com/samtools/samtools)/Picard (version 2.5;
broadinstitute.github.io/picard), the final BAM files were used for
variant calling. Single nucleotide polymorphisms and small
insertions/deletions were detected using GATK-haplotypeCaller
(version 3.6; gatk.broadinstitute.org/hc/en-us). ANNOVAR was then
used to annotate the detected variants. The present study focused
on previously reported PCD-associated genes (4) and found that two probands (patients 1
and 2) carried DNAAF1 (NM_178452.6) mutations. Sanger
sequencing was then used to confirm the WES results and analyze the
DNAAF1 sequences of the probands' parents. The following
primers (Generay Biotech Co., Ltd.) were used for DNAAF1
sequencing: Exon 7, forward: 5′-GTCTGATGCTCACTTTGCTTTGA-3′ and
reverse: 5′-AAGGAACTCTGGGGCTGTTGT-3′; exon 1, forward:
5′-GTTGGGCTGTAAAGACTAGGGC-3′ and reverse:
5′-TCACTGACTAGCCGAGGGTTA-3′; and exon 4, forward:
5′-TAGGCAAAAACAAGGGTGACCG-3′ and reverse:
5′-TGCTGGGTACCCTTACAGAGG-3′. The DNA polymerase (Taq EXtra) was
purchased from Kapa Biosystems. The PCR cycling conditions were as
follows: 94°C for 25 sec, 55°C for 15 sec and 72°C for, 1 min for
35 cycles. These mutations were then analyzed to predict their
functional effects on DNAAF1. SIFT (http://sift.jcvi.org) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2)
were used to predict the effects of amino acid substitutions, and
the Human Splicing Finder (HSF) tool and MaxEnt scan (MES) were
used to evaluate splice-site mutations (9,10).
AutoPVS1 (http://autopvs1.genetics.bgi.com) was used to
interpret the pathogenicity of the variants, according to the
