Introduction
Vein grafts are currently the preferred method for
coronary artery bypass grafting (1). However, because of atherosclerosis,
atherosclerotic plaque rupture and intimal hyperplasia (2), the 10-year primary patency rate is
only 60% (3–5). Few interventions are effective in
preventing vein graft failure (1).
No-touch techniques, antiplatelet (aspirin) therapy and
lipid-lowering agents are effective treatments for atherosclerosis,
but no intervention is unequivocally clinically effective in
intimal hyperplasia, which is the primary reason for vein graft
failure (1). Therefore, a novel
treatment strategy for intimal hyperplasia is essential for
long-term surgical success.
S100 calcium binding protein A8 (S100A8) and A9
(S100A9) belong to the S100 calcium-binding protein family, which
is characterized by the presence of two Ca2+-binding
sites of the EF-hand type (6).
S100A8 and S100A9 normally form a heterodimer called calprotectin
that, when secreted, binds receptor for advanced glycation end
products (RAGE) (7,8) and Toll-like receptor 4 on different
cell types (9,10). In inflammation, S100A8/9 are
primarily secreted by neutrophils and induce a broad range of
processes, including cellular differentiation, apoptosis,
proliferation, migration, calcium homeostasis, energy metabolism
and inflammation (11–14). S100A8/9 are increased in a number
of cardiovascular diseases. The expression levels of S100A8/9 are
increased in coronary artery blood in patients with acute coronary
syndrome where they co-localize with leukocytes and are correlated
with leukocyte activation (15).
This activation induces their adhesion to the endothelium and
increases transendothelial migration (16). Serum S100A8/9 expression levels are
significantly promoted 1 day after percutaneous coronary
intervention in patients with acute myocardial infarction (17). As inflammatory molecules, S100A8/9
have an important role in inducing vascular inflammation (18). Previous research revealed that
S100A8/9 promoted vessel formation (19). However, endothelial cell injury or
exposure of vascular smooth muscle cells to circulating blood
components triggers the development of neointimal hyperplasia
(1). Venous graft failure after
coronary artery bypass graft is caused by the loss of endothelial
cell integrity (20).
Extensive studies have focused on the role of
S100A8/9 in proinflammation (9,18,19);
however, previous findings indicated that a low concentration of
S100A8/9 might be associated with cell growth (21,22)
and angiogenesis-related activity (19). Xu et al (21) observed cell proliferation in a
concentration- and time-dependent manner with maximal effects
observed with 500 ng/ml S100A9. Li et al (19) demonstrated that following treatment
with S100A8 and S100A9, angiogenesis was promoted for endothelial
cells; both proteins increased vessel development in gel plugs that
were subcutaneously injected in BALB/c mice. These findings
indicate that S100A8/9 are associated with endothelial cell growth
and angiogenesis, which might protect vein grafts from neointimal
hyperplasia. However, the downstream signaling mechanisms are not
fully understood, which led the present study to explore their
potential mechanisms. The phosphatidylinositol 3-phosphate kinase
(PI3K)/Akt/mTOR signaling pathway is a regulator of cell growth,
proliferation, metastasis, lipogenesis, angiogenesis and
lipogenesis. In the early stage of neointimal hyperplasia, the
activation of PI3K/Akt/mTOR promotes the proliferation of vascular
smooth muscle cells and increased extracellular matrix (23). The present study focused on
overexpressed S100A8 and S100A9 genes in a vein graft model and
used in vitro human umbilical vein endothelial cells
(HUVECs) to evaluate the effects of exogenous S100A8/9 on
proliferation, migration and angiogenesis, and to explore the
signaling pathways involved.
Materials and methods
Cell culture and viability assay
HUVECs were isolated from umbilical cord veins of
normal pregnancies following a previously described protocol
(24). The cells were cultured in
Endothelial Cell Medium (ScienCell Research Laboratories, Inc.)
containing 100 U/ml penicillin, 100 µg/ml streptomycin and 5% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc.) and 1%
vascular Endothelial Cell Growth Factor (ECGF; ScienCell Research
Laboratories, Inc.) at 37°C with 5% CO2. HUVECs were
pretreated with 10 µg/ml RAGE blocking antibody (cat. no. MAB11451;
R&D Systems) or 100 nM rapamycin for 1 h prior to treating with
S100A8/9 for 48 h at 37°C with 5% CO2. Cell viability
was measured using a Cell Counting Kit-8 (CCK-8) assay
(Sigma-Aldrich; Merck KGaA) according to the manufacturer's
protocol. Briefly, cells (3×104 cells/100 µl per well)
were plated in 96-well microplates, cultured overnight in complete
culture medium, washed and starved in serum-free DMEM (ScienCell
Research Laboratories, Inc.) for 6 h. Cells were then treated with
S100A8/9 mixture (0, 1, 2, 4 and 8 µg/ml) in 5% serum DMEM for 24,
48 and 72 h. The mixture contained the same concentration of
recombinant low endotoxin-grade human S100A8 and S100A9 (CycLex:
Medical & Biological Laboratories Co., Ltd). A total of 10 µl
CCK-8 reagent was added to each well and incubated for 1 h in the
dark and absorbance was quantified at 450 nm using a
SpectraMax® 190 spectrophotometer (Molecular Devices,
LLC).
Cell transfection
RAGE-specific small interfering (si)RNA (si-RAGE)
and rapamycin-insensitive companion of mTOR (Rictor)-specific siRNA
(si-Rictor) (25 nM, final concentration) were transfected into
HUVECs (3×104) in 6-well plates using 10 nM
Lipofectamine® RNAiMAX Transfection Reagent (Thermo
Fisher Scientific, Inc.) for 24 h at 37°C, according to the
manufacturer's instructions. All gene-specific siRNAs (RAGE and
Rictor) and negative controls (cat. no. A06001) were purchased from
Suzhou GenePharma Co., Ltd.. After treatment with 8 µg/ml S100A8/9
for 24 h, cell proliferation, Transwell assays and vascular tube
formation assays were subsequently performed and transfected cells
were harvested at 24 h for reverse transcription-quantitative PCR
(RT-qPCR) or western blotting.
Western blotting
RIPA lysis buffer (TransGen Biotech Co., Ltd.) and
protease inhibitor cocktail (TransGen Biotech Co., Ltd.) were used
to extract total protein from HUVECs. Total protein was quantified
using a bicinchoninic acid assay kit (TransGen Biotech Co., Ltd.).
Proteins (30 µg protein/lane) were separated via 10% SDS-PAGE and
electroblotted onto polyvinylidene difluoride membranes.
Subsequently, the membranes were blocked with TBS containing 5%
non-fat dry milk at 37°C for 1 h. Membranes were incubated
overnight at 4°C with the following antibodies: Anti-β-actin
(1:1,000; Abcam; cat. no. ab6276), anti-phosphorylated (p)-PI3K
(1:1,000; Cell Signaling Technology, Inc.; cat. no. 4257) and total
(t)-PI3K (1:1,000; Abcam; cat. no. ab40776), anti-p-Akt (1:5,000;
Abcam; cat. no. ab81283) and t-Akt (1:1,000; Cell Signaling
Technology, Inc.; cat. no. 4691), anti-p-mTOR (1:1,000; Cell
Signaling Technology, Inc.; cat. no. 5536) and anti-t-mTOR
(1:1,000; Cell Signaling Technology, Inc.; cat. no. 2983), Bcl-2
(1:1,000; Abcam; cat. no. ab32124), Rictor (1:500; Abcam; cat. no.
ab104838), anti-protein kinase Cα (PKCα; 1:1,000; Abcam; cat. no.
