Introduction
X-linked mental disability (MRX) is a form of
intellectual disability (ID) associated with the X chromosome. It
is one of the most frequent forms of ID with an incidence rate of
3% in the human population.
One of the X-linked loci associated with MRX is the
α thalassemia X-linked intellectual disability (ATRX) gene
(OMIM #301040; Xq21.1); characterized by a complex phenotype and
the primary elements consist of a serious impairment of psychomotor
development, facial dysmorphism, genital anomalies and α
thalassemia. Inherited mutations of ATRX are associated with
an X-linked mental disability syndrome (XLMR), and the majority of
cases also have α-thalassemia syndrome (ATR-X). ATRX syndrome,
which has been associated with ATRX mutations is
characterized by a complex phenotype, including serious impairment
of psychomotor development, facial dysmorphism, genital anomalies
and α thalassemia (1). Female
carriers of ATRX mutations are intellectually normal and
rarely show physical manifestations, due to an extreme skewing of
X-inactivation in favor of the chromosome containing the non-mutant
allele (1). There are >200
cases of ATR-X syndrome that have been identified using molecular
diagnostics (2–5).
ATRX encodes a protein, consisting of 2,492
amino acids. It has multiple functions, including regulation of
transcription, chromatin remodeling and ATP-dependent helicase
activity (https://www.uniprot.org/uniprot/P46100). Alternative
splicing of the gene results in distinct protein isoforms.
The protein has two primary functional domains, the
N-terminal ADD domain and the C-terminal helicase/adenosine
triphosphatase domain (ATPase), where the majority of reported
mutations are located. Most of the reported mutations are missense,
with c.536A>G and c.736C>T, both in the ADD domain, being the
most common mutations found in different families. Mutations in the
ADD domain are associated with greater impairment of psychomotor
development, while most cases with mild/moderate mental disability
have mutations in the helicase domain. The common mutation,
c.736C>T, has been associated with low frequency of Hemoglobin H
HbH (6).
In the present case report, a novel, likely
pathogenic, ATRX variant c.5295C>G associated with mental
disability has been identified. The family in the report shows a
strong transmission bias in favor of the mutant allele: All 4
offspring inherited the mutant ATRX allele from their
mother.
Case report
Whole blood (3 ml) was collected for exome analysis
after informed consent was provided. DNA was extracted using the
Qiagen BioRobot DNA extraction kit (Qiagen Benelux B.V.) according
to the manufacturer's instructions and quantified using NanoDrop
spectral analysis (Thermo Fischer Scientific, Inc.). DNA integrity
was evaluated using standard agarose gel electrophoresis (100 V; 30
min; 1.5% agarose gel in TBE buffer). DNA library preparation and
whole exon enrichment were performed using Agilent All Exon V.6 kit
(Agilent Technologies, Inc.). The library was sequenced using the
HiSeq2500 Illumina Sequencer (125-bp paired end sequence mode;
Illumina, Inc.). Bioinformatics analysis included the following: i)
Next generation sequencing reads were mapped to whole genomes using
the Burrows-Wheeler Alignment tool with the default parameters, ii)
PCR duplicate removal using Picard (http://picard.sourceforge.net), iii) identification of
single nucleotide polymorphisms and insertions/deletions using the
Genome Analysis Toolkit (GATK) UnifiedGenotyper, iv) variant
annotation using snpEff (http://snpeff.sourceforge.net) and v) false positive
variant filtration using the GATK VariantFiltration module. Exome
sequencing data and read alignment analysis was checked for
coverage depth and alignment quality using the Bedtools software
package. Variant classification was performed in accordance with
the guidelines from the American College of Medical Genetics and
Genomics (ACMG).
Phenotype driven analysis coupled with the
employment of in silico multigene panels specific for ID and
neurodevelopmental disorders was used to filter, select and
interpret genetic variants obtained following exome sequencing. The
presence of significant variants was confirmed using Sanger
sequencing.