refined criteria (PVS1) of the ClinGen Sequence Variant
Interpretation Working Group (10). Subsequently, the 1,000 genomes
database (www.ncbi.nlm.nih.gov/variation/tools/1000genomes) was
searched to identift the frequency of the variations in the healthy
population.
Results
Clinical data for patients PCD with
DNAAF1 mutations
The clinical data for all subjects are summarized in
Table I. Given that DNAAF1
mutations were found in two probands, a more detailed account of
their clinical characteristics are described below.
 | Table I.Dynein axonemal assembly factor 1
mutations of the two patients with primary ciliary dyskinesia. |
Table I.
Dynein axonemal assembly factor 1
mutations of the two patients with primary ciliary dyskinesia.
| Patient no. | Allele | Location | Gene variation | Protein change | Type of
variant |
|---|
| 1 | Paternal | Exon 1 | c.[3G>A;
c.124+1G>C] | p.Met1Ile | Start codon and
donor splicing site mutation |
| 1 | Maternal | Exon 4 | c.509delG | p.Glu126Lysfs ×
35 | Frame-shift
mutation |
| 2 | Both | Exon 7 | c.943A>T | p.Lys315 × | Nonsense
mutation |
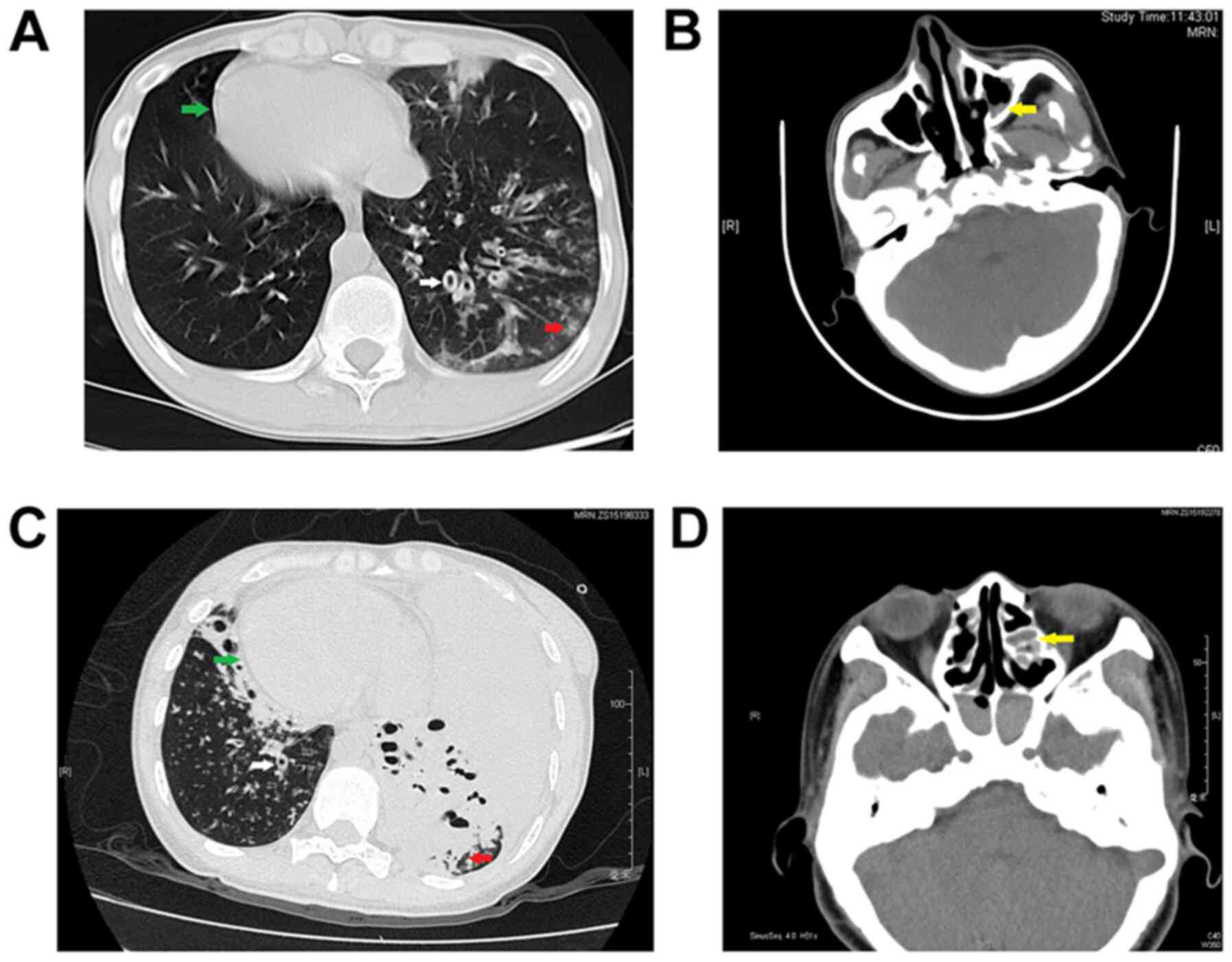
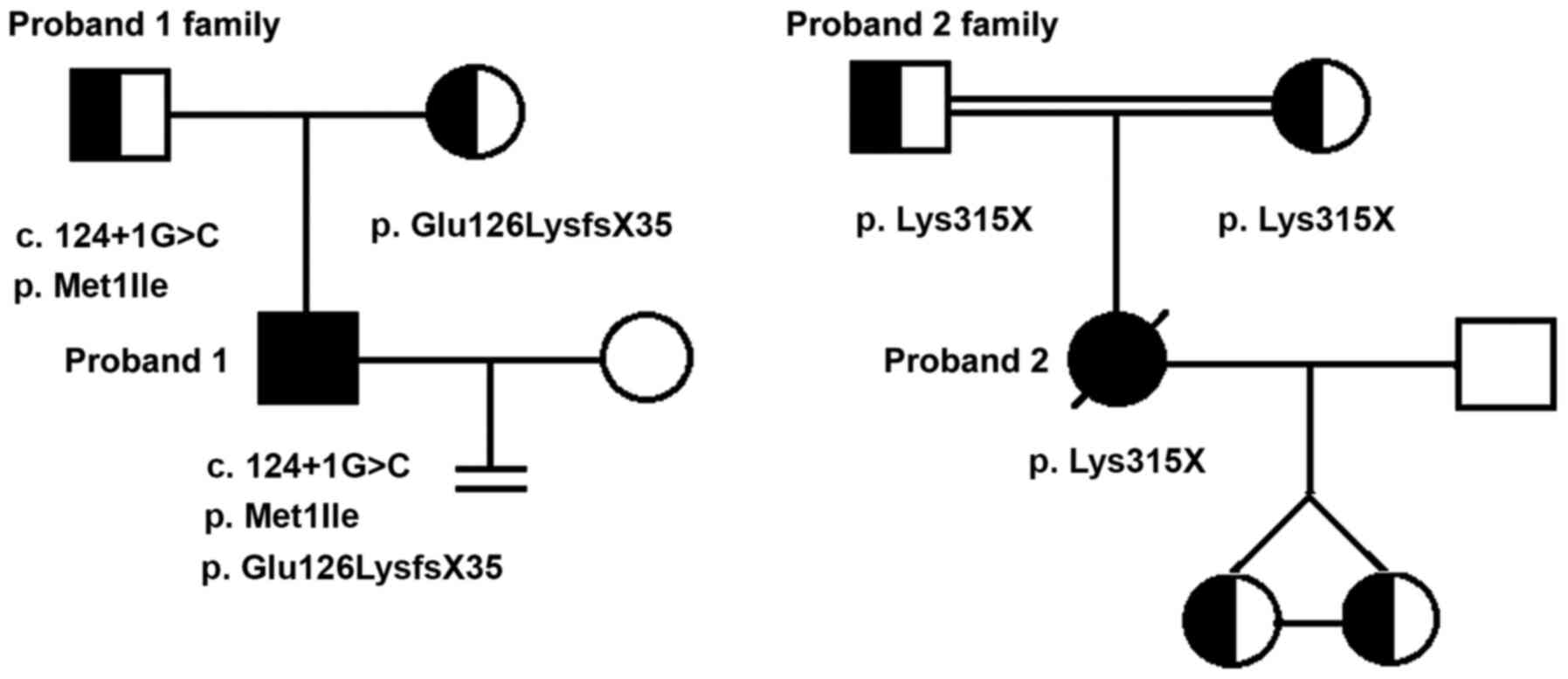
Proband 1 was a 32-year old, male, non-smoker who
had a chronic cough and sputum for over 20 years. He showed
recurrent hemoptysis and shortness of breath. He also had a long
history of nasosinusitis and primary infertility. Chest CT results
showed bronchiectasis in multiple lobes of the lung and situs
inversus. Paranasal CT results revealed bilateral maxillary
sinusitis and ethmoid sinusitis (Fig.