32376) and anti-GAPDH (1:1,000; Abcam; cat. no. ab8245). Membranes
were incubated with HRP-conjugated secondary antibodies (cat. nos.
ab6721 and ab6728; 1:3,000; Abcam) at 37°C for 1 h. Finally, the
immunoreactive bands were developed using Supersignal West Femto
Maximum Sensitivity Substrate (Thermo Fisher Scientific, Inc.).
Protein expression levels were semi-quantified using Quantity One
software (version 4.6.6; Bio-Rad Laboratories, Inc.).
Flow cytometry
HUVECs were stimulated with 8 µg/ml S100A8/9 for 24
h. After two washes with PBS, harvested cells were fixed with 75%
ice-cold ethanol in 4°C for 8 h. HUVECs were incubated with Cell
Cycle Staining Kit (Nanjing KeyGen Biotech Co., Ltd.) for 30 min in
the dark according to the manufacturer's instructions. Then, cells
were analyzed using a BD FACSCanto™ II flow cytometer (BD
Biosciences). Data were analyzed using FlowJo software (version
7.6.1; Tree Star, Inc.).
Cell apoptosis
In the present study, the induction of early and
late apoptosis was evaluated by Annexin V-FITC and PI staining and
flow cytometry. In brief, HUVECs were seeded (1.2×106)
in 6-well plates and treated as described in the previous sections.
After cell serum-starvation, 8 µg/ml S100A8/9 was added into the
culture medium and the cells were cultured for another 24 h.
Subsequently, the apoptosis of HUVECs was evaluated using an
Annexin V-FITC/PI kit (BD Biosciences), according to the
manufacturer's protocol. Cells were gently washed with ice-cold PBS
by centrifugation (120 × g for 5 min at 4°C), and resuspended in
500 µl 1X binding buffer supplemented with 5 µl Annexin V-FITC and
5 µl PI each cell suspension and incubated for 15 min at room
temperature in the dark. The samples were analyzed using a BD
FACSCalibur™ flow cytometer (BD Biosciences) and FlowJo software
(version 7.6.1; Tree Star, Inc.).
Transwell migration assay
HUVECs were cultured, transfected with RAGE siRNA
(si-RAGE), Rictor siRNA (si-Rictor) or negative controls (NC), and
then serum-starved as aforementioned. Transfected cells were
harvested by trypsinization. Cells (3×104) in DMEM
supplemented with 5% FBS (ScienCell Research Laboratories, Inc.)
were seeded into the Transwell insert (8 µm pore size; Corning,
Inc.). DMEM supplemented with 5% FBS with or without S100A8/9
protein (2, 4 or 8 µg/ml) was placed in the lower chamber. The
Transwell plates were incubated at 37°C in 5% CO2 for 20
h. Non-migrated cells in the top insert were carefully removed
using a cotton swab, and the cells that had migrated to the lower
surface of the filter were stained with crystal violet dye for 20
min in room temperature. Images were captured from three random
fields per well using a light microscope (magnification, ×200). The
results are expressed as the number of migrated cells per
field.
In vitro vascular tube formation
assay
Transfection of cultured cells was performed as
aforementioned, with modifications. Microplates (48-well) were
coated with 100 µl ice-cold growth factor reduced Matrigel (BD
Biosciences) at 37°C for 1 h. Cells (3×104) were plated
in each well with media with or without S100A8/A9 proteins. The
plates were then incubated at 37°C in 5% CO2 for 4 h.
Cultures were then photographed in three randomly selected fields
using a fluorescence inverted microscope (magnification, ×100) and
tube-like structures were evaluated using ImageJ software (version
1.49; National Institutes of Health).
RT-qPCR
HUVECs were adjusted to a density of
1×105 cells per cm2 in culture media and
incubated with S100A8/9 for 24 h. Total RNA was then extracted from
the cells using TRIzol® (Beijing Transgen Biotech Co.,
Ltd.). Single-stranded complementary DNA was synthesized from total
RNA by reverse transcription using the PrimeScript RT Reagent kit
(Takara Bio, Inc.). RT-qPCR was performed using SYBR-Green master
mix (Takara Bio, Inc.) in 96-well reaction plates using the ABI
StepOnePlus™ Real-Time PCR system (Thermo Fisher Scientific, Inc.).
Optimal reaction conditions for amplification of the target genes
were used according to the manufacturer's recommendations. β-actin
was used as an internal control. Each experiment was performed in
duplicate and repeated independently at least three times.
Expression levels were quantified using the 2−ΔΔCq
method (25). The primers used for
RT-qPCR are presented in Table
I.
 | Table I.Primers used for reverse
transcription-quantitative PCR. |
Table I.
Primers used for reverse
transcription-quantitative PCR.
| Gene | Sequence
(5′→3′) |
|---|
| β-actin | F:
TCACCAACTGGGACGACAT |
|
| R:
GCACAGCCTGGATAGCAAC |
| Bcl-2 | F:
CCGTGGAATGGAATGAGAT |
|
| R:
TGGTCAAACTTGTTGTCCC |
| Rictor | F:
CTGGAAATTCTGGGATACAGTCTCT |
|
| R:
GGGCTTCTATGAACTCATCCGT |
Microarray and bioinformatics analyses
of gene expression
Gene expression data from our previous study of
rabbit vein grafts was used (26).
Briefly, New Zealand white rabbits (2.5–3.0 kg) were subjected to
the induction of anesthesia with 3% sodium pentobarbital solution
(30 mg/kg). Rabbits underwent unilateral jugular vein into common
carotid artery interposition grafting. This procedure involved 2 cm
segments of jugular vein being harvested using a no-touch method.
Segments were stored in an iso-osmotic sodium chloride solution
(0.9 g/l) containing 2 IU/ml of heparin and 50 µg/ml of glyceryl
trinitrate at 23°C until needed. Animals were sacrificed with an
overdose of sodium pentobarbital solution (900 mg/kg), cardiac and
respiratory arrest as a mark of animal death. A total of six
samples were selected from each of the peaks of apoptosis (day 1)
and the peak of PCNA proliferation (day 7) and four samples from
normal veins (Control group). Total RNA from each sample was
quantified using the NanoDrop ND-1,000 system (NanoDrop
Technologies; Thermo Fisher Scientific, Inc.). RNA integrity was
assessed using standard electrophoresis on a denaturing agarose
gel. Sample labelling and array hybridization were carried out
according to the protocol provided by a One-color Microarray-based
Gene Expression Analysis kit by Agilent Technologies, Inc. A
bioinformatics analysis of the gene expression data was performed.
All the animal experiments were carried out according to ethical
principles and protocols approved by Medical Ethical Committee of
The First Affiliated Hospital of Nanchang University.
Statistical analysis
The data are presented as means ± standard deviation
from at least three independent experiments. Significant
between-group differences were analyzed by one-way analysis of
variance followed by a Tukey's post hoc test. The analyses were
performed with SPSS version 22.0 software (IBM Corp.). P<0.05
was considered to indicate a statistically significant
difference.
