Introduction
Cystic fibrosis (CF) is an autosomal recessive
disease that can be attributed to the disrupted function of the CF
transmembrane conductance regulator (CFTR) gene
(1,2). Although CF predominantly affects the
lungs, it is a multiorgan disease (3), affecting the pancreas, liver, kidneys
(4) and intestine (5). CFTR mutation is the cause of the
pathogenesis of CF (6) and CF is
generally a result of the deletion of the phenylalanine at the
508th position of CFTR, which is induced by the loss of three
nucleotides (7). In vertebrates,
CFTR serves as a membrane protein and participates in the functions
of Cl− channels (8,9). Due
to its important regulatory functions, CFTR is ubiquitous
throughout the body and is expressed in epithelial cells in the
kidney, pancreas, airway, intestine (4), sweat glands and the male reproductive
tract, where it serves a fundamental role in the transepithelial
fluid (10). The number of
identified CF-associated mutations are increasing, with ~1,700 CFTR
mutations being previously recognized to be CF-prone (11); however, this number was
re-estimated at 383 in 2019 according to the Clinical and
Functional Translation of CFTR website (www.cftr2.org; date of access: 05/08/20). These
potential mutations were screened by a specific criteria that
determines the mutations responsible for the onset of CF: Firstly,
the mutation could cause changes in the amino acid sequence,
affecting both the expression level and functions of CFTR (12); secondly, the mutation introduces
premature signals and exhibits a novel amino acid sequence that is
absent in the normal CFTR gene (13).
The prevalence of CF varies with ethnicity (14,15).
The relatively high incidence of CF among Caucasians may be
attributed to their increased number (>1,400) of CFTR mutations
(16). Furthermore, while 1/3,000
of Caucasians will develop CF, the incidence is lowered to 1/15,000
among the African population, further decreasing in Asian
populations to 1/30,000, compared to the aforementioned two ethnic
groups (15). The ratio of CF
incidence between male and female is 1:1; however, the mortality
rate of CF-associated lung infections is higher among female
patients as they are subjected to greater deterioration of
pulmonary function at puberty. These gender/age gaps have been
proposed to be a result of the elevation in the hormone secretion
(including estrogen) in adults, which may disrupt airway ion
transport in lungs (17).
There are two major molecular subtypes of CF:
Classic CF and non-classic CF. Non-classic CF refers to CF with
better prognostic outcome, as certain functions of the CFTR protein
are preserved, providing advantages for survival. Non-classic
patients with CF have ≥1 copy of a defect CFTR gene with
partially conserved CFTR protein functions. Due to the partial
preservation of pancreatic exocrine functions, the symptoms of
digestion disorders are less common among patients suffering from
this milder type of CF. In contrast, patients suffering from
classic CF have completely lost their functional CFTR protein. This
subtype is characterized by persistent bacterial infection in the
airways and sinuses, disrupted fat digestion due to the lack of
pancreatic exocrine, male dysgenesis due to obstructive azoospermi
and increased sweat Cl− levels (18–20).
The original description of CF can be dated back to
1938 (21). Since then, progress
in the understanding of CF has been made in a step-by-step manner
and the following 50 years has witnessed remarkable improvement in
life expectancy and life quality among patients with CF (22), which may be attributed to
technological innovations. In the late 1950s, a stimulated sweat
test to diagnose patients with CF through Cl− or Na
levels was developed (23) based
on the recognition of the altered electrolyte composition in sweat
(24). The preliminary works
contributed markedly to the diagnosis of CF and the understanding
of CF was further promoted nearly 30 years later due to the
discovery of the CFTR gene, a key mediator of CF (9), which enabled the diagnosis of CF by
directly identifying 2 mutated CF alleles (25). Aside from improved diagnostics,
numerous therapies have been applied to treat CF, including
antibiotics against infections, nutritional supplementation and/or
lung transplantation, through which the life expectancy of patients
with CF can be significantly prolonged (26).
Despite prognostic improvements of CF, the median
survival of patients with CF is <50 years (22). As described above, as the molecular
mechanisms in CF are associated with ethnical and sex differences
in terms of incidence rate, they can also be used to determine
phenotypes of CF. Therefore, the innovation of methods for the
detection and identification of CF at the molecular level will be
beneficial to the diagnosis and prognosis of CF. The current review
aimed to summarize the recent research advances of CF, including
technical improvement in the understanding of the molecular
mechanism of CF. Additionally, the increasing number of molecular
markers that have the potential to improve diagnostic and
prognostic outcomes of CF are discussed. Briefly, a PubMed
(pubmed.ncbi.nlm.nih.gov; date of access: 13/08/2020) search was
conducted using the following key words: ‘Cystic fibrosis’,
‘molecular’, ‘diagnosis’, ‘prognosis’ and ‘therapy’. Examples were
chosen as long as they fulfilled one of the following eligibility
criteria: i) Provided genetic information regarding the pivotal
role of CFTR in CF; ii) described the latest progresses in parsing
the molecular mechanisms underlying CF using novel techniques; iii)
demonstrated the association between CF and other bioactive
molecule (molecular chaperone) and the potential clinical
implementations, including CF diagnosis and treatment.
Molecular mechanism underlying the CFTR
mutation in CF
Molecular structure of CFTR
The molecular weight and length of the CFTR protein
are 1,480 amino acids and 168,173 Da, respectively (7,12,27).