1A and B). A pulmonary function test yielded the following
results: FEV1, 1.80 L/s; FEV1/pred, 53.1% and FEV1/FVC, 63.43%,
which were significantly decreased compared with healthy
individuals. A lung biopsy obtained using a bronchoscope showed
bronchiolitis. As few cilia were found in the nasal mucosa biopsy
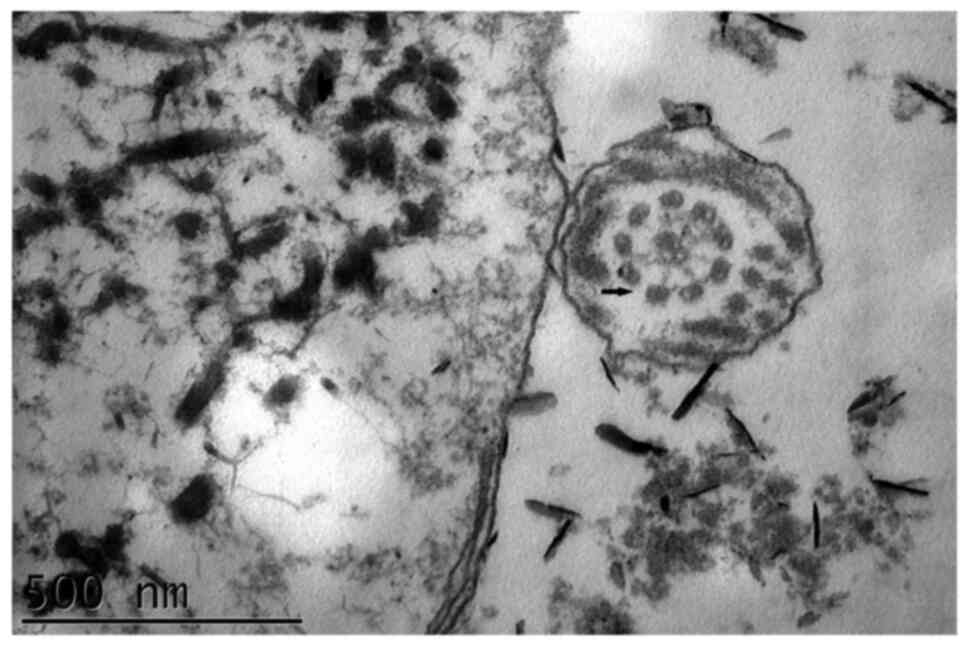
sample, the sperm flagella were analyzed using TEM, demonstrating
both IDA and ODA defects (Fig. 2)
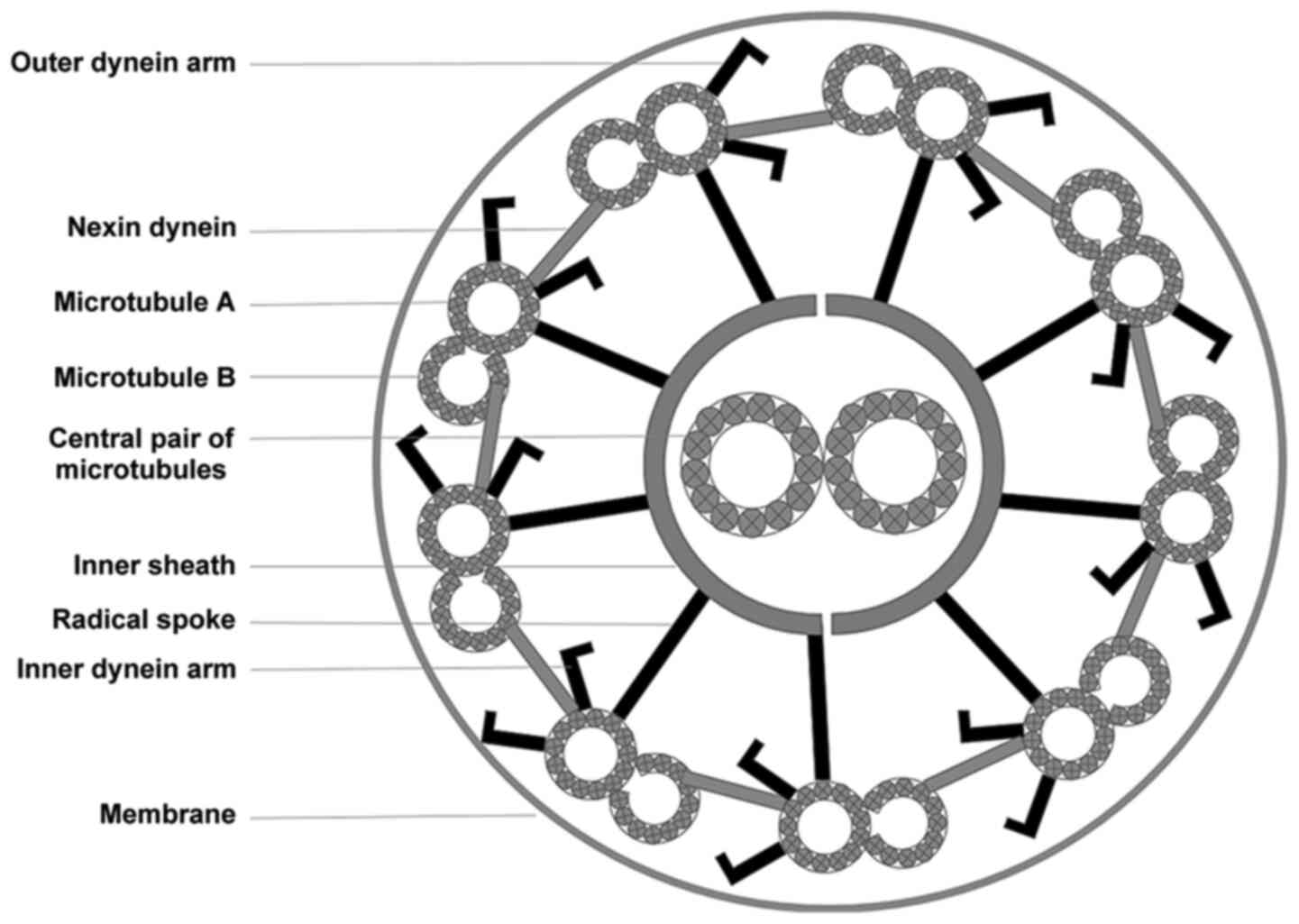
compared with the normal cilia construction (Fig. 3). The proband's parents were not
consanguineous. His mother had a history of asthma, and his father
was healthy.