Results
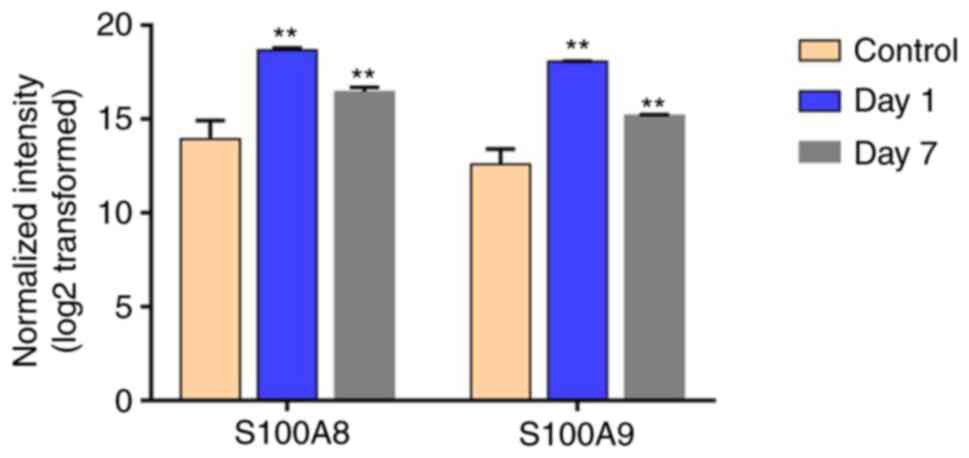
S100A8 and S100A9 expression is
upregulated in a rabbit vein graft model
A total of 1 day after surgery, cell apoptosis
reached a peak and cell proliferation reached a peak 7 days after
surgery (26). To discover the
potential genes in vein grafts whose upregulation was dysregulated
during environmental rehabilitation in arteries, these two time
points were selected to analyze differential gene expression in the
experimental vessel wall relative to the control group. As shown in
Fig. 1, S100A8 and S100A9 were
significantly upregulated genes among the differentially expressed
genes compared with the control group.
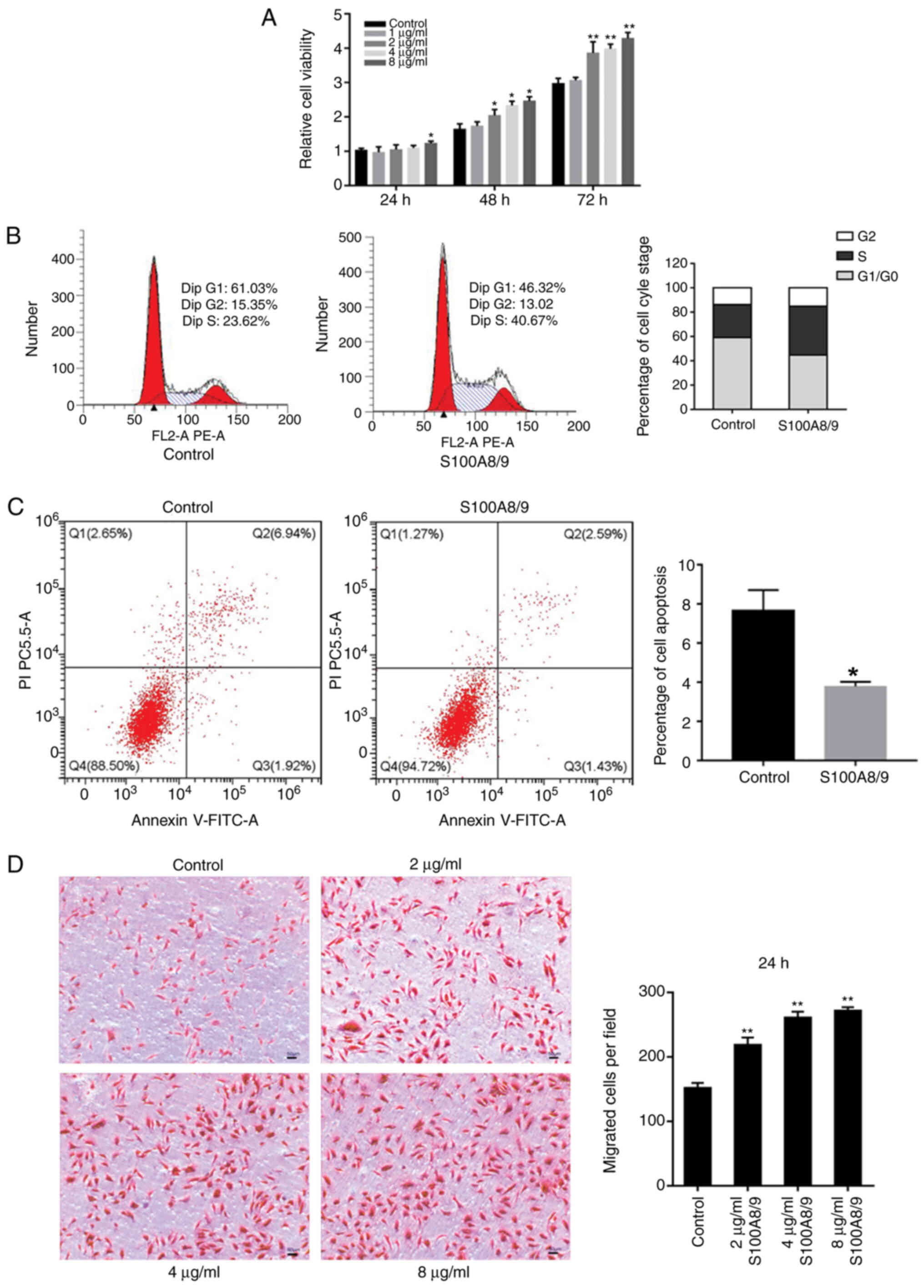
S100A8 and S100A9 promote HUVEC
viability, proliferation and migration
To investigate the influence of S100A8/9 on cell
viability, HUVECs were starved in serum-free DMEM for 6 h before
treatment with S100A8/9 at different times. As shown in Fig. 2A, S100A8/9 exerted a distinct
effect on HUVECs. When S100A8/9 was added to culture media at 1, 2,
4 and 8 µg/ml, it increased HUVEC viability in dose-dependent and
time-dependent manners. The greatest effects were observed with 8
µg/ml at 72 h. As the cell cycle distribution is directly connected
to proliferation, the present study further investigated the cell
cycle distribution of HUVECs incubated with S100A8/9. As shown in
Fig. 2B, the S100A8/9 group
displayed the following distribution: G0/G1
phase, 44.76±1.10%; S phase, 40.10±0.73%; G2/M phase,
15.14±1.59%. The control group displayed the following
distribution: G0/G1 phase, 59.13±1.52%; S
phase, 27.16±5.31%; G2/M phase, 13.70±4.12%. The results
showed that S100A8/9 increased the G1 phase cells to
enter the S phase, thereby improving the cell proliferation
ability. Furthermore, the effects of S100A8/9 on HUVEC apoptosis
were detected using flow cytometry. After treatment with 8 µg/ml
S100A8/9 for 24 h, the cell apoptosis rate was significantly
decreased from 7.39±1.23 to 4.17±0.89 (P<0.05; Fig. 2C). Next, the effects of S100A8/9 on
migration were evaluated using a Transwell chamber system. As shown
in Fig. 2D, DMEM with S100A8/9 at
2, 4 and 8 µg/ml in the lower chamber stimulated cell migration and
maximal effects were observed with 8 µg/ml S100A8/9. These results
indicated that a S100A8/9 promoted cell viability and
migration.