Clinical description
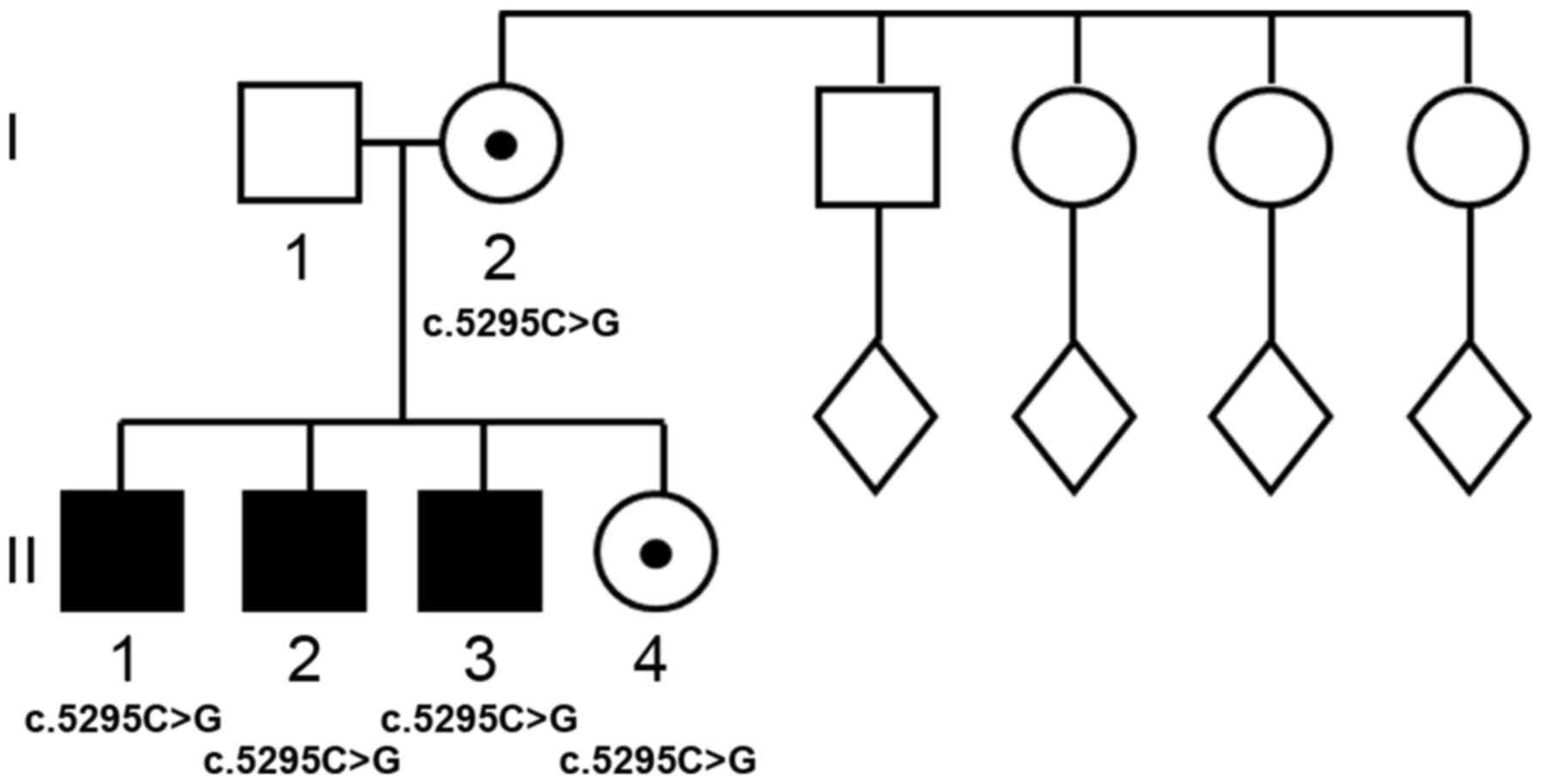
In the Italian family A (Fig. 1) with healthy and
non-consanguineous parents (I-1 and I-2), all three male probands
(II-1, II-2, II-3) showed medium to severe mental disability and
mild dysmorphism, while the heterozygous females (I-2 and II-4) are
phenotypically and intellectually normal. The mother was 16, 18, 32
and 35-years-old when all of her children were born, respectively.