Proband 2 was a 37-year old female non-smoker who
was diagnosed with bronchiectasis at 12 years of age. She had a
long history of nasosinusitis. Since she was unable to achieve
pregnancy naturally for 5 years, she had utilized in vitro
fertilization and embryonic implantation using her own ova, which
had resulted in healthy twin daughters. She presented with a fever
and wheezing when she was admitted. Chest CT results showed
bronchiectasis, a severe lung infection and situs inversus.
Paranasal CT revealed bilateral maxillary sinusitis, ethmoid
sinusitis, sphenoid sinusitis and frontal sinusitis (Fig. 1C and D). Culture of a sputum sample
acquired using a bronchoscope showed the presence of an imipenem-
and cilastatin sodium-resistant Streptococcus viridans
infection. Although she agreed to treatment with ceftazidime
combined with amikacin and mechanical ventilation through tracheal
intubation, she died due to a severe lung infection and poor heart
function, which are the main causes of death for the majority of
patients with PCD (11). Her
parents were cousins and had no history of respiratory disease.
WES and Sanger sequencing
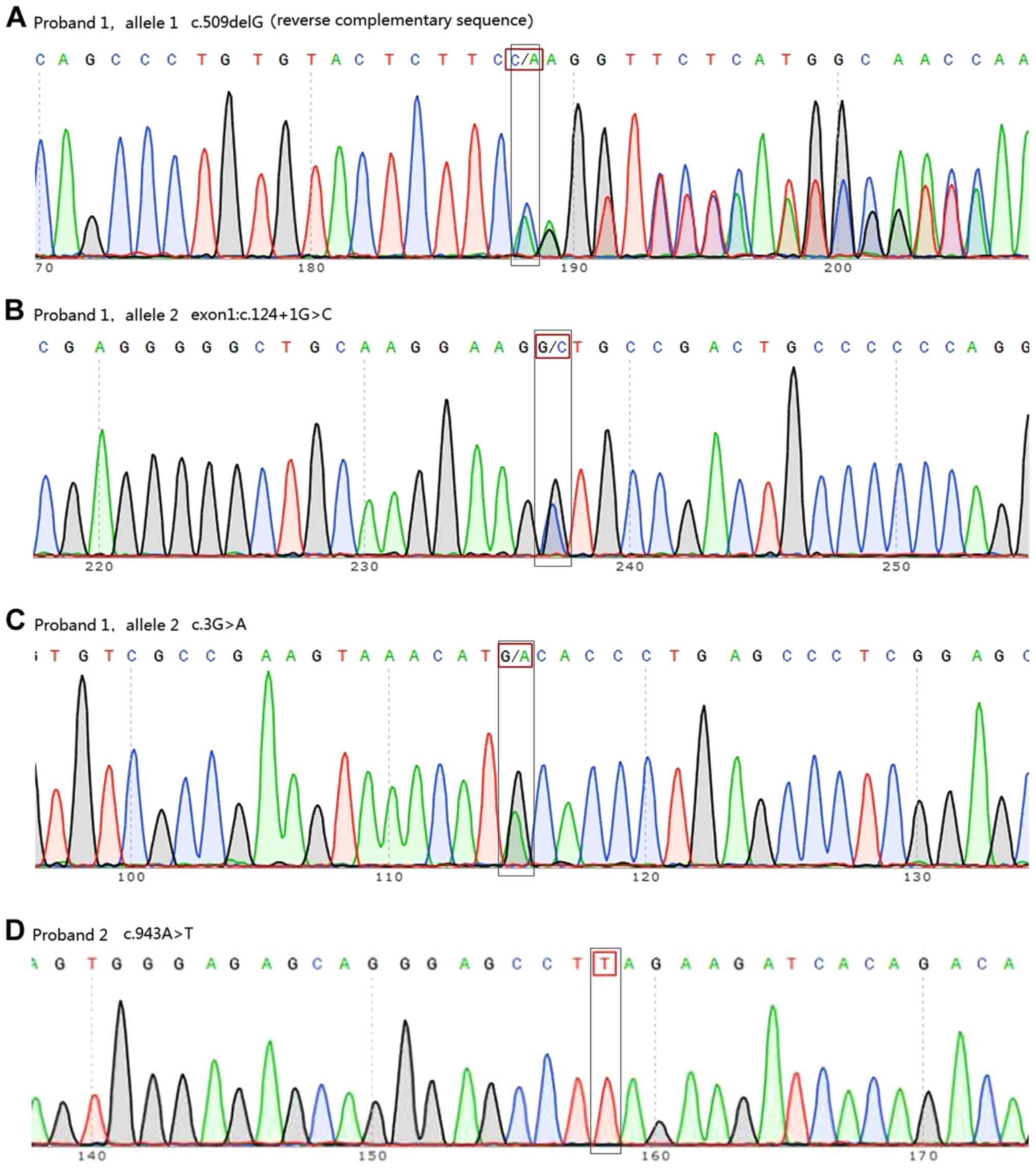
WES analysis showed that two of the seven patients
carried DNAAF1 mutations. Proband 1 carried three point
mutations (exon 4: c.509delG and exon 1: c.[3G>A;124+1G>C])
and proband 2 was homozygous for exon 7: c.943A>T (Fig. 4, Table
I). Information on all seven patients is listed in Table II. Out of the seven patients, TEM
tests were conducted for five patients. The cilia of one patient
was damaged due to severe infection. Another patient displayed
normal cilia structure. The three other patients displayed IDA
defects, central pair components defects or IDA and ODA defects,
respectively. Among the four mutations identified, one (exon 4:
c.509delG) was already registered in the dbSNP database as
rs756239623. However, to the best of our knowledge, the present
study was the first to identify the four mutations identified in
patients with PCD. Sanger sequencing confirmed these mutations and
revealed that proband 1 inherited exon 1: c.[3G>A;c.124+1G>C]
from his father and exon 4: c.509delG from his mother. Proband 2
was found to have inherited the homozygous DNAAF1 mutation
exon 7: c.943A>T from her parents, both of whom were
heterozygous carriers (Fig. 5).