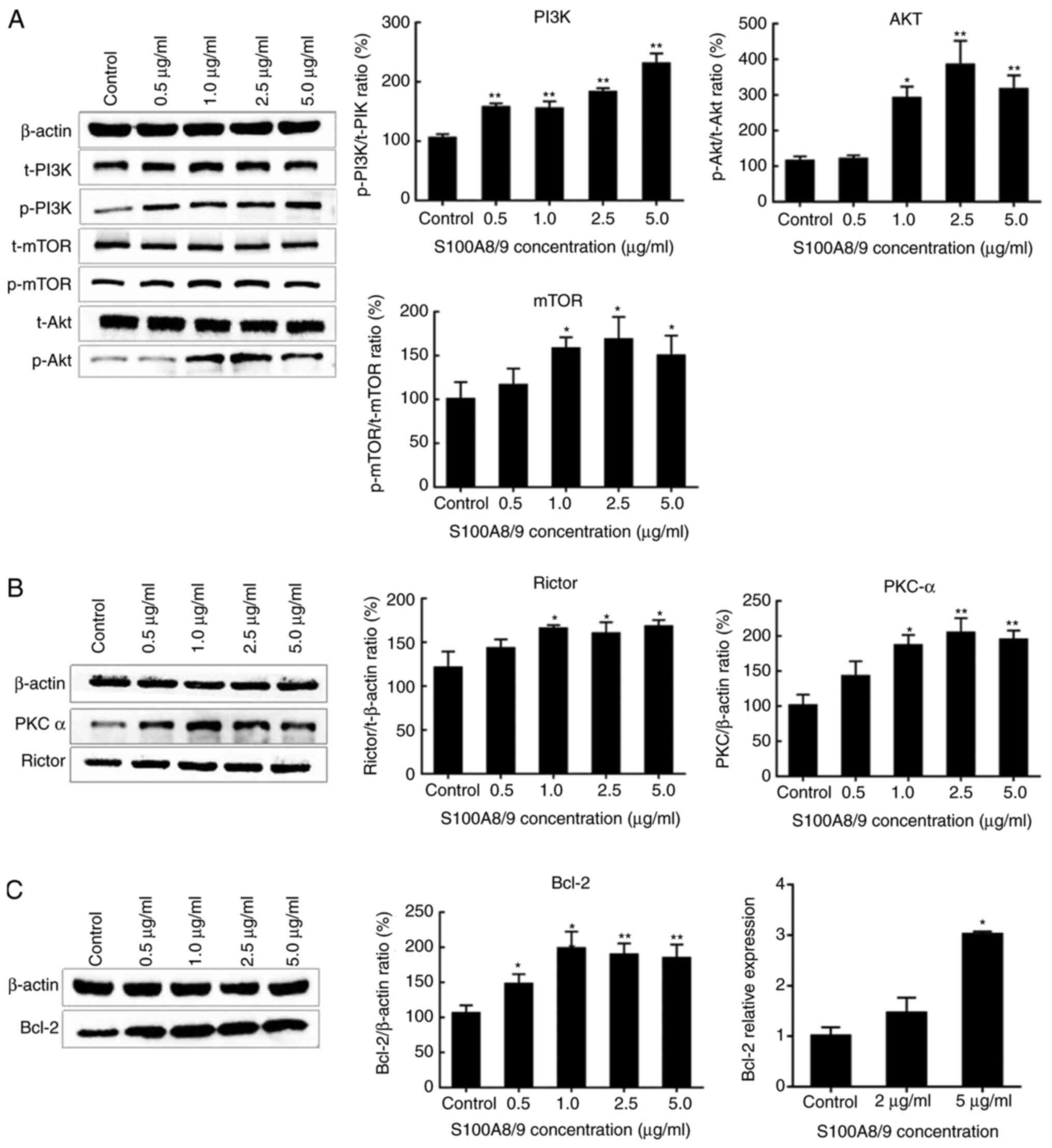
S100A8 and S100A9 stimulate the
PI3K/Akt/mTOR signaling pathway
It was previously demonstrated, in an experimental
rabbit vein graft model, that the mTOR signaling pathway was
upregulated 1 and 7 days after transplantation, when the vein was
in the arterialization process (26). It was hypothesized that this could
be the mechanism leading to remodeling in the vessel wall. To
explore the pathway involved in the effect of S100A8/9 on HUVECs,
HUVECs were cultured with 0.5–5 µg/ml S100A8/9 for 48 h. Western
blot assays (Fig. 3A) showed that
the PI3K/Akt/mTOR pathway was activated, as indicated by the
increased expression ratios of p-PI3K/t-PI3K, p-Akt/t-Akt and
p-mTOR/t-mTOR. The Akt signaling pathway regulates Bcl-2, which is
an important anti-apoptotic protein, therefore, the expression
levels of Bcl-2 protein and mRNA were determined. As shown in
Fig. 3C, S100A8/9 promoted the
expression of Bcl-2 mRNA and protein. mTOR is involved in two
distinct multi-protein complexes, mTORC1/2. mTORC2 contains mTOR,
target of rapamycin complex subunit LST8, Rictor, target of
rapamycin complex 2 subunit MAPKAP1, proline-rich protein 5 and DEP
domain containing mTOR-interacting protein, and regulates Akt and
PKCα phosphorylation and actin cytoskeleton formation (27,28).
The expression of Rictor was examined to determine whether mTORC2
was activated. S100A8/9 significantly upregulated Rictor, similarly
to its downstream factor PKCα (Fig.
3B). This suggested that PI3K/Akt/mTOR and mTORC2 signaling
pathways may participate in S100A8/9-induced HUVEC activation.