The length of its coding sequence, which encodes the amino acid
sequence for protein products, is 4,443 bp (28). The intron-free sequence of the CFTR
transcript is 6,129 bp in length (12,28),
whereas the normal allelic variant for CFTR is ~250,000 bp in
length and contains 27 exons (12,28).
CFTR is comprised of 5 functional domains (12): Two domains (MSD1 and MSD2)
controlling membrane-spanning, which constitute the ion channel for
Cl− transportation; an R domain, which exerts regulatory
roles; and two domains (NBD1 and NBD2) that bind and catalyze the
hydrolysis of adenosine triphosphate.
Biological functions of CFTR
The CFTR protein is positioned in the cell membrane
(29) and is associated with
proteins involved in the active transportation of material through
the cell membrane (12,30). Specifically, CFTR regulates the
movement of Cl−. Therefore, defects in CFTR gene
can render the CFTR protein absent or dysfunctional, thereby
blocking the transportation of Cl− to the cell surface
(29,30). Additionally, aside from
Cl−, CFTR regulates the epithelial Na channel (31). Abnormalities in the CFTR protein
disrupt the balance between Na and Cl− ions (30,32),
which leads to changes in mucous constituents and abnormal
reabsorption of H2O. This produces a layer of thick,
sticky mucus that cannot be removed by cilia, which eventually
causes inadequate mucociliary function and chronic infections
(33). This can be fatal. In the
lungs, accumulated mucus can become infested with bacteria and the
chronic inflammation leads to pneumonia, resulting in deterioration
with life-threatening difficulties in breathing. Given the
molecular mechanisms of the deficiency of CFTR, the common symptoms
of CF include severe cough and shortness of breath; however, CF can
also lead to abnormal bowel movements, difficulty in gaining weight
and infertility (12,29).
Classification of CFTR mutations
Based on the effects on protein translation,
cellular processing or channel gating of CFTR (28,30),
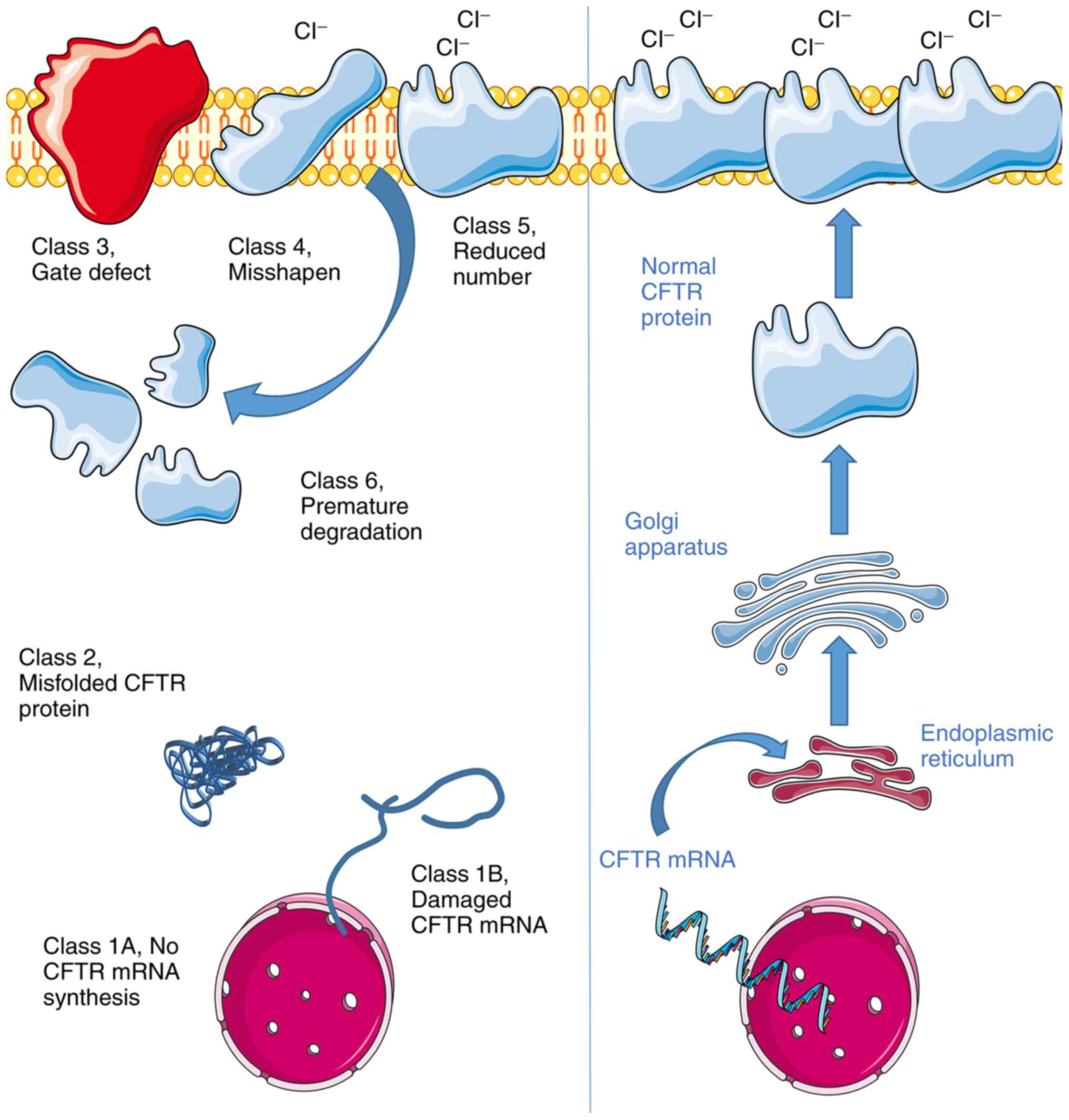
several different classification systems (Fig. 1) have been proposed over the years.