WES and Sanger sequencing data are available on request.
| Table II.Clinical data of all patients with
primary ciliary dyskinesia. |
Table II.
Clinical data of all patients with
primary ciliary dyskinesia.
| Subject number | Age, years | Sex | Chest CT | Paranasal CT | Pulmonary function
test | TEM test |
|---|
| 1 | 32 | Male | Bronchiectasis
in | Bilateral
maxillary | FEV1: 1.80 L,
FEV1/ | IDA and ODA |
|
|
|
| multi-lobes of the
lung | sinusitis,
ethmoid | pred: 53.1%,
FEV1/ | defects |
|
|
|
| and situs
inversus | sinusitis | FVC: 63.43% |
|
| 2 | 37 | Female | Bronchiectasis,
severe | Bilateral
maxillary | FEV1: 0.77 L,
FEV1/ | Nasal
epithelium |
|
|
|
| lung infections
and | sinusitis,
ethmoid | pred: 23.78%,
FEV1/ | cilia damaged |
|
|
|
| situs
inversus | sinusitis,
sphenoid | FVC: 59.58% | because of |
|
|
|
|
| sinusitis and
frontal |
| severe
infection |
|
|
|
|
| sinusitis |
|
|
| 3 | 41 | Female | Bronchiectasis,
and | Bilateral
maxillary | FEV1: 0.71 L;
FEV1/ | Normal ODA |
|
|
|
| situs
inversus, the | sinusitis,
ethmoid | pred: 28.80%;
FEV1/ | and IDA |
|
|
|
| middle lobe of
left | sinusitis,
sphenoid | FVC: 48.39% | structures |
|
|
|
| lung resected
because | sinusitis and
frontal |
|
|
|
|
|
| of bronchiectasis
hemorrhage | sinusitis |
|
|
| 4 | 8 | Male | Bronchiectasis,
and | Bilateral
ethmoid | FEV1: 0.70 L;
FEV1/ | Refused TEM |
|
|
|
| situs
inversus | sinusitis,
sphenoid | pred: 33.92%;
FEV1/ | analysis |
|
|
|
|
| sinusitis and
frontal sinusitis | FVC: 89.44% |
|
| 5 | 28 | Male | Bronchiectasis,
and | Paranasal
sinusitis | FEV1: 2.83 L;
FEV1/ | Central pair |
|
|
|
| situs
inversus |
| pred: 78.98%;
FEV1/ | components |
|
|
|
|
|
| FVC: 76.65% | defect or
dislocation |
| 6 | 34 | Male | Bilateral
bronchiectasis, and situs inversus | Paranasal
sinusitis | FEV1: 2.57 L;
FEV1/ | Refused TEM |
|
|
|
|
|
| pred: 76.24%;
FEV1/ | analysis |
|
|
|
|
|
| FVC: 69.48% |
|
| 7 | 18 | Female | Bronchiectasis
in | Paranasal
sinusitis | Not be able to | IDA defects |
|
|
|
| multi-lobes, and
situs inversus |
| perform PFT |
|
Bioinformatics analysis
Bioinformatics analysis (data not shown) predicted
that exon 1: c.3G>A in proband 1 would cause the amino acid
substitution NP_848547.4:p.Met1Ile, leading to the loss of the
DNAAF1 start codon. This was predicted to be ‘damaging’ by
SIFT and PolyPhen-2, and the PVS1 strength level was found to be
‘moderate’. Exon 1: c.124+1G>C is a splice-site mutation that
was predicted by Human HSF and MES software to disrupt a highly
conserved donor splice site in the mRNA and thus affect splicing.
The PVS1 strength level of the variant containing this mutation was
found to be ‘very strong’. Together, exon 1: c.3G>A and
c.124+1G>C were hypothesized to cause DNAAF1 loss of
function. Exon 4: c.509delG was predicted to cause a frameshift and
create a premature stop codon (p.Glu126Lysfs × 35), leading to the
loss of function of the protein in the other allele of proband 1.
The PVS1 strength level of this variant was also ‘very strong’.
Exon 7: c.943A>T, carried by proband 2, is a nonsense mutation
(p.Lys315×) that was predicted to create a premature stop codon
(Fig. 6), and the variant with
this mutation also had a ‘very strong’ PVS1 strength level.
Finally, data for the healthy Chinese population was obtained from
the 1000 Genomes Database and found that none of the aforementioned
mutations had been registered. As the results of the functional
analyses showed a ‘significantly damaging’ status for these
mutations, it was proposed that these mutations may be the cause of
the cilial defects in these two patients.
Discussion
Based on their structure and function, cilia can be
subdivided into two categories: Motile and immotile. The airway
epithelium contains mainly motile cilia, each of which consists of
nine peripheral microtubule doublets and two central microtubules
(12). IDA and ODA, which stretch
out from the nine peripheral doublets, are multisubunit ATPase
complexes. Their coordinated activation and inactivation generates
a wave of beating cilia. ODA influences ciliary beat frequency and
IDA determines the ciliary waveform (13). A deficiency in ODA and/or IDA is
the main cause of PCD (5). It is
estimated that ~30% of PCD patients are both ODA- and IDA-deficient
(14,15). DNAAF1 mutations are
responsible for cilia dysfunction in 4–5% of PCD cases (7).