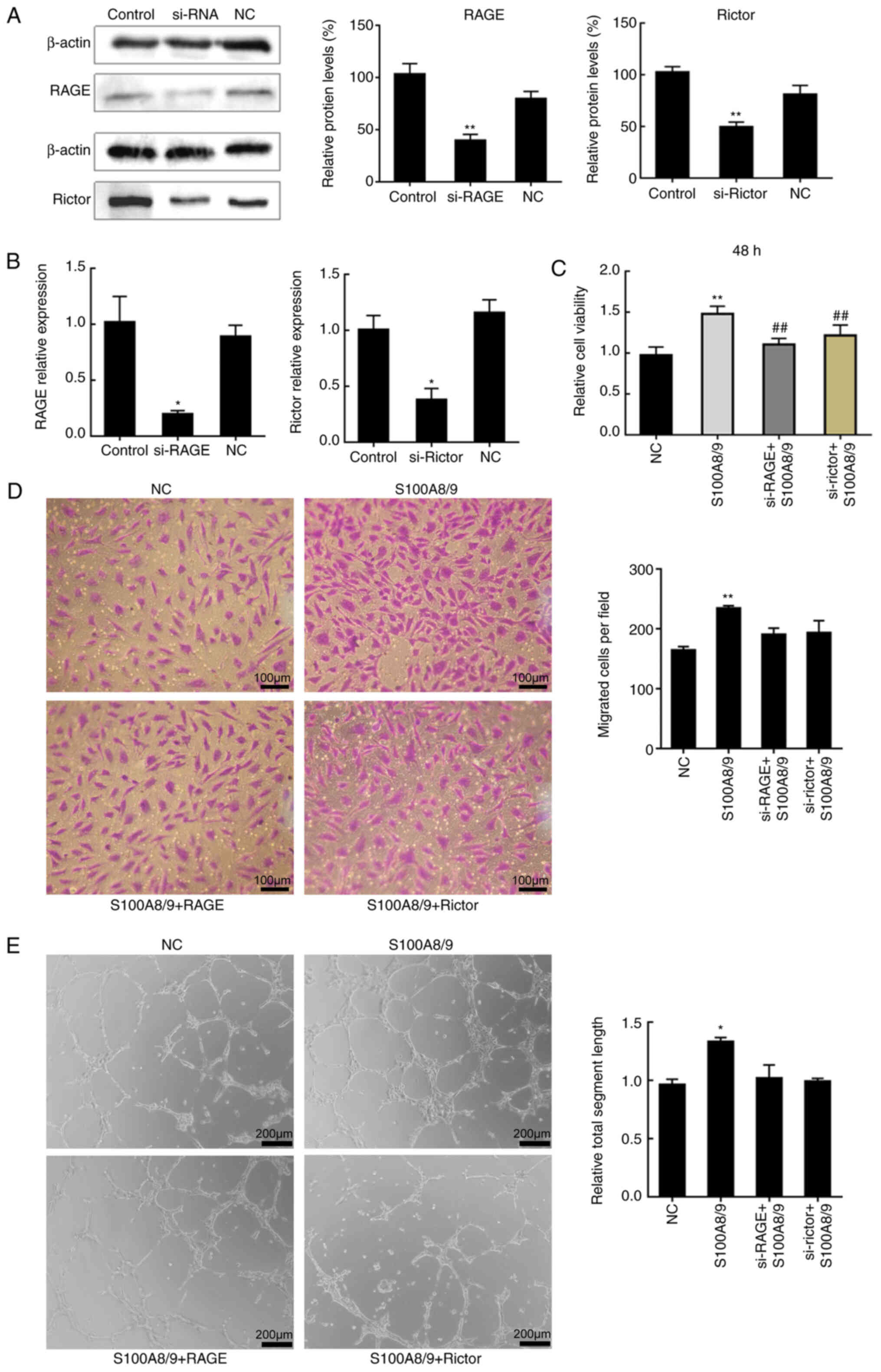
Effects of RAGE blockade on S100A8/9
stimulation of the PI3K/Akt/mTOR and mTORC2 signaling pathways
RAGE is a primary extracellular-membrane receptor of
S100 proteins. Therefore, the influence of RAGE was investigated on
the S100A8/9 signaling pathway, HUVECs were pretreated with a RAGE
blocking antibody or the mTOR inhibitor, 100 nM rapamycin. Fig. 4A shows that the RAGE blocking
antibody decreased the expression ratios of p-PI3K/t-PI3K,
p-Akt/t-Akt and p-mTOR/t-mTOR. Rapamycin also decreased the
expression ratios of p-mTOR/t-mTOR induced by S100A8/9, similarly
to the RAGE blocking antibody. As a main component of mTORC2,
Rictor protein expression was determined. As shown in Fig. 4B, at the protein level, only the
RAGE blocking antibody inhibited expression of Rictor and PKCα. At
the mRNA expression level, Rictor expression was decreased when
cells were pretreated with a RAGE blocking antibody before
stimulation with S100A8/9 (Fig.
4C). Therefore, RAGE appears to be a candidate receptor not
only of the PI3K/Akt/mTOR signaling pathway but also mTORC2
signaling in HUVECs.
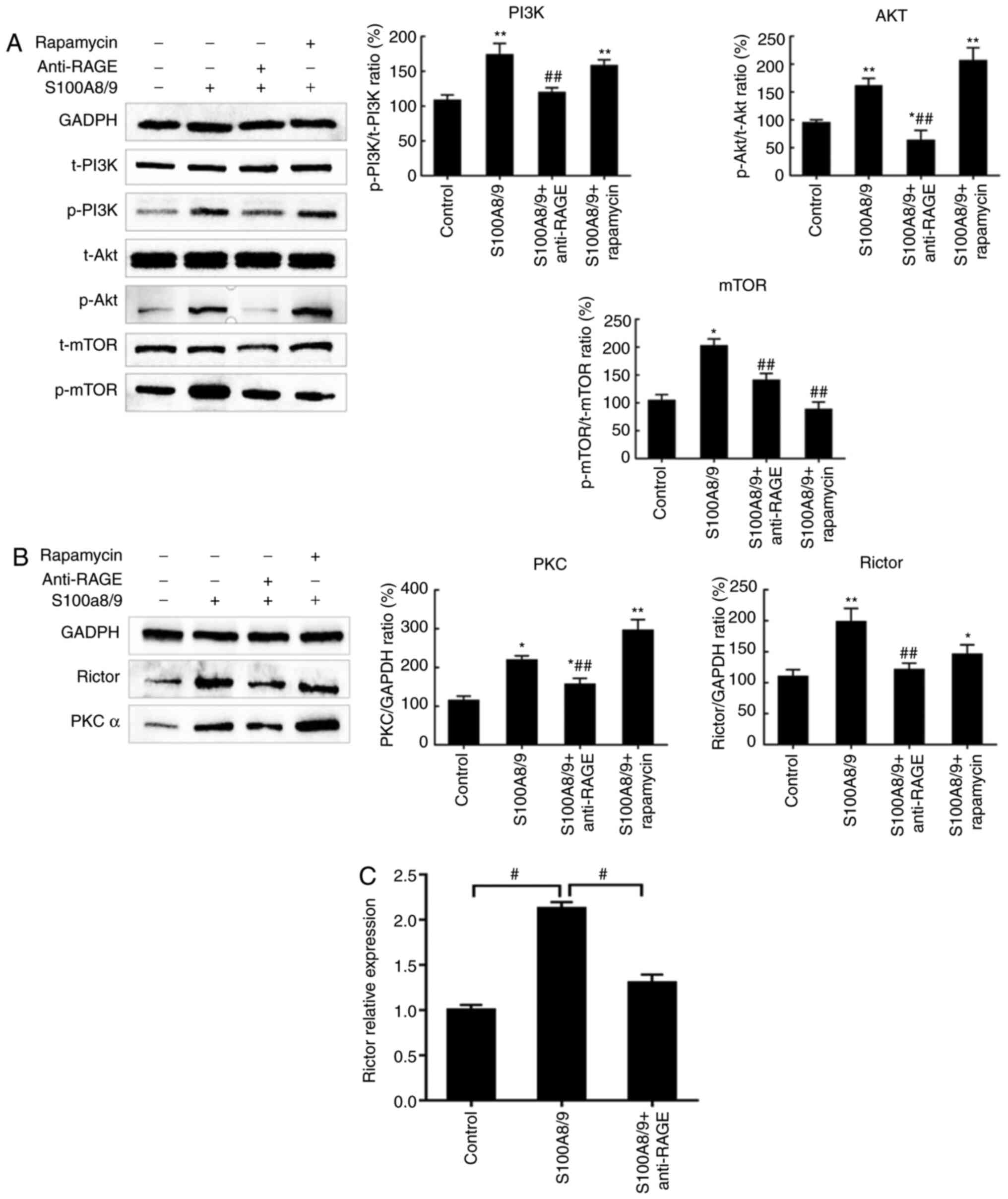 | Figure 4.S100A8/9-induced HUVEC activation is
RAGE-dependent. HUVECs were pretreated with 10 µg/ml RAGE blocking
antibody or 100 nM rapamycin for 1 h prior to treating with
S100A8/9 for 48 h. Total protein was harvested and subjected to
western blotting. (A) RAGE antibody pretreatment blocked the
PI3K/Akt/mTOR pathway, whereas rapamycin only reduced mTOR
phosphorylation. (B) RAGE antibody pretreatment blocked Rictor and
PKCα, whereas rapamycin made no difference. (C) The change in
Rictor mRNA expression was measured by reverse
transcription-quantitative PCR. Data and error bars represent the
mean ± SEM (n=3). *P<0.05, **P<0.01 vs. control group;
#P<0.05, ##P<0.01 vs. S100A8/9 group.