Generally, the Class 1 mutation results in severe disease, as this
mutation prevents the CFTR protein from being generated. Patients
with Class 1A mutation do not synthesize any CFTR mRNA.
Furthermore, patients with Class 1B produce damaged CFTR mRNA,
which cannot be converted into protein (28). In Class 2 mutations, the CFTR
protein is produced; however, it is misfolded. The misfolded
protein will be prevented from migrating to the cell membrane. In
Class 3 mutations, channels in the CFTR protein are not properly
opened due to gate defect (29,32,34).
For the Class 4 mutation, while the CFTR protein is responsive to
cell signaling, it is misshapen, resulting in a limited flow of
Cl− ions. Furthermore, in Class 5 mutations,
insufficient CFTR protein is produced, leading to a reduction in
the number of CFTR protein channels at the cell membrane (35). In class 6 mutations, less stable
protein is prematurely degraded after it reaches the cell surface.
Relatively, this mutation is less severe compared with the other
mutations and, therefore, is a milder subtype. Generally, the Class
1, 2 and 3 mutations are more common and responsible for
insufficiency in the organs suffering from CF.
Technical advances and implementations in
CF
PCR analysis of CF
PCR is a widely used laboratory technique that
allows for the semi-quantification of mRNAs. As early as 1992,
allelic specific-PCR was used for detecting F508del mutation in
CFTR (36). In the following
decades, expression of CFTR had been verified by various models by
reverse transcription-quantitative PCR (RT-qPCR). Certain
implementations of RT-qPCR in CF include the following: i) In CF
cell IB3-1 transduced with CFTR vectors, CFTR mRNA expression was
detected using RT-qPCR, whereby the efficiency of transduction was
measured (37); and ii) multiplex
fluorescent RT-qPCR was used for scanning the exons to detect large
CFTR rearrangements (38).
Aside from the aforementioned utilizations in CFTR
detection, PCR is frequently performed to examine bacterial
infection in CF; for instance, PCR was used to detect
Aspergillus fumigatus DNA, which commonly infects the
airways of patients with CF (39),
in samples collected from patients with CF. Preimplantation genetic
testing (PGT) is an important method to detect CF before or at
pre-embryonic stages (40). The
updated version of the PGT guidelines regarding CF proposed that
PCR analysis should be performed to detect the causative
mutation(s), along with associated polymorphisms within or near to
the CFTR gene (41).
Nevertheless, PCR has its own limitations. For instance, during
unequal allelic PCR amplification, allele dropout can hamper the
detection of CFTR mutations, as the annealing of a primer to the
matched allelic sequence is predominated as compared to its
mismatched counterpart (42).
Implementation of next-generation
sequencing (NGS) in CF
The revolutionary innovation of sequencing
technologies, including NGS platforms, allows for the detection of
a broader spectrum of potential mutations in CF, particularly
single-nucleotide polymorphism (43), which are hypothesized to be a cause
of CF (44). NGS outperformed
whole genome sequencing in terms of cost-efficiency and its
accuracy is guaranteed by stringent thresholds during data
processing (43). Therefore, NGS
has been widely used in carrier (at-risk individual) screening,
including CFTR mutation screening, to improve genetic counseling
and reduce the incidence of CF among carriers' (at-risk-couples)
descendants (43). In another
study of methodology establishment, NGS-based expanded carrier
screening, which determines variants through hybridization capture
gene enrichment, identified several genetic alterations, including
copy-number variants in the CFTR gene. The combination of
NGS and variant interpretation achieved higher accuracy in
identifying CF-associated phenotypes compared with the traditional
method (23 variants screening) (45). Furthermore, by retrospectively
performing NGS assays on patients with single CF mutated screened
by sweat tests, all CFTR mutations were correctly detected,
indicating that NGS assays were completely concordant with
traditional methods (46). These
reports demonstrated the effective implementation of NGS in CF
detection, particularly at the early stage of the disease.
Gene editing for CF
Clustered regularly interspaced short palindromic
repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) is an emerging
technology by which Cas9 proteins work in conjunction with guide
RNA molecules and locate the site of target DNA sequence prior to
cutting it out (47), following
which the gap can be filled with the corrected gene sequence
through the endogenous cellular regeneration. Therefore, this
technology can be implemented in different single-gene-driven
heritable deficiencies. With the advances in such gene editing
technology, a promising future has been demonstrated in regard to
the replacement of defective CFTR gene at the DNA level,
through which normal CFTR function could be fundamentally restored
(48). Although gene editing is
still at its infant stage due to the relatively high off-target
rate (49), it has demonstrated
greater potential when compared to traditional CF therapies
targeting (instead of editing) DNA, RNA or proteins. For instance,
the functional repair of CFTR has been successfully performed in an
in vitro model derived from stem cells of patients with CF,
namely intestinal organoids (50).