The DNAAF1 protein plays an essential role in the
preassembly of IDA and ODA. DNAAF1 is mainly expressed in the
airways, lungs and testicles in adult humans (7). These tissues and organs are covered
with a ciliated epithelium or are rich in sperm flagella. DNAAF1 is
distributed diffusely in the cytoplasm and cilia of ciliated cells
and is concentrated at the spindle pole in mitotic cells. The
DNAAF1 gene is located on the chromosomal band 16p24.1 and
produces two predicted transcripts, encoding two isoforms of 673
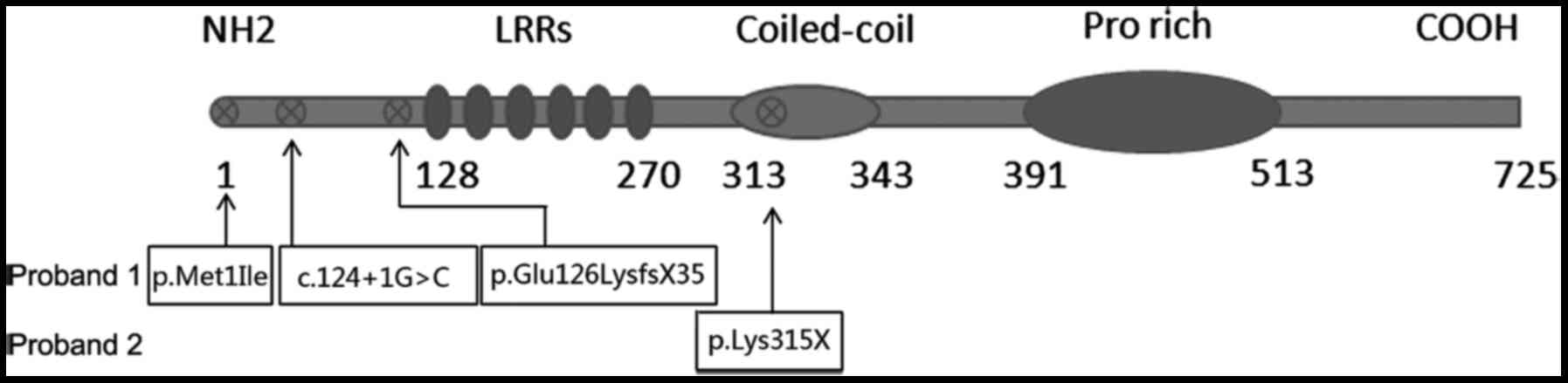
and 725 amino acid residues, respectively (16). Both isoforms contain six highly
conserved N-terminal leucine-rich repeats (LRRs), a coiled coil, an
LRR cap and a non-conserved proline-rich domain (16). The detailed mechanism of action of
DNAAF1 is unclear; however, the evidence obtained thus far suggests
that DNAAF1 is essential for the preassembly of dynein arms
(17). The loss of DNAAF1
results in the absence of ODA heavy chains dynein heavy chain
(DNAH)5 and DNAH9, ODA intermediate chain dynein intermediate chain
2 and IDA light chain dynein axonemal light intermediate chain 1 in
cilia (6,18). Instead, these dynein arm components
accumulate in the cytoplasm as cilium assembly is a continuous
process (18). These observations
explain why both ODA and IDA defects are present in individuals
with DNAAF1 mutations.
A total of 15 causative DNAAF1 mutations have
been reported thus far, in 13 PCD patients from ten families
(Table III) (6,7,17).
Meanwhile, the present study describes three novel DNAAF1
mutations in two Chinese Han patients with PCD. TEM analysis showed
that proband 1, a 37-year old patient with ODA and IDA defects in
his sperm flagella, inherited compound heterozygous mutations from
his parents. The maternal allele included c.509delG, which
introduced a frameshift and created a premature stop codon
(p.Glu126Lysfs × 35). As a result, ~75% of the normal amount of
DNAAF1 protein was lost, resulting in a partial, but substantial,
loss of function of DNAAF1. The paternal allele included a missense
point mutation (c.3G>A) and a splice-site mutation
(c.124+1G>C). The missense mutation, which resulted in the amino
acid substitution p.Met1Ile, is a start codon mutation, which
prevented the transcription of the entire DNAAF1 gene and
was predicted to be damaging by SIFT and PolyPhen-2. However, the
splice-site mutation disrupted a highly conserved donor splice site
at the 5′ end of the mRNA and affected the correct splicing of exon
1. HSF and MES algorithms predicted that the effect of this
mutation was possibly severe damage. A similar donor splice-site
mutation has been reported in mouse DNAAF1, leading to the
absence of the entire exon 4 sequence in the transcript (19). On the basis of these observations,
it was propose that both of these mutations contributed to the loss
of DNAAF1 protein function in this proband.
| Table III.Dynein axonemal assembly factor 1
mutations in patients with primary ciliary dyskinesia. |
Table III.
Dynein axonemal assembly factor 1
mutations in patients with primary ciliary dyskinesia.