RAGE, receptor for advanced glycation end products; S100A, S100
calcium binding protein A; Rictor, rapamycin-insensitive companion
of mTOR; PKCα, protein kinase Cα; p-, phosphorylated; t-,
total. |
Effect of RAGE and mTORC2 on
S100A8/9-stimulation of angiogenesis and cell migration
mTORC2 is essential for VEGF-induced angiogenic
responses, whereas mTORC1 is known to have a relatively modest
effect on vascular endothelial cell function (29). In addition, PKCα, a distinct
signaling protein downstream of mTORC2, is required for
proliferation of vascular endothelial cells (30). To further demonstrate the role of
RAGE and mTORC2 in S100A8/9 stimulation, a CCK-8 assay was
performed to measure cell viability. HUVECs were transfected with
siRNAs targeting the expression of Rictor or RAGE or a negative
control siRNA. First, transfection efficiency of si-Rictor and
si-RAGE was determined (Fig. 5A and
B). Reduction of Rictor or RAGE expression decreased the
viability of S100A8/9-treated cells by similar amounts (Fig. 5C). Then, HUVEC migration (Fig. 5D) and angiogenesis (Fig. 5E) were measured. As expected,
S100A8/9 increased angiogenesis, whereas depletion of RAGE and
Rictor produced the opposite result (Fig. 5E). Similarly, the increase in
migration was abrogated upon depletion of RAGE or Rictor (Fig. 5D); however, the results were not
significant.
Discussion
In the present study, S100A8/9 was found to enhance
cell growth and angiogenesis. Inhibition of RAGE with si-RAGE
decreased HUVEC viability and migration. S100A8/9 stimulated the
mTORC2 pathway, which was significantly suppressed by rapamycin and
a RAGE-blocking antibody; Rictor deficiency partly reduced cell
viability and angiogenesis induced by S100A8/9.
Recently, the failure of vein grafts induced by
neointimal hyperplasia has attracted attention in research
(5). Neointimal hyperplasia is an
exaggerated wound-healing process in the vessel wall resulting from
surgical injury, arterial environment changes or inflammation
post-surgery (1). It is initiated
by injury to vascular endothelial cells and exposure of vascular
smooth muscle cells to circulating blood molecules (23). A study indicated that
reendothelialization is beneficial to prevent the failure of vein
transplantation, but so far there is no treatment to promote this
process (20). Our previous
studies demonstrated that, in the rabbit vein transplant model, a
high level of cell proliferation was observed in the vascular wall
of vein graft on day 7 after surgery (26). The present study found that S100A8
and S100A9 genes in vein grafts were upregulated during
environmental rehabilitation in arteries 7 days after surgery, when
a high level of cell proliferation is observed in the vascular wall
of vein grafts. At present, primary umbilical vein endothelial
cells are widely used in experiments to explore the function of
venous endothelial cells (31).
Therefore, umbilical vein endothelial cells were selected as the
research subject in the present study. In vitro, the
promotion of proliferation and angiogenesis was observed with
S100A8/9 treatment. These results were consistent with those of Li
et al (19) who showed that
low concentrations of S100A8 and S100A9, either alone or in
combination, promoted angiogenesis-related activity in vascular
endothelial cells. This evidence confirmed the proliferative
function of S100A8/9 on endothelial cells. It was further
speculated that, in preventing neointimal hyperplasia, S100A8/9
maintained the endothelial integrity of vein grafts, which
protected vascular smooth muscle cells from circulating blood
inflammatory molecules.
PI3K/Akt signaling has a key role in multiple
cellular processes, such as promotion of survival and growth in
response to extracellular signals. Activated Akt mediates
downstream responses, including cell survival, growth,
proliferation, cell migration and angiogenesis (16), by phosphorylating a range of
intracellular proteins, which includes mTOR. mTOR links with other
proteins and serves as a core component of two distinct protein
complexes, mTORC21 and mTORC2, which regulate different cellular
processes (32). In the rabbit
vein grafts model, mTORC2 function was increased in the week after
surgery and remained upregulated (26), which could be a major reason behind
the formation of neointimal hyperplasia. In the present study,
S100A8 and S100A9 genes were found to be overexpressed. In
vitro experiments showed that PI3K/Akt/mTOR and mTORC2 pathways
were activated by S100A8/9. This result was consistent with our
previous animal experimental model data (26), which indicated that mTORC2
signaling pathways are activated by S100A8/9. Depleting the core
molecule Rictor from the mTORC2 complex, attenuated the cell
proliferation caused by S100A8/9. These results further indicated
that S100A8/9 promoted cell proliferation, migration and
angiogenesis via the mTORC2 signaling pathway. Considering this
evidence, it was speculated that S100A8/9 protects the integrity of
endothelial cells through mTORC2 signaling pathways.
The present study also investigated the role of RAGE
in S100A8/9-stimulated activation of the mTORC2 pathway and
promotion of angiogenesis. The multi-ligand receptor, RAGE is a
strong candidate for several pathways mediating arterial
inflammation (33). RAGE ligands
include advanced glycation end products, high mobility group box 1
and S100/calgranulins, which includes S10012, S100A8 and S100A9
(34,35). It has been demonstrated that
S100A8/9 interacts with RAGE in HUVECs (36,37).
Furthermore, RAGE regulates the phosphorylation of mTOR and
promotes Beclin-1/VPS34 autophagosome formation (38). Advanced glycation end products
induce cell autophagy by inhibiting the PI3K/Akt/mTOR pathway via
RAGE (39,40). The signaling pathway of RAGE in
endothelial cells has not yet been fully elucidated. The present
results showed that the RAGE blocking antibody reduced activation
of the S100A8/9-upregulated mTORC2 pathway, whereas Rapamycin had
little effect; RAGE deficiency partly reversed the promotion of
angiogenesis. These results provided evidence that RAGE is the
receptor for S100A8/9, and that RAGE-dependent signaling is
involved in mediating the effects of S100A8/9, which is consistent
with the findings of Xu et al (21).
A previous study demonstrated that hypoxia causes a
transient increase in mTORC1 activity and a sustained increase in
mTORC2 activity in vascular endothelial cells, which indicates that
proliferation in vascular endothelium is correlated with mTORC2
activity (41). The knockout
regulatory-associated protein of mTOR (Raptor)/Rictor gene mouse
model demonstrated that absence of Rictor inhibited endothelial
cell proliferation and angiogenesis induced by VEGF in vivo,
which could be achieved by decreasing downstream factor-PKCα,
whereas Raptor knockout had little effect on vascular endothelial
cells (29). Previous evidence
suggested that the chemokine stromal cell-derived factor 1, through
its receptor C-X-C chemokine receptor type 4, increased endothelial
cell migration and microangiogenesis to promote angiogenesis
(27). To block angiogenesis,
mTORC2 signaling is the correct blocking target, rather than mTORC1
(27), however, while rapamycin is
a potent inhibitor of mTORC1, mTORC2 is only partially affected
after long-term exposure (42).