The family history was unremarkable for ID or birth defects. In the
mother's family, the male sibling was intellectually normal, as
were the seven nephews born to the three female siblings (data not
shown).
The three affected male probands have a similar
clinical history and phenotype with greater intellectual impairment
in one of the probands. They were delivered at term by caesarean
section, following an uneventful pregnancy. Their birth weights
were 3,200, 3,000, and 3,050 g for II-1, II-2 and II-3,
respectively. The auxological parameters were within the limits of
the normal range, while gross motor, cognitive and social
milestones were delayed in early childhood. None of the probands
had epileptic seizures. With respect to the characteristics
associated with the pathology of ATRX syndrome, specific signs of
facial dysmorphism (facial hypotonia and hypertelorism) were
absent, while other signs (mouth ‘carp’ and depressed nasal bridge)
were present. However, genital anomalies (cryptorchidism and/or
ambiguous genitalia), epileptic seizures, autistic behavior, and
α-thalassemia (microcytic anemia and Heinz bodies) were absent.
The first-born, II-1 (48 years old) lives at home
with his family. Currently, the patient is autonomous in his
movements. Partial presence of linguistic acquisitions and gestural
communication is also present. The patient is very calm and has no
problems with breathing or swallowing. There are no ‘snap’
movements of the upper limbs and enjoys care and physical contact.
The other two probands have been hospitalized at a facility. The
cranial magnetic resonance imaging performed on II-1 revealed
cerebral hypoplasia without signs of cortical dysplasia, important
dilatation of the subarachnoid spaces in the temporal and frontal
regions, and large cisternal spaces in front of the trunk.
The second born, II-2 (46 years old) is the most
intellectually and neurologically compromised. He does not control
sphincters and physiological stimuli, and has no linguistic
acquisition and gestural communication. Therefore, assessment of
his intellectual level is not possible.
The youngest of the three male siblings, II-3 (32
years old) presents acquisition of language and gestural
communication, as well as learning related to social and relational
skills.
Laboratory analysis
The standard karyotypes of all the patients were
normal. Fragile X screening, which was performed in the mother, all
3 male probands and the female sibling highlighted the presence of
two alleles with values in the normal range. Genome analysis using
the comparative genomic hybridization-array technique with
SurePrintG3 ISCA V2 GCH60K microarrays (Cytogenomics v3.0.0.019;
Agilent Technologies, Inc.) was performed on the mother and one of
the probands (II-2). No imbalances in the number of copies of the
analyzed genomic sequences, i.e. no deletions or duplications, were
detected (data not shown).
Sequencing analysis of the coding exons of the genes
included in the genetic panel for the molecular diagnosis of ID and
neurodevelopmental disorders showed the presence of the hemizygous
variant, c.5295C>G (resulting in the amino acid change
p.Ile1765Met) in the ATRX gene in all three probands, while
the unaffected mother and female sibling were heterozygous carriers
of this variant.
This mutation was not present in allele frequency
databases (ExAC, EVS and GnomeAD). Bioinformatics predictors
indicate that isoleucine at position 1,765 is highly conserved
through higher vertebrates (PhyloP-Primates, 2.70/6.42;
PhyloP-Primate, 0.60/0.65) and the missense change, isoleucine to
methionine is likely to be deleterious (Polyphen2, 1.00/1.00; SIFT,
0.001/0.00; CADD-Phred, 18). The analysis of the quaternary
structure of the protein (pfam database) showed that the novel
mutation, at position 1,765, falls within the SNF2 N helicase
domain (Table I) located between
the ADD and the helicase C domains. The SNF domain is found in
proteins involved in a variety of different functions, including
transcriptional regulation, recombination, and DNA repair (7).
 | Table I.Table pfam domains. |
Table I.