| Author, year | Number of
cases | DNA change | Location on
DNAAF1 | Protein change | (Refs.) |
|---|
| Loges et al,
2009 | 1 |
[c.1349_1350insC]a | Exon 8 | p.Pro451Alafs ×
5 | (6) |
| Loges et al,
2009 | 1 | [11 kb del]+[640 kb
del]b |
16q24.1/16q23.3–16q24.1 | Loss of protein
DNAAF1, HSDL1, et al | (6) |
| Loges et al,
2009 | 1 | [c.811C>T]+[220
kb del]b | Exon 6/16q24.1 | p.Arg271 ×/Loss
of | (6) |
|
|
|
|
| protein DNAAF1,
HSDL1, et al |
|
| Duquesnoy et
al, 2009 | 1 |
[c.792C>A]+[c.508dupG]b | Exon 6/4 | p.Tyr264 ×
/p.Glu170Glyfs x 10 | (7) |
| Duquesnoy et
al, 2009 | 1 |
[c.115dupT]+[c.1300_1322del23bp]b | Exon 1/8 | p.Cys39Leufs ×
44/p. Gly434Profsx 4 | (7) |
| Duquesnoy et
al, 2009 | 1 |
[c.1198_1199insTCGC]+
[c.124+1536_353-2102del5376bp]b | Exon 8/ 2–3 | p.Pro400Leufs ×
6/p. Glu42_Lys117del | (7) |
| Duquesnoy et
al, 2009 | 2 |
[c.524T>G]a | Exon 4 | p.Leu175Arg | (7) |
| Hartill, et
al, 2018 | 3 |
[c.281delA]+[c.1484delC]b | Exon 3/ 8 | Lys95Asnfs ×
14/Pro495Glnfs × 40 | (17) |
| Hartill, et
al, 2018 | 1 |
[c.1484delC]a | Exon 8 | Pro495Glnfs ×
40 | (17) |
| Hartill, et
al, 2018 | 1 | [Exon 6-Exon 7
deletion]a | Exon 6–7 | Incomplete
protein | (17) |
Proband 2, who was a female patient born to
consanguineous parents, carried a homozygous nonsense point
mutation (c.943A>T), which led to a premature translation stop
codon at p.315, resulting in the deletion of half of the normal
protein. This was expected to result in a complete loss of function
of the DNAAF1 protein. Due to the severe infection in her airways,
there were not enough cilia to examine in the biopsy samples
collected from her nose or bronchi. Hence, her cilial phenotype
remains undetermined. Nevertheless, genetic analysis confirmed the
PCD diagnosis in this patient. Unfortunately, this patient died due
to the severe lung infection and poor lung and heart function,
which are a common complications of chronic lung disease (20). Compared with mutations in the human
orthologue of medaka kintoun gene, another PCD-associated gene that
can also cause ODA and IDA defects (21), patients with DNAAF1-mutant
PCD tend to have more severe cell defects (6,21).
For example, both of the patients in the present study developed
bronchiectasis and infection in both lungs, against a background of
situs inversus and infertility. Infertility is another
common complication of PCD, especially in male patients. The
immobility of sperm flagella can cause an impairment in or the
complete absence of the ability of the flagella to swim, which
ultimately results in male infertility (22). Since coordinated ciliary beating,
in conjunction with muscle contractions, plays a role in directing
the oocyte through the fallopian tubes to the uterus, PCD is also
thought to be an important cause of female subfertility (23). However, accumulating evidence has
shown that, compared with male patients, female patients with PCD
have a greater chance of achieving spontaneous conception and
fertility (24). All reported
female patients with DNAAF1 mutations have not had offspring
naturally (6,7,17);
however, due to the small number of known patients, an association
between DNAAF1 mutations and female infertility has not been
established. Fortunately, most patients with PCD have healthy
offspring with the help of assisted reproductive technology
(22).
Aside from ODA and IDA, DNAAF1 also contributes to
the construction of the axoneme. Inactivation of DNAAF1
expression significantly reduces the brush border or decreases
cilial length in the proximal tubule epithelium cell line HK-2
(16). Considering that normal
human kidneys do not contain motile cilia or dynein arms, these
data suggest that DNAAF1 mutation interrupts the
construction of the cilial axoneme backbone instead of dynein
arms.
DNAAF1 may play a role in the regulation of gene
expression. Miao et al (25) evaluated the mRNA expression
profiles of 227 neural tube closure-associated genes in a patient
with DNAAF1 mutations and neural tube defects and healthy
controls. The results demonstrated that three genes were
upregulated and 19 were downregulated in this patient. Moreover,
changes in DNAAF1 expression levels led to a corresponding
change in the expression levels of left-right patterning genes and
the sonic hedgehog signaling-associated genes, Lefty1, Lefty2 and
Gli2, in NE-4C neuroectodermal stem cells. Nonetheless, it
remains unknown whether DNAAF1 regulates gene expression in human
airways.
PCD is a genetically heterogeneous disorder, and the
genetic causes of ~30% of PCD cases remain to be elucidated
(5). Patients with DNAAF1
mutations constitute only a small proportion of all PCD cases;
however, DNAAF1 mutation usually causes the misalignment of
both ODA and IDA and thus induces severe clinical symptoms. The
present study reported two patients with DNAAF1 mutations,
one of whom died due to severe lung infection and poor lung and
heart function. An effective strategy to treat genetic defects in
PCD patients has not yet been devised. The injection of
DNAAF1 mRNA into DNAAF1-mutant zebrafish can reverse
their cilial defects, but it remains unclear whether exogenous
DNAAF1 can promote cilial motility in humans (16). To further explore the role of
DNAAF1 in ciliated epithelial cells, as well as the mechanism
underlying bronchiectasis, a DNAAF1 mutant mouse model should be
established in future studies, which could be used to investigate
novel treatment strategies for targeting DNAAF1.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China Key grant (grant nos. 81630001,
81490533, 81770055, 81500026, 81570028 and 81600056), the State Key
Basic Research Program Project (grant no. 2015CB553404), the
Shanghai Science and Technology Committee (grant nos. 15DZ1930600,
15DZ1930602 and 16ZR1405700) and Shanghai Municipal Commission of
Health and Family Planning (grant no. 201540370).
Availability of materials and data
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
LZ carried out the molecular genetic studies and
wrote the original draft. ZL, CC carried out the molecular and
genetic bioinformatic studies. CD enrolled the subjects and
conducted their treatments, performed the literature searching and
analysis. LG, YinS and FZ participated in phenotyping and clinical
data collection. YuaS designed this study, wrote, reviewed and
edited the final version of the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Zhongshan Hospital Qingpu Branch (approval no.
2018-12; Shanghai, China). All participants or their guardians
provided written informed consent.
Patient consent for publication
Informed written consent was obtained from the
patient or their guardian for the publication of this case report
and any accompanying images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Olm MA, Caldini EG and Mauad T: Diagnosis
of primary ciliary dyskinesia. J Bras Pneumol. 41:251–263. 2015.(In
English, Portuguese). View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Frommer A, Hjeij R, Loges NT, Edelbusch C,
Jahnke C, Raidt J, Werner C, Wallmeier J, Große-Onnebrink J,
Olbrich H, et al: Immunofluorescence analysis and diagnosis of
primary ciliary dyskinesia with radial spoke defects. Am J Respir
Cell Mol Biol. 53:563–573. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brown J and Witman G: Cilia and Diseases.