Another approach, Zinc finger nucleases-mediated gene editing, was
used to correct defective CFTR in induced pluripotent stem cells
(51). Notably, a mutation which
could cause β thalassemia was corrected in human embryos using
CRISPR/Cas9, indicating the capability of embryonic gene editing
with this technique (52). Thus,
we hypothesized that CRISPR/Cas9-induced gene editing in embryos
threatened with potential mutations is a promising for the future
treatment of CF.
Molecular regulators in CF
Non-coding RNAs (ncRNAs) in CF
Although CF is monogenic, the phenotypes of patients
with CF are heterogeneous, which may be attributed to multiple
regulators that contribute to CF pathogenesis (53). Non-coding (nc)RNAs are a type of
RNA molecule that do not translate into a protein. Instead, ncRNAs
exerts regulatory roles in multiple biological processes, such as
translation (54), RNA splicing
(55) and gene regulation
(56), among which microRNA (miRNA
or miR), long non-coding RNA (lncRNA) and circular RNA (circRNA)
have been extensively studied. The current review discusses several
ncRNAs that participate in CF pathogenesis.
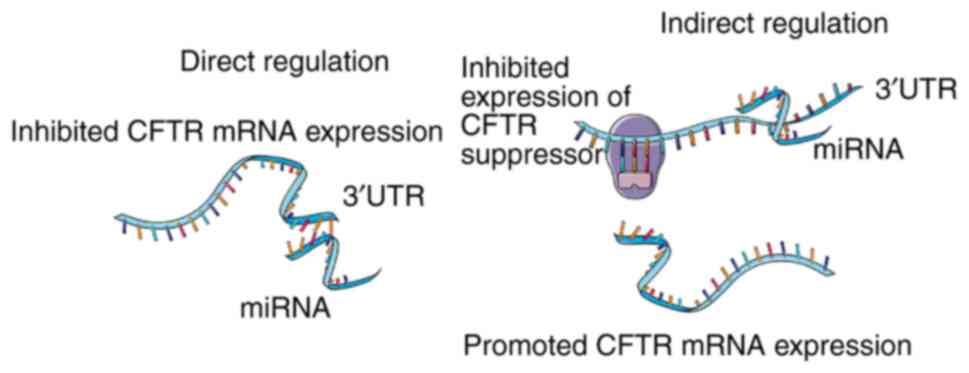
miRNA and CF
Previous studies have demonstrated that miR-145,
miR-494 and miR-101 directly or indirectly target and regulate CFTR
(53,57–59).
The inhibition of miR-145 through peptide nucleic acids was
reported to promote the expression of CFTR, as miR-145 binds to the
3′-untranslated region (3′UTR) of the CFTR gene (57). Additionally, the interaction of
miR-494/miR-101 and CFTR was verified and confirmed through
luciferase reporter assays (58).
Considering evidence that has demonstrated the inhibitory effects
of these miRNAs on CFTR, the exacerbated pulmonary condition caused
by air pollution or cigarette smoke was studied and attributed to
elevated miR-101 and miR-144 (60). Although most miRNAs that directly
target CFTR serve as suppressors, certain miRNAs exert indirect
regulatory roles on CFTR to promote its expression (Fig. 2). For instance, in primary
epithelial cells derived from CF bronchia (CFTR defect phenotype),
miR-138 was reported to downregulate the expression of SIN3
transcription regulator family member A (SIN3A), a negative
transcriptional regulator of CFTR (61). In an indirect manner, miR-138
promoted the expression of CFTR through alleviating the repression
that SIN3A imposed on CFTR.
Furthermore, miRNAs participate in other biological
processes. In CF lung epithelial cells, miR-155 promoted
inflammation through the inositol 5′-phosphatase 1-PI3K/Akt
cascade. Moreover, as chronic bacterial lung infection is a major
cause of CF morbidity, exhaled breath condensate was used for miRNA
profile analysis of patients with CF with microbial infection. The
results demonstrated that 6 miRNAs (has-miRNA-432-5p,
hsa-miRNA-3170, hsa-miR449c, hsa-miR-1276, hsa-miR-1247 and
hsa-miR-548) were identified as potential biomarkers for patients
with CF and chronic Pseudomonas infection (62). These reports indicated that miRNAs
serve crucial functions in the pathogenesis of CF and are key
diagnosis and prognosis markers for CF.
lncRNA and CF
Dysregulation of lncRNAs have been reported to be
associated with chronic pulmonary infection, adaptive immune
responses and inflammation in patients with CF (63). By working concurrently with several
proteins, lncRNA BGas (a novel long noncoding RNA located in the
intron 11 of the CFTR gene) regulated CFTR by regulating its
local chromatin and DNA structure (53). Microarray profiling of lncRNAs
revealed one upregulated (X-inactive specific transcript) and
several downregulated (HOX Transcript Antisense RNA, Metastasis
Associated Lung Adenocarcinoma Transcript 1 and Toll Like Receptor
8 Antisense RNA 1) lncRNAs in the patients with CF compared with
the matched non-CF controls (64).