Table pfam domains.
| Source | Domain | Start | End |
|---|
| Disorder | n/a | 1 | 159 |
| Low_complexity | n/a | 24 | 33 |
| Low_complexity | n/a | 59 | 83 |
| Low_complexity | n/a | 103 | 114 |
| Pfam | ADD ATRX | 158 | 213 |
| Disorder | n/a | 311 | 312 |
| Disorder | n/a | 415 | 440 |
| Disorder | n/a | 445 | 581 |
| Disorder | n/a | 586 | 588 |
| n/a | n/a | n/a | n/a |
| n/a | n/a | n/a | n/a |
| n/a | n/a | n/a | n/a |
| Pfam | SNF 2 N | 1536 | 1889a |
| Disorder | n/a | 1544 | 1545 |
| Disorder | n/a | 1551 | 1553 |
| Low_complexity | n/a | 1912 | 1925 |
| Disorder | n/a | 1913 | 1915 |
| n/a | n/a | n/a | n/a |
| n/a | n/a | n/a | n/a |
| n/a | n/a | n/a | n/a |
| Pfam | Helicase C | 2018 | 2155 |
| Disorder | n/a | 2218 | 2229 |
| Disorder | n/a | 2254 | 2266 |
| Low_complexity | n/a | 2261 | 2269 |
| Low_complexity | n/a | 2267 | 2282 |
| n/a | n/a | n/a | n/a |
| n/a | n/a | n/a | n/a |
| n/a | n/a | n/a | n/a |
| Low_complexity | n/a | 2466 | 2479 |
Therefore, in the absence of further information,
and in agreement with guidelines by ACMG (2015), the mutation
should be considered of uncertain significance. We propose that,
based on the overlap within the phenotypes of the probands, with
those previously described, the concordance with the mode of
inheritance, together with the result of the analysis of family
segregation, the variant p.Ile1765Met (c.5295C>G) reported in
the present study and found in a hemizygous state in all the
affected subjects of the same family, may be considered ‘likely
pathogenic’.
Meta-analysis of maternal transmission
of ATRX mutations
Family A shows a striking bias in favor of
transmission of the mutant allele from the mother who is a carrier
to all four of her living children. Such bias may be due to i) a
preferential transmission of mutant ATRX alleles; ii) the
presence of a deleterious mutation on the X chromosome that carries
the non-mutant ATRX allele in the mother; or iii) it could
be purely stochastic.
To investigate the possibility that mutant
ATRX alleles were preferentially transmitted to offspring, a
meta-analysis of published pedigrees with complete reporting of
family structures was conducted. Data on transmission of
ATRX alleles for 42 mothers were extracted from papers
published between 1991 and 2019 (8–21).
Probands and index cases were excluded from the calculations, as
well as offspring from one mosaic mother. A modest bias was found
in favor of ATRX mutations being transmitted to male
offspring. Among the total of 127 offspring, 54% inherited the
mutation, while 58% of the 70 males inherited the mutant allele.
However, the bias did not reach statistical significance (P=0.29;
χ2 test). All data from families with ATRX were analyzed
using the chi square test (χ2 test). A chi-square test
is a statistical hypothesis test, used to determine whether there
is a statistically significant difference between the expected
frequencies and the observed frequencies in one or more categories
of a contingency table. The chi-square test applied to our sample
was P=0.29, which does not reach statistical significance.
Next, the analysis was limited to a subset of
families where the mutations were identified, and all members of
the family were genotyped (Table
II) (8–12,14–17).
A modest sex-ratio distortion was observed in favor of the male
offspring, as well as transmission-ratio distortion in favor of the
ATRX mutant alleles, but these did not reach statistical
significance either (Table
II).
| Table II.Meta-analysis of ATRX allele
transmission from carrier mother to offspring in families with
confirmed mutations. |
Table II.