The present results showed that rapamycin failed to decrease
activation of mTORC2 stimulated by S100A8/9, whereas RAGE had a
notable effect on the inhibition of mTORC2 stimulation. These
findings indicated that RAGE could be a potential target in the
mTORC2 pathway.
There are some limitations of the present study. No
clinical evidence has been presented concerning the relationship
between serum expression levels of S100A8/9 and intimal hyperplasia
after vein graft surgery in patients with coronary atherosclerosis.
The results of this study may not be directly applicable to
neointimal hyperplasia in an in vivo coronary artery bypass
grafting model because the findings in this study are from a HUVEC
in vitro model only. Therefore, a new human-mouse chimeric
model based on human-mouse chimeric model reported by Yi et
al (43) is in development. In
future studies, human saphenous vein segments should be
transplanted as abdominal aorta interposition grafts into an
immunodeficient mouse host. In addition, the present study did not
investigate the interaction between the PI3K/Akt/mTOR signaling
pathway and mTORC2. Although, no time-dependent activity of
S100A8/9 was provided for PI3K/Akt/mTOR, the potential mechanisms
underlying the effects of S100A8/9 on promoting cell viability were
investigated. Furthermore, while the western blotting results
(Fig. 3A) may not be optimal, they
are sufficient to draw the conclusion that S100A8/9 stimulates
activation of the PI3K/Akt/mTOR pathway.
In summary, S100A8/9 promoted cell viability,
proliferation and migration via RAGE signaling and activation of
mTORC2 pathways. S100A8 and S100A9 genes in vein grafts were
upregulated in a rabbit vein grafts model. Thus, it was speculated
that S100A8/9 protected the endothelial integrity of vein grafts,
which play an important role in preventing intimal hyperplasia
(20). In addition, RAGE could
represent a potential target in the mTORC2 pathway. The present
study therefore offers a preliminary understanding of the potential
role of S100A8/9 in intimal hyperplasia in vein graft surgery.
Further studies are required to fully understand the role of
S100A8/9 in the pathogenesis of intimal hyperplasia, and vascular
smooth muscle cells are an ideal tool for this so these cells will
be used for future studies.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural
Science Foundation of China (grant no. 81760078) and Jiangxi
Provincial Natural Science Foundation of China (grant no.
20192ACBL21039).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XZ performed the experiments. FX, LC and ZL analyzed
the data and revised the manuscript. FX collected data and wrote
the manuscript. QW conceived and designed the experiments. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Medical
Ethical Committee of The First Affiliated Hospital of Nanchang
University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
de Vries MR, Simons KH, Jukema JW, Braun J
and Quax PH: Vein graft failure: From pathophysiology to clinical
outcomes. Nat Rev Cardiol. 13:451–470. 2016.PubMed/NCBI
|
|
2
|
Owens CD: Adaptive changes in autogenous
vein grafts for arterial reconstruction: Clinical implications. J
Vasc Surg. 51:736–746. 2010.PubMed/NCBI
|
|
3
|
Gasper WJ, Owens CD, Kim JM, Hills N,
Belkin M, Creager MA and Conte MS: Thirty-day vein remodeling is
predictive of midterm graft patency after lower extremity bypass. J
Vasc Surg. 57:9–18. 2013.PubMed/NCBI
|
|
4
|
Conte MS, Owens CD, Belkin M, Creager MA,
Edwards KL, Gasper WJ, Kenagy RD, LeBoeuf RC, Sobel M and Clowes A:
A single nucleotide polymorphism in the p27(Kip1) gene is
associated with primary patency of lower extremity vein bypass
grafts. J Vasc Surg. 57:1179–1185.e1-e2. 2013.PubMed/NCBI
|
|
5
|
Lu DY, Chen EY, Wong DJ, Yamamoto K,
Protack CD, Williams WT, Assi R, Hall MR, Sadaghianloo N and Dardik
A: Vein graft adaptation and fistula maturation in the arterial
environment. J Surg Res. 188:162–173. 2014.PubMed/NCBI
|
|
6
|
Hunter MJ and Chazin WJ: High level
expression and dimer characterization of the S100 EF-hand proteins,
migration inhibitory factor-related proteins 8 and 14. J Biol Chem.
273:12427–12435. 1998.PubMed/NCBI
|
|
7
|
Boyd JH, Kan B, Roberts H, Wang Y and
Walley KR: S100A8 and S100A9 mediate endotoxin-induced
cardiomyocyte dysfunction via the receptor for advanced glycation
end products. Circ Res. 102:1239–1246. 2008.PubMed/NCBI
|
|
8
|
Chang CC, Khan I, Tsai KL, Li H, Yang LW,
Chou RH and Yu C: Blocking the interaction between S100A9 and RAGE
V domain using CHAPS molecule: A novel route to drug development
against cell proliferation. Biochim Biophys Acta. 1864:1558–1569.
2016.PubMed/NCBI
|
|
9
|
Averill MM, Kerkhoff C and Bornfeldt KE:
S100A8 and S100A9 in cardiovascular biology and disease.
Arterioscler Thromb Vasc Biol. 32:223–229. 2012.PubMed/NCBI
|
|
10
|
Vogl T, Propper C, Hartmann M, Strey A,
Strupat K, van den Bos C, Sorg C and Roth J: S100A12 is expressed
exclusively by granulocytes and acts independently from MRP8 and
MRP14. J Biol Chem. 274:25291–25296. 1999.PubMed/NCBI
|
|
11
|
Marenholz I, Heizmann CW and Fritz G: S100
proteins in mouse and man: From evolution to function and pathology
(including an update of the nomenclature). Biochem Biophys Res
Commun. 322:1111–1122. 2004.PubMed/NCBI
|
|
12
|
Leclerc E, Fritz G, Vetter SW and Heizmann
CW: Binding of S100 proteins to RAGE: An update. Biochim Biophys
Acta. 1793:993–1007. 2009.PubMed/NCBI
|
|
13
|
Donato R, Cannon BR, Sorci G, Riuzzi F,
Hsu K, Weber DJ and Geczy CL: Functions of S100 proteins. Curr Mol
Med. 13:24–57. 2013.PubMed/NCBI
|
|
14
|
Gross SR, Sin CG, Barraclough R and
Rudland PS: Joining S100 proteins and migration: For better or for
worse, in sickness and in health. Cell Mol Life Sci. 71:1551–1579.
2014.PubMed/NCBI
|
|
15
|
Buyukterzi Z, Can U, Alpaydin S, Guzelant
A, Karaarslan S, Kocyigit D and Gurses KM: Enhanced S100A9 and
S100A12 expression in acute coronary syndrome. Biomark Med.
11:229–237. 2017.PubMed/NCBI
|
|
16
|
Wang S, Song R, Wang Z, Jing Z, Wang S and
Ma J: S100A8/A9 in Inflammation. Front Immunol.
9:12982018.PubMed/NCBI
|
|
17
|
Li Y, Chen B, Yang X, Zhang C, Jiao Y, Li
P, Liu Y, Li Z, Qiao B, Bond Lau W, et al: S100a8/a9 signaling
causes mitochondrial dysfunction and cardiomyocyte death in
response to ischemic/reperfusion injury. Circulation. 140:751–764.