Although the current evidence regarding the association between
lncRNAs and CF is relatively limited, it is notable that in a
comparative study, 636 and 1,974 differentially expressed lncRNAs
were found in two groups of patients suffering from CF airway
epithelium or CF lung parenchyma (compared with the matched non-CF
counterparts), respectively. By analyzing these lncRNAs in a
comprehensive manner, the antisense lncRNA RN7SK Pseudogene 237 was
found to be significantly altered in CF airway tissues, whereas the
downregulation of two intergenic lncRNAs, long intergenic
non-protein coding RNA (LINC)01023 and LINC00176 were confirmed in
CF parenchyma tissues (65).
Additionally, in silico analysis of RNA-seq data
demonstrated that Pseudomonas aeruginosa infections lead to
108 altered lncRNAs expression between respiratory epithelial cells
derived from patients with CF and non-CF donors. Among these
lncRNAs, LINC00862 and CTD-2619J13 were significantly altered at
different time points throughout the process (0 h prior to
infection and 2, 4 and 6 h following infection) (63). These studies indicated that lncRNAs
exerted important regulatory roles in CF, which still remain to be
fully elucidated.
circRNA and CF
In the field of RNA, circRNAs are endogenous ncRNAs.
These RNAs have been identified in organisms, including eukaryotes,
archaea, bacteria and viruses, and act as a sponge for certain
miRNAs in pulmonary diseases (66). Currently, reports regarding the
potential roles of circRNAs in CF are relatively limited. However,
due to their close regulatory association with miRNAs, it is likely
that circRNAs participate in the progression of CF. Specifically,
in bladder cancer, circ-solute carrier family 8 member A1 was
reported to be a sponge of miR-494 (67), whereas direct interactions between
circ-baculoviral IAP repeat containing 6 and miR-145 were indicated
in embryonic stem cells (68).
Reports concerning miR-494 and miR-145 and their involvement in
CFTR were revealed (57,58), indicating that these regulatory
circRNAs may regulate CFTR through these miRNAs. Analogously, it
has been hypothesized that numerous miRNAs regulating CFTR may be
targets for various circRNAs, which might be involved in CF. This
is a novel research avenue to consider in the future since the
elucidation of the mechanisms driven by cirRNAs will provide a
further understanding on the pathogenesis of CF and its
complications.
Epigenetic modifications in CF
Epigenetics is a mechanism that alters gene
expression without changing the fundamental DNA sequence. The two
mostly known mechanisms underlying epigenetic modifications include
histone modifications and DNA methylation (69), both of which are involved in
regulation of CFTR. It had been hypothesized that aside from
ncRNAs, epigenetics is a contributing factor in the disease
variability of CF (70).
Additionally, epigenetic mechanisms have been proposed to be an
activator of host defenses that induce a robust immune response
(71). The association between
immunity and epigenetics has demonstrated that DNA methylation at
numerous gene loci in lung macrophages was responsible for the
malfunction of innate immune cells in lungs with CF (72). Additionally, differentially altered
DNA methylation at CpG sites was associated with lung function and
their overexpression was demonstrated in numerous regulatory genes
responsible for cell adhesion (for example, ETS homologous factor)
and inflammatory responses (for example, baculoviral IAP
repeat-containing protein 1) (73)
in nasal epithelial samples from patients with CF (74). Furthermore, acetylation has been
proposed to be associated with CF. For instance, the inhibition of
histone deacetylase (HDAC)7 was demonstrated to restore the
function of F508del (75) and
HDAC2 was reported to be responsive to defective CFTR function
(76). A previous study
demonstrated that microtubule deacetylase regulated cholesterol
accumulation and NF-κB activation in CF cells through the
HDAC6-Ac-tub cascade, which corroborated with the findings that
HDAC6 may be a therapeutic site for various CF phenotypes (77). Collectively, therapeutic approaches
for CF that target epigenetic mechanisms have been considered
promising, as epigenetic alterations are dynamic and reversible.
However, epigenetic therapy of CF disease is still at its infant
stage (78).
Clinical applications of CF-associated
molecules
Molecular diagnosis of CF
The association between clinical presentations and
residual CFTR function has been established (79). Congenital absence of the vas
deferens is established when the proportion of normal CFTR function
is <10%, as this number decreases (<5%), positive sweat test
results could be supported and patients might suffer from pulmonary
infection when CFTR function further drops to <4.5%. The worst
cases (CFTR function <1%) lead to pancreatic insufficiency,
aside from the aforementioned symptoms (80). As varying molecular subtypes are
associated with different phenotypes, experts from the ‘Cystic
Fibrosis Foundation’ convened a panel of criteria for diagnosing CF
in 1996 (81,82). Several tests, including the sweat
test (83,84), nasal potential difference (NPD)
(85–87), DNA screening (88,89)
and a ciliary test, were recommended. The current review discussed
traditional (regular) approaches (sweat test and NPD measurement)
and novel methods.
The sweat test is an effective method for detecting
CF, covering all age ranges (83,84).
However, the application of creams and lotions within 1 day prior
to sweat collection can disrupt the precision of diagnosis. The
criteria for determining CF varies based on different ages. In
infants up to 6 months of age, a CF diagnosis is very unlikely if
the level of Cl− is not >29 mmol/l. However, the
possibility of establishing a CF diagnosis increases when
Cl− levels range 30–59 mmol/l. Generally, the diagnosis
of CF can be confirmed when this level is >60 mmol/l. The
criteria vary slightly in patients aged >6 months. CF cannot be
diagnosed when the Cl− concentration is <39 mmol/l.