Meta-analysis of ATRX allele
transmission from carrier mother to offspring in families with
confirmed mutations.
|
|
|
|
| Sons | Daughters |
|
|
|---|
|
|
|
|
|
|
|
|
|
|---|
| No. | Mutation | Pedigree | Mother | Carrier | Non-carrier | Carrier | Non-carrier | Proband/index
Caseexcluded |
Authors/(Refs.) |
|---|
| 1 | c.5295C>G |
|
| 2 | 0 | 1 | 0 | Yes | Current report |
| 2 | c.6130C>T |
|
| 1 | 0 | 0 | 0 | Yes | Altiner and Raymond
2019 (9) |
| 3 | c.515C>T |
| II-1 | 0 | 0 | 1 | 0 |
| Li et al
2020 (14) |
| 4 | c.6257T >C | 3 | III-1 | 1 | 0 | 0 | 0 |
| Yan et al
2019 (21). |
| 5 | c.6257T >C | 3 | III-4 | 1 | 0 | 0 | 1 |
| Yan et al
2019 (21) |
| 6 | c.6718C>T |
|
| 1 | 0 | 0 | 0 | Yes | Thakur et al
2011 (17) |
| 7 | c.6740A>C |
|
| 0 | 0 | 2 | 0 | Yes | Bouazzi et
al 2016 (11) |
| 8 | c.109C>T | K8035 | I-2 | 1 | 4 | 2 | 0 |
| Basehore et
al 2015 (10) |
| 9 | c.109C>T | K8035 | II-1 | 2 | 2 | 1 | 1 |
| Basehore et
al 2015 (10) |
| 10 | c.109C>T | K8035 | III-1 | 1 | 2 | 1 | 1 |
| Basehore et
al 2015 (10) |
| 11 | c.109C>T | K8035 | II-4 | 1 | 0 | 0 | 1 | Yes | Basehore et
al 2015 (10) |
| 12 | c.109C>T | K8360 | I-2 | 2 | 2 | 2 | 1 | Yes | Basehore et
al 2015 (10) |
| 13 | c.109C>T | K8360 | II-3 | 1 | 0 | 0 | 0 |
| Basehore et
al 2015 (10) |
| 14 | c.109C>T | K8820 | I-2 | 4 | 4 | 1 | 1 |
| Basehore et
al 2015 (10) |
| 15 | c.109C>T | K8820 | II-10 | 1 | 1 | 0 | 1 | Yes | Basehore et
al 2015 (10) |
| 16 | c.109C>T | K9574/MRX77 | II-2 | 1 | 1 | 5 | 0 |
| Basehore et
al 2015 (10) |
| 17 | c.109C>T | K9574/MRX77 | III-1 | 1 | 0 | 0 | 2 |
| Basehore et
al 2015 (10) |
| 18 | c.109C>T | K9574/MRX77 | III-3 | 1 | 0 | 1 | 0 |
| Basehore et
al 2015 (10) |
| 19 | c.109C>T | K9574/MRX77 | III-4 | 2 | 0 | 0 | 0 |
| Basehore et
al 2015 (10) |
| 20 | c.109C>T | K9574/MRX77 | III-5 | 0 | 1 | 0 | 1 |
| Basehore et
al 2015 (10) |
| 21 | c.109C>T | K9574/MRX77 | III-6 | 2 | 0 | 0 | 0 |
| Basehore et
al 2015 (10) |
| 22 | c.109C>T | JHH2445-6 |
| 1 | 0 | 0 | 0 | Yes | Basehore et
al 2015 (10) |
| 23 | c.109C>T |
| II | 1 | 0 | 0 | 0 | Yes | Moncini et
al 2013 (16) |
| 24 | c.109C>T |
| IV-2 | 0 | 1 | 1 | 0 | Yes | Abidi et al
2005 (8) |
| 25 | c.7156C>T |
|
| 0 | 1 | 0 | 1 | Yes | Masliah-Planchon
et al |
|
| Total |
|
| 28 | 20 | 18 | 11 |
| 2018 (15) |
It was proposed that the non-mutant X chromosome of
the mother in family A was affected by a deleterious aberration
limiting its transmission to offspring. However, no evidence of
deletions or duplications, or X-autosomal translocations were
found. Nevertheless, the possibility that she carries an
unidentified deleterious mutation on the chromosome harboring the
non-mutant ATRX allele cannot be ruled out.