2019.PubMed/NCBI
|
|
18
|
Stocca A, O'Toole D, Hynes N, Hynes SO,
Mashayekhi K, McGinley L, O'Connell E, Coleman C, Sultan S, Duffy
A, et al: A role for MRP8 in in stent restenosis in diabetes.
Atherosclerosis. 221:325–332. 2012.PubMed/NCBI
|
|
19
|
Li C, Li S, Jia C, Yang L, Song Z and Wang
Y: Low concentration of S100A8/9 promotes angiogenesis-related
activity of vascular endothelial cells: Bridges among inflammation,
angiogenesis, and tumorigenesis? Mediators Inflamm.
2012:2485742012.PubMed/NCBI
|
|
20
|
Ben Ali W, Bouhout I and Perrault LP: The
effect of storage solutions, gene therapy, and antiproliferative
agents on endothelial function and saphenous vein graft patency. J
Card Surg. 33:235–242. 2018.PubMed/NCBI
|
|
21
|
Xu X, Chen H, Zhu X, Ma Y, Liu Q, Xue Y,
Chu H, Wu W, Wang J and Zou H: S100A9 promotes human lung
fibroblast cells activation through receptor for advanced glycation
end-product-mediated extracellular-regulated kinase 1/2,
mitogen-activated protein-kinase and nuclear factor-κB-dependent
pathways. Clin Exp Immunol. 173:523–535. 2013.PubMed/NCBI
|
|
22
|
Ghavami S, Rashedi I, Dattilo BM, Eshraghi
M, Chazin WJ, Hashemi M, Wesselborg S, Kerkhoff C and Los M:
S100A8/A9 at low concentration promotes tumor cell growth via RAGE
ligation and MAP kinase-dependent pathway. J Leukoc Biol.
83:1484–1492. 2008.PubMed/NCBI
|
|
23
|
Owens CD, Gasper WJ, Rahman AS and Conte
MS: Vein graft failure. J Vasc Surg. 61:203–216. 2015.PubMed/NCBI
|
|
24
|
Jaffe EA, Nachman RL, Becker CG and Minick
CR: Culture of human endothelial cells derived from umbilical
veins. Identification by morphologic and immunologic criteria. J
Clin Invest. 52:2745–2756. 1973.PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI
|
|
26
|
Wang Q, Wan L, Liu L and Liu J: Role of
the mTOR signalling pathway in experimental rabbit vein grafts.
Heart Lung Circ. 25:1124–1132. 2016.PubMed/NCBI
|
|
27
|
Ziegler ME, Hatch MM, Wu N, Muawad SA and
Hughes CC: mTORC2 mediates CXCL12-induced angiogenesis.
Angiogenesis. 19:359–371. 2016.PubMed/NCBI
|
|
28
|
Lieberthal W and Levine JS: Mammalian
target of rapamycin and the kidney. I. The signaling pathway. Am J
Physiol Renal Physiol. 303:F1–F10. 2012.PubMed/NCBI
|
|
29
|
Wang S, Amato KR, Song W, Youngblood V,
Lee K, Boothby M, Brantley-Sieders DM and Chen J: Regulation of
endothelial cell proliferation and vascular assembly through
distinct mTORC2 signaling pathways. Mol Cell Biol. 35:1299–1313.
2015.PubMed/NCBI
|
|
30
|
Tandon M, Chen Z and Pratap J: Runx2
activates PI3K/Akt signaling via mTORC2 regulation in invasive
breast cancer cells. Breast Cancer Res. 16:R162014.PubMed/NCBI
|
|
31
|
Li X, Sun Y, Huang S, Chen Y, Chen X, Li
M, Si X, He X, Zheng H, Zhong L, et al: Inhibition of AZIN2-sv
induces neovascularization and improves prognosis after myocardial
infarction by blocking ubiquitin-dependent talin1 degradation and
activating the Akt pathway. EBioMedicine. 39:69–82. 2019.PubMed/NCBI
|
|
32
|
Lipton JO and Sahin M: The neurology of
mTOR. Neuron. 84:275–291. 2014.PubMed/NCBI
|
|
33
|
Yan SF, Ramasamy R and Schmidt AM: The
RAGE axis: A fundamental mechanism signaling danger to the
vulnerable vasculature. Circ Res. 106:842–853. 2010.PubMed/NCBI
|
|
34
|
Palanissami G and Paul SFD: RAGE and its
ligands: Molecular interplay between glycation, inflammation, and
hallmarks of cancer-a review. Horm Cancer. 9:295–325.
2018.PubMed/NCBI
|
|
35
|
Yamagishi SI and Matsui T: Role of ligands
of receptor for advanced glycation end products (RAGE) in
peripheral artery disease. Rejuvenation Res. 21:456–463.
2018.PubMed/NCBI
|
|
36
|
Pruenster M, Vogl T, Roth J and Sperandio
M: S100A8/A9: From basic science to clinical application. Pharmacol
Ther. 167:120–131. 2016.PubMed/NCBI
|
|
37
|
Roth J, Vogl T, Sorg C and Sunderkötter C:
Phagocyte-specific S100 proteins: A novel group of proinflammatory
molecules. Trends Immunol. 24:155–158. 2003.PubMed/NCBI
|
|
38
|
Kang R, Tang D, Schapiro NE, Livesey KM,
Farkas A, Loughran P, Bierhaus A, Lotze MT and Zeh HJ: The receptor
for advanced glycation end products (RAGE) sustains autophagy and
limits apoptosis, promoting pancreatic tumor cell survival. Cell
Death Differ. 17:666–676. 2010.PubMed/NCBI
|
|
39
|
Hou X, Hu Z, Xu H, Xu J, Zhang S, Zhong Y,
He X and Wang N: Advanced glycation endproducts trigger autophagy
in cadiomyocyte via RAGE/PI3K/AKT/mTOR pathway. Cardiovasc
Diabetol. 13:782014.PubMed/NCBI
|
|
40
|
Chen J, Zhao D, Zhu M, Zhang M, Hou X,
Ding W, Sun S, Bu W, Feng L, Ma S and Jia X: Paeoniflorin
ameliorates AGEs-induced mesangial cell injury through inhibiting
RAGE/mTOR/autophagy pathway. Biomed Pharmacother. 89:1362–1369.
2017.PubMed/NCBI
|
|
41
|
Li W, Petrimpol M, Molle KD, Hall MN,
Battegay EJ and Humar R: Hypoxia-induced endothelial proliferation
requires both mTORC1 and mTORC2. Circ Res. 100:79–87.
2007.PubMed/NCBI
|
|
42
|
Sarbassov DD, Ali SM, Sengupta S, Sheen
JH, Hsu PP, Bagley AF, Markhard AL and Sabatini DM: Prolonged
rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell.
22:159–168. 2006.PubMed/NCBI
|
|
43
|
Yi T, Fogal B, Hao Z, Tobiasova Z, Wang C,
Rao DA, Al-Lamki RS, Kirkiles-Smith NC, Kulkarni S, Bradley JR, et
al: Reperfusion injury intensifies the adaptive human T cell
alloresponse in a human-mouse chimeric artery model. Arterioscler
Thromb Vasc Biol. 32:353–360. 2011.PubMed/NCBI
|