When levels range 40–59 mmol/l, a higher probability of CF is
expected. The diagnosis of CF can be established if the
Cl− levels are >60 mmol/l. Collection of a sufficient
volume of sweat is required for laboratory assays, through which Na
and Cl− concentrations are determined. Incorrect results
occur due to contamination, evaporation, insufficient sample and
technical errors (83,84).
NPD measurement is used to follow-up patients with
CF (85–87). NPD is generally used to evaluate
the voltage between the reference electrode and the exploring
electrode, which is sensitive and specific (85–87).
In vivo, NPD provides data about incorrect ion transport due
to CFTR protein dysfunction in the nasal epithelial cells of
patients. Ancillary test is used to verify the phenotype of
patients and identify ion channel abnormality. However, specific
skills are required to perform the test and interpret the
results.
DNA screening can detect severe mutations, including
F508del and minor mutations such as the 5T variant, and is
particularly useful to detect CF in patients who are unable to
perform the sweat test (88,89).
This method can provide a general idea associated with the severity
of the illness and can detect less severe CF variants, including
azoospermia and congenital bilateral absence of the vas deferens
(90). Previously, children
suffering from CFTR-associated metabolic disorders were classified
into non-typical or moderate type of CF. However, these indistinct
categories lack stringent criteria. Therefore, this resulted in
ambiguities in subtype stratification. Currently, DNA screening is
widely used in newborn screening for improved stratification of the
different subtypes of CF. Nevertheless, regular evaluation remains
important (42). The most
significant benefit of newborn screening and early diagnosis of CF
is the possibility to treat disease-prone patients prior to the
occurrence of serious symptoms (91).
One of the consequences of developing CF is the
chronic pulmonary infection caused by colonized bacteria at an
early age. Phenotypic features associated with CF diagnosis
provides information about chronic sinopulmonary disease
manifestation due to many microorganism, including
Staphylococcus aureus, nontypeable Haemophilus
influenzae, mucoid/non-mucoid Stenotrophomonas maltophilia,
Pseudomonas aeruginosa and Burkholderia cepacian
(92). These pathogenic bacteria
can provoke gastrointestinal dysfunction responsible for
intestinal, pancreatic, hepatic and nutritional troubles.
Identification of the microorganism in patients with CF guides the
path for subsequent antibiotic therapy (93). As mentioned in previous sections,
microorganism detection can be performed by analyzing the
expression profile of miRNAs (62)
or other novel biomarkers (63).
Therefore, traditional PCR analysis, microarray methods and NGS are
capable of biomarkers profiling.
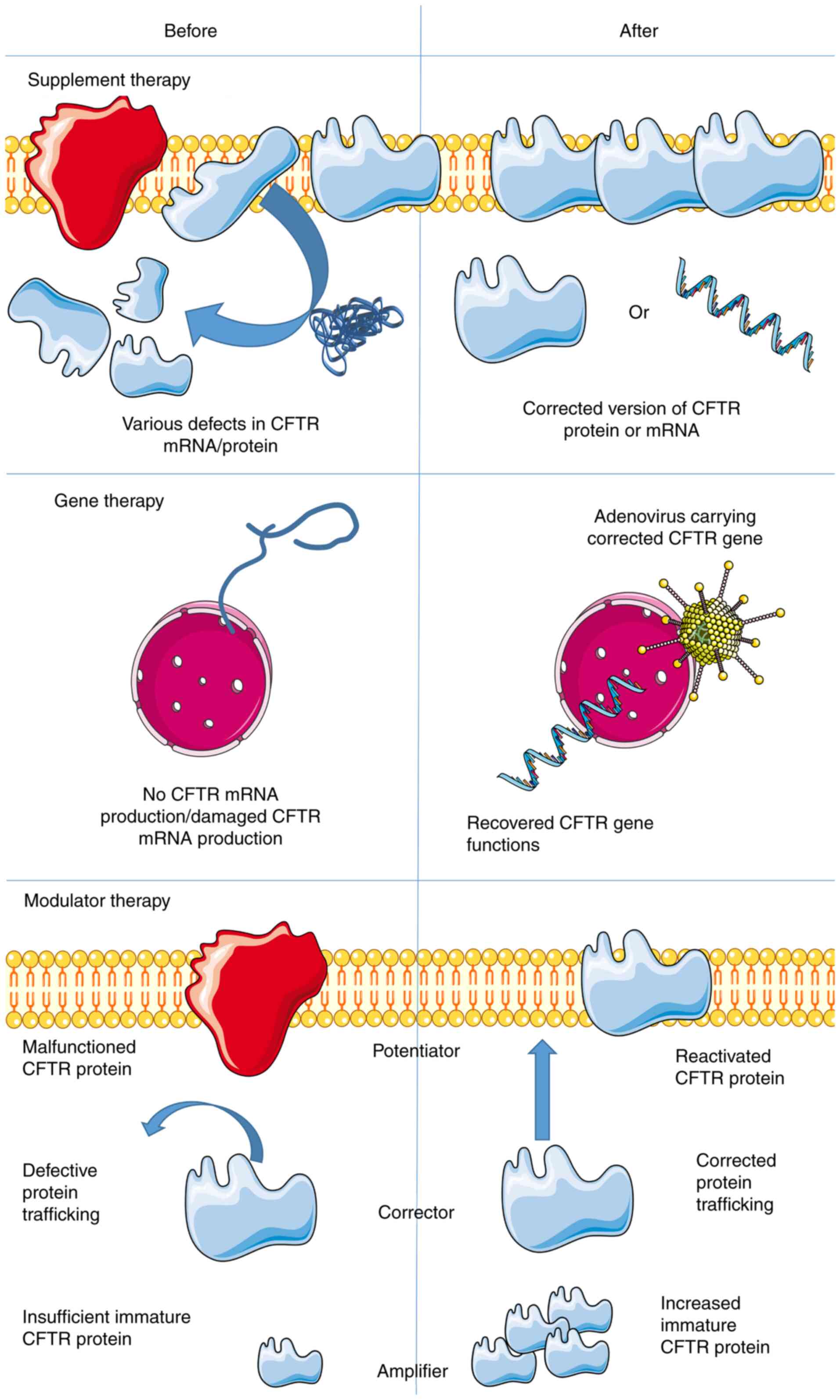
Molecular therapy for CF
Molecular therapy serves a crucial role in CF
treatment. The current review discussed several alternatives, which
are summarized in Fig. 3. In 1993,
a gene therapy clinical trial was performed. The first trial
focused on the nasal epithelium and adenovirus vectors containing
the CFTR gene was used in an attempt to restore CFTR
function (1,94–97).