Discussion
The ATRX gene encodes a protein that belongs
to the SNF2 chromatin remodeling protein family. Members of this
large family of proteins modify the accessibility of DNA wrapped
around a nucleosome, ensuring the nucleic acid is more or less
accessible to proteins, which regulate the transcription process.
To date, the exact role of ATRX is still under debate (22). ATRX contains two highly conserved
domains, namely the ADD domain at the N-terminus and a C-terminal
ATPase/helicase domain, the latter is involved in ATP hydrolysis.
The ADD domain was named ARTX-DNMT3-DNMT3L, based on the sequence
homology with a family of DNA methyltransferases. Argentaro et
al (7) reported the solution
structure of the ADD domain of ARTX, which consisted of an
N-terminal GATA-like zinc finger, a plant homeodomain (PHD), and a
long C-terminal α-helix. All the pathogenic mutations associated
with X-linked mental disability map to the ADD and helicase
domains.
In the present case report, a novel, likely
pathogenic, missense mutation located in exon 24 of the major
splice form of ATRX was identified. The novel p.lle1765Met
variant changes the amino acid sequence of the SNF2 N helicase
domain in the protein. This region is often affected by mutations
associated with ATRX syndrome (23). Methionine is an α-amino acid and
termed as a non-polar, aliphatic amino acid due to the S-methyl
thioether side chain. Isoleucine is a non-polar, uncharged (at
physiological pH) aliphatic amino acid with a branched chain.
Isoleucine and methionine are both non-polar amino acids; however,
due to their different structures, a modification of the tertiary
structure and function of the SNF2 N domain cannot be excluded. The
novel pIle1765Met mutation is also close to the highly conserved
ATPase active site motif III. Another mutation, Leu1746Ser, which
is located very close but outside of motif III, has been shown to
uncouple the ATPase activity of ATRX from its ability to bind and
translocate along DNA (23). In
in vitro experiments, the mutant Leu1746Ser was more
efficient at hydrolyzing ATP compared with that in the wild type
variant, but failed to displace the third DNA strand in the triple
helix displacement assay (23).
Considering the family tree of family A, a notable
observation is that the ATRX mutation was transmitted to all
four offspring from the carrier mother, while the composite
probability of such a transmission is only 6.25%. We therefore
hypothesized if the mutant ATRX allele was more likely to be
transmitted to offspring by carrier mothers and conducted a
meta-analysis of the segregation of ATRX mutations in
published pedigrees (Table II).
There was a tendency towards preferential transmission of the
mutant allele to male offspring (58% of males inherited the mutant
allele); however, the bias was not statistically significant
(P=0.29, χ2 test). The transmission-ratio distortion and
its underlying mechanisms are still poorly understood, with
explanations ranging from meiotic drive (alleles preferentially
retained in the oocyte vs. the polar body) to differences in embryo
survival (24–26). It is worth noting that ATRX is
essential for oogenesis and its loss affects normal chromosome
segregation (27). However, even
without full understanding of the underlying mechanisms,
preferential transmission of mutant alleles may be an important
aspect to be considered in genetic counseling.
Acknowledgements
Not applicable.