Bioscience. 64:1126–1137. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cao Y, Shao C, Song Y, Bai C and He L:
Clinical analysis of patients with primary ciliary dyskinesia in
mainland China. Clin Respir J. 10:765–771. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Leigh MW, Horani A, Kinghorn B, O'Connor
MG, Zariwala MA and Knowles MR: Primary Ciliary Dyskinesia (PCD): A
genetic disorder of motile cilia. Transl Sci Rare Dis. 4:51–75.
2019.PubMed/NCBI
|
|
6
|
Loges NT, Olbrich H, Becker-Heck A,
Häffner K, Heer A, Reinhard C, Schmidts M, Kispert A, Zariwala MA,
Leigh MW, et al: Deletions and point mutations of LRRC50 cause
primary ciliary dyskinesia due to dynein arm defects. Am J Hum
Genet. 85:883–889. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Duquesnoy P, Escudier E, Vincensini L,
Freshour J, Bridoux AM, Coste A, Deschildre A, de Blic J, Legendre
M, Montantin G, et al: Loss-of-function mutations in the human
ortholog of Chlamydomonas reinhardtii ODA7 disrupt dynein arm
assembly and cause primary ciliary dyskinesia. Am J Hum Genet.
85:890–896. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Marshall CR, Scherer SW, Zariwala MA, Lau
L, Paton TA, Stockley T, Jobling RK, Ray PN, Knowles MR; FORGE
Canada Consortium, ; Hall DA, et al: Whole-exome sequencing and
targeted copy number analysis in primary ciliary dyskinesia. G3
(Bethesda). 5:1775–1781. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Desmet FO, Hamroun D, Lalande M,
Collod-Béroud G, Claustres M and Béroud C: Human Splicing Finder:
An online bioinformatics tool to predict splicing signals. Nucleic
Acids Res. 37:e672009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yeo G and Burge CB: Maximum entropy
modeling of short sequence motifs with applications to RNA splicing
signals. J Comput Biol. 11:377–394. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kennedy MP, Omran H, Leigh MW, Dell S,
Morgan L, Molina PL, Robinson BV, Minnix SL, Olbrich H, Severin T,
et al: Congenital heart disease and other heterotaxic defects in a
large cohort of patients with primary ciliary dyskinesia.
Circulation. 115:2814–2821. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Satir P and Christensen ST: Overview of
structure and function of mammalian cilia. Annu Rev Physiol.
69:377–400. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fliegauf M, Benzing T and Omran H: When
cilia go bad: Cilia defects and ciliopathies. Nat Rev Mol Cell
Biol. 8:880–893. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jorissen M, Willems T, Van der Schueren B,
Verbeken E and De Boeck K: Ultrastructural expression of primary
ciliary dyskinesia after ciliogenesis in culture. Acta
Otorhinolaryngol Belg. 54:343–356. 2000.PubMed/NCBI
|
|
15
|
Papon JF, Coste A, Roudot-Thoraval F,
Boucherat M, Roger G, Tamalet A, Vojtek AM, Amselem S and Escudier
E: A 20-year experience of electron microscopy in the diagnosis of
primary ciliary dyskinesia. Eur Respir J. 35:1057–1063. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
van Rooijen E, Giles RH, Voest EE, van
Rooijen C, Schulte-Merker S and van Eeden FJ: LRRC50, a conserved
ciliary protein implicated in polycystic kidney disease. J Am Soc
Nephrol. 19:1128–1138. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hartill VL, Hoek G, Patel MP, Little R,
Watson CM, Berry IR, Shoemark A, Abdelmottaleb D, Parkes E,
Bacchelli C, et al: DNAAF1 links heart laterality with the AAA1
ATPase RUVBL1 and ciliary intraflflagellar transport. Hum Mol
Genet. 27:529–545. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fliegauf M, Olbrich H, Horvath J,
Wildhaber JH, Zariwala MA, Kennedy M, Knowles MR and Omran H:
Mislocalization of DNAH5 and DNAH9 in respiratory cells from
patients with primary ciliary dyskinesia. Am J Respir Crit Care
Med. 171:1343–1349. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ha S, Lindsay AM, Timms AE and Beier DR:
Mutations in DNAAF1 and LRRC48 cause hydrocephalus, laterality
defects, and sinusitis in mice. G3 (Bethesda). 6:2479–2487. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lobo LJ, Zariwala MA and Noone PG: Primary
ciliary dyskinesia. QJM. 107:691–699. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Omran H, Kobayashi D, Olbrich H, Tsukahara
T, Loges NT, Hagiwara H, Zhang Q, Leblond G, O'Toole E, Hara C, et
al: Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal
dyneins. Nature. 456:611–616. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sha Y, Ding L and Li P: Management of
primary ciliary dyskinesia/Kartagener's syndrome in infertile male
patients and current progress in defining the underlying genetic
mechanism. Asian J Androl. 16:101–106. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lyons RA, Saridogan E and Djahanbakhch O:
The reproductive signifificance of human Fallopian tube cilia. Hum
Reprod Update. 12:363–372. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Raidt J, Werner C, Menchen T, Dougherty
GW, Olbrich H, Loges NT, Schmitz R, Pennekamp P and Omran H:
Ciliary function and motor protein composition of human fallopian
tubes. Hum Reprod. 30:2871–2880. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Miao C, Jiang Q, Li H, Zhang Q, Bai B, Bao
Y and Zhang T: Mutations in the motile cilia gene DNAAF1 are
associated with neural tube defects in humans. G3 (Bethesda).
6:3307–3316. 2016. View Article : Google Scholar : PubMed/NCBI
|