The rationale behind this method was to restore the dysfunctional
gene or to supplement the patient with the corrected version of the
protein prior to irreversible damage (95,98,99).
For this technique, the DNA has to penetrate the nucleus to be
transcribed, which is the major barrier in gene therapy. In
practice, gene therapy entails inhalation of a spray which delivers
therapeutic DNA to the lungs. During the therapeutic process,
either viral vectors (including adenovirus, lentivirus and herpes
virus) or non-viral vectors (such as plasmids) were used. The best
therapeutic outcome would be the successful replacement of the
defective gene in the lungs to cure CF fundamentally. In other
outcomes, CF symptoms are alleviated by decelerated disease
progression; specifically, to clear aberrant and excessive
secretions, combat pulmonary infections and to prevent intestinal
obstruction (99). Additionally,
gene therapy is the first and most advanced vector system using
recombinant retroviruses ex vivo. In vivo gene
therapy uses vectors based on the recombinant form of adenovirus.
The recent virus-based system is an adeno-associated system and
numerous vector systems have been validated in clinical trials
involving human participants. Among them, adenoviruses and
adeno-related viruses have been widely used (37,100). Aside from virus vectors, cationic
lipids-based vectors are also popular (99).
Transcript supplementation therapy using the correct
version of CFTR mRNA transfected or transduced into the respective
target cells has been documented (95). In this therapy, mRNA is actively
producing CFTR in the cytoplasm, thereby circumventing the nuclear
membrane. However, protein delivery is often ineffective and it is
difficult to include natural posttranscriptional protein
modifications. Additionally, RNA antisense therapy is taken into
account in CF treatment. The hypothesis is to use inhaled RNA
antisense to produce functional CFTR protein by inducing RNA to
work more efficiently (53).
Notably, nanotechnology using package miRNAs to treat CF was proven
to be safe and effective. However, more research is required before
applying this model to other diseases (66).
Another alternative for CFTR treatment includes
modulator therapies (101), which
can be categorized into two groups: Potentiators and correctors.
The potentiators act on the CFTR ion channels. Therefore, these
modulators are geared toward the class III subject group (gate
defect), among which ivacaftor prolonged the opening of the CFTR
channel, thereby facilitating Cl− ion flow (102). In January 2012, the U.S. Food and
Drug Association approved ivacaftor use and, currently, ivacaftor
is the only licensed CFTR potentiator (103). Observational data based on
clinical and in vitro studies have indicated that ivacaftor
is efficient for several mutations within classes III, IV and V in
rat thyroid cell lines (102–104).
The correctors serve a key role in the
transportation of nascent proteins (104). For instance, corrector lumacaftor
is considered a stabilizer that increases the stability of mutated
CFTR proteins, through which these proteins could be transported to
the cell membrane more effectively and remain there for an extended
period of time (105). These
stable substrates could be further enhanced by potentiators
(106). Monotherapy with
lumacaftor, as a corrector, failed to demonstrate significant
results in homozygous patients (106,107). Furthermore, another type of
corrector, tezacaftor, demonstrated great result, improving the
processing and trafficking of mutated CFTR, and promoting chloride
transportation in bronchial epithelial cells derived from
F508del/F508del donors, which were achieved without the problems
associated with lumacaftor (for example, pulmonary exacerbation and
increment in weight) (108). The
underlying mechanism and propriety of tezacaftor are very close to
those of lumacaftor (107), as
tezacaftor therapy increases Cl− transport. When
combined with ivacaftor, tezacaftor is efficient in transporting
the CFTR protein to its correct position on cell surfaces (109–111). Therefore, potentiators and
correctors are often combined to treat patients with CF.
Specifically, CFTR potentiators increase the activity of CFTR on
epithelial surfaces, whereas CFTR correctors promote processing and
trafficking of mutated protein. In order to restore the
availability and functionality of CFTR protein in the epithelium,
CFTR modulator drugs are taken orally (106).