Funding
The current study was supported by the ‘Zigote
Center’ and by ‘Research & Innovation srl’.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MS contributed in conceiving and designing the
study; DC and EDG performed the sequencing analysis, AFR and MCI
performed standard and molecular cytogenetics. AKN contributed in
meta-analysis of maternal transmission of ATRX mutations and
conducted χ2 test. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
The guardian of the patient provided written
informed consent for the publication of any associated data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Gibbons RJ, Brueton L, Buckle VJ, Burn J,
Clayton-Smith J, Davison BC, Gardner RJ, Homfray T, Kearney L,
Kingston HM, et al: Clinical and hematologic aspects of the
X-linked alpha-thalassemia/mental disability syndrome (ATR-X). Am J
Med Genet. 55:288–299. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gibbons RJ: Alpha thalassaemia-mental
disability, X linked. Orphanet J Rare Dis. 4:15Review2006.
View Article : Google Scholar
|
|
3
|
Stevenson RE: Alpha-Thalassemia X-linked
Intellectual Disability Syndrome. GeneReviews((R). Adam MP,
Ardinger HH and Pagon RA: University of Washington; Seattle, WA:
1993
|
|
4
|
Stevenson RE: Alpha-Thalassemia X-Linked
Intellectual Disability Syndrome. 2000 Jun 19 [Updated 2020 May
28]. GeneReviews® [Internet]. Adam MP, Ardinger HH,
Pagon RA, et al: University of Washington; Seattle, WA: 1993-2020,
simplehttps://www.ncbi.nlm.nih.gov/books/NBK1449/February
5–2020
|
|
5
|
Garber KB, Warren ST and Visootsak J:
Chapter 107-Fragile X Syndrome and X-linked intellectual
disability. Emery and Rimoin's Principles and Practice of Medical
Genetics (6th edition). Rimoin. 1–27. 2013. View Article : Google Scholar
|
|
6
|
Lin SB, Sun HY, Song XM, Chen LM, Du ML
and Chen Z: Mutation analysis for a Chinese family featuring
X-linked alpha thalassemia/mental disability syndrome. Zhonghua Yi
Xue Yi Chuan Xue Za Zhi. 654–658. 2013.(In Chinese). PubMed/NCBI
|
|
7
|
Argentaro A, Yang JC, Chapman L, Kowalczyk
MS, Gibbson RJ, Higgs DR, Neuhaus D and Rhodes D: Structural
consequences of disease-causing mutations in the ATRX-DNMT3-DNMT3L
(ADD) domain of the chromatin-associated protein ATRX. Proc Natl
Acad Sci USA. 104:11939–11944. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Abidi FE, Cardoso C, Lossi AM, Lowry RB,
Depetris D, Mattéi MG, Lubs HA, Stevenson RE, Fontes M, Chudley AE
and Schwartz CE: Mutation in the 5′ alternatively spliced region of
the XNP/ATR-X gene causes Chudley-Lowry syndrome. Eur J Hum Genet.
13:176–183. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Altiner Ş and Raymond L: A novel ATRX
mutation presenting with intellectual disability and severe
kyphoscoliosis. Fetal Pediatr Pathol. 1–5. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Basehore MJ, Michaelson-Cohen R,
Levy-Lahad E, Sismani C, Bird LM, Friez MJ, Walsh T, Abidi F,
Holloway L, Skinner C, et al: Alpha-thalassemia intellectual
disability: Variable phenotypic expression among males with a
recurrent nonsense mutation-c.109C>T (p.R37X). Clin Genet.
87:461–466. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bouazzi H, Thakur S, Trujillo C, Alwasiyah
MK and Munnich A: Novel ATRX gene damaging missense mutation
c.6740A>C segregates with profound to severe intellectual
deficiency without alpha thalassemia. Indian J Med Res. 143:43–48.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hamzeh AR, Nair P, Mohamed M, Saif F,
Tawfiq N, Al-Ali MT and Bastaki F: A novel missense mutation in
ATRX uncovered in a Yemeni family leads to alpha-thalassemia/mental
disability syndrome without alpha-thalassemia. Ir J Med Sci.
186:333–337. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ion A, Telvi L, Chaussain JL, Galacteros
F, Valayer J, Fellous M and McElreavey K: A novel mutation in the
putative DNA helicase XH2 is responsible for male-to-female sex
reversal associated with an atypical form of the ATR-X syndrome. Am
J Hum Genet. 58:1185–1191. 1996.PubMed/NCBI
|
|
14
|
Li L, Yu J, Zhang X, Han M, Liu W, Li H
and Liu S: A novel ATRX mutation causes SmithFinemanMyers syndrome
in a Chinese family. Mol Med Rep. 21:387–392. 2020.PubMed/NCBI
|
|
15
|
Masliah-Planchon J, Lévy D, Héron D,
Giuliano F, Badens C, Fréneaux P, Galmiche L, Guinebretierre JM,
Cellier C, Waterfall JJ, et al: Does ATRX germline variation
predispose to osteosarcoma? Three additional cases of osteosarcoma
in two ATR-X syndrome patients. Eur J Hum Genet. 26:1217–1221.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Moncini S, Bedeschi MF, Castronovo P,
Crippa M, Calvello M, Garghentino RR, Scuvera G, Finelli P and
Venturin M: ATRX mutation in two adult brothers with non-specific
moderate intellectual disability identified by exome sequencing.
Meta Gene. 1:102–108. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Thakur S, Ishrie M, Saxena R, Danda S,
Linda R, Viswabandaya A and Verma IC: ATR-X syndrome in two
siblings with a novel mutation (c.6718C>T mutation in exon 31).
Indian J Med Res. 134:483–486. 2011.PubMed/NCBI
|
|
18
|
Villard L, Toutlain A, Lossi AM, Gecz J,
Houdayer C, Moraine C and Fontès M: Splicing mutation in the ATR-X
gene can lead to a dysmorphic mental disability phenotype without
alpha-thalassemia. Am J Hum Genet. 58:499–505. 1996.PubMed/NCBI
|
|
19
|
Villard L, Fontés M, Adès LC and Gecz:
Identification of a mutation in the XNP/ATR-X gene in a family
reported as Smith-Fineman-Myers syndrome. Am J Med Genet. 91:83–85.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wilkie AO, Gibbons RG, Higgs DR and
Pembrey ME: X linked alpha thalassaemia/mental disability: Spectrum
of clinical features in three related males. J Med Genet.
28:738–741. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yan H, Shi Z, Wu Y, Xiao J, Gu Q, Yang Y,
Li M, Gao K, Chen Y, Yang X, et al: Targeted next generation
sequencing in 112 Chinese patients with intellectual
disability/developmental delay: Novel mutations and candidate gene.
BMC Med Genet. 20:802019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dyer MA, Qadeer ZA, Valle-Garcia D and
Bernstein E: ASTRX and DAXX: Mechanisms and mutation. Cold Spring
Harb Perspect Med. 7:a0265672017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mitson M, Kelley LA, Sternberg MJ, Higgs
DR and Gibbson RJ: Functional significance of mutations in the Snf2
domain of ATRX. Hum Mol Genet. 20:2603–2610. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Naumova AK, Greenwood CM and Morgan K:
Imprinting and deviation from Mendelian transmission ratios.
Genome. 44:311–320. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang LO, Labbe A and Infante-Rivard C:
Transmission ratio distortion: Review of concept and implications
for genetic association studies. Hum Genet. 132:245-263. 2013.
View Article : Google Scholar
|
|
26
|
LeMair-Adkins R and Hunt PA: Nonrandom
segregation of the mouse univalent X chromosome: Evidence of
spindle-mediated meiotic drive. Genetics. 156:775–783.
2000.PubMed/NCBI
|
|
27
|
De La Fuente R, Baumann C and Viveiros MM:
Chromatin structure and ATRX function in mouse oocytes. Results
Probl Cell Differ. 55:45–68. 2012. View Article : Google Scholar : PubMed/NCBI
|