Furthermore, the third type of modulator, which is
still in development, is termed the amplifier. These modulators
selectively promote cellular immature CFTR protein production,
supplying correctors and potentiator with sufficient substrate
(112). For instance, patients
with CF and F508del mutations received gentamicin nasal drops for
14 days, which led to a 22% increase in their wild-type CFTR
function (113). Additionally,
curcumin was used to treat CF by potentiating the activation of
CFTR (114). Currently, triple
combination therapy (elexacaftor, tezacaftor and ivacaftor)
outperformed dual combination therapy (elexacaftor and tezacaftor)
as it promoted the Cl− and fluid transportation, thereby
further increasing the beat frequency of cilia, as manifested by
in vitro efficacy in F508Del/F508Del human bronchial
epithelial cells (115).
Challenges and perspectives
Despite the fact that considerable data have been
obtained in regard to the molecular mechanisms of CF, challenges
still remain. Further research is required concerning the following
aspects: i) Although 3-base-pair deletion and >100 related
variants have been reported to account for CF pathogenesis,
phenotypes of other variants, particularly those with single amino
acid alterations, remain to be elucidated (41); ii) interpretation regarding
molecular and genetic results of CFTR (whether specific variation
should be defined as ‘disease-prone’ or ‘neutral’) has remained
controversial, mainly due to the one-to-many association between CF
genotypes to phenotypes (116),
which result in difficulties in associating genetic information
with clinical traits; iii) while gene therapies (gene editing)
exhibit potential in CF treatment, the efficiency is decreased by
high off-target effects (117);
iv) another defect due to technical restriction is that prior to
being intracellularly de-packaged, the transferred gene can be
severely damaged by multiple natural barriers, including mucus and
the immune response (118); and
v) the spectrum of treatable mutations should be extended (119).
Conclusions
The current review summarizes the advances in the
understanding of the molecular mechanisms underlying CF, the
corresponding molecular regulators and their clinical
implementations. Emerging technology, including NGS analysis and
gene therapy, will improve the understanding of the underlying
molecular mechanisms. Increasing numbers of novel molecular
regulators, such as miRNAs and lncRNAs, have been reported, some of
which displayed potential to be biomarkers of CF. CF diagnosis was
improved by carrier screening, while newborn screening facilitates
the prognostic outcome via the timely intervention of CF at the
early stage. The developed understanding of molecular variants
(genotypes) of CF defects have enabled the development of
increasingly precise and customized CF treatments, which
significantly prolonged the survival of patients with CF with novel
therapies, including gene, supplementation and modulator therapies.
These have demonstrated promising future for CF treatment. Although
rapid progresses have been reported in the understanding and
treatment of CF, improvements are required and challenges remain to
be overcome.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural
Science Foundation of China (grant no. 81670004), the Guangxi
Natural Science Fund Project (grant nos. 2017GXNSFAA198163,
2017GXNSFAA198149 and 2020GXNSFAA159099), the Nanning Scientific
Research and Technology Development Project (grant nos. 20153124,
20163138 and 20153011), the Youth Science Foundation of Guangxi
Medical University (grant no. GXMUYSF201307), the Scientific
Research Project of Health Committee of Guangxi (grant nos.
Z2014456 and Z2015197), the Guangxi Key Laboratory of Bio-targeting
of Theranostics Fund (grant no. GXSWBX2020001), the Key R&D
plan of Qingxiu District Science and Technology Planning Project
(grant no. 2020025) and the Academic Promotion Programme of
Shandong First Medical University (grant no. 2019QL013).
Availability of data and materials
Not applicable.
Authors' contributions
TW, HS and YS wrote and revised the manuscript. WC,
YL, ZH and QJ contributed in drafting the manuscript. CX designed
the work. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CF
|
cystic fibrosis
|
|
CFTR
|
cystic fibrosis transmembrane
conductance regulator
|
References
|
1
|
Klimova B, Kuca K, Novotny M and Maresova
P: Cystic fibrosis revisited-a review study. Med Chem. 13:102–109.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brewington JJ, Filbrandt ET, LaRosa FJ
III, Ostmann AJ, Strecker LM, Szczesniak RD and Clancy JP:
Detection of CFTR function and modulation in primary human nasal
cell spheroids. J Cyst Fibros. 17:26–33. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Keiser NW, Birket SE, Evans IA, Tyler SR,
Crooke AK, Sun X, Zhou W, Nellis JR, Stroebele EK, Chu KK, et al:
Defective innate immunity and hyperinflammation in newborn cystic
fibrosis transmembrane conductance regulator-knockout ferret lungs.
Am J Respir Cell Mol Biol. 52:683–694. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Santoro D, Postorino A, Lucanto C, Costa
S, Cristadoro S, Pellegrino S, Conti G, Buemi M, Magazzù G and
Bellinghieri G: Cystic fibrosis: A risk condition for renal
disease. J Ren Nutr. 27:470–473. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dekkers JF, Wiegerinck CL, de Jonge HR,
Bronsveld I, Janssens HM, de Winter-de Groot KM, Brandsma AM, de
Jong NW, Bijvelds MJ, Scholte BJ, et al: A functional CFTR assay
using primary cystic fibrosis intestinal organoids. Nat Med.
19:939–945. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Harutyunyan M, Huang Y, Mun KS, Yang F,
Arora K and Naren AP: Personalized medicine in CF: From modulator
development to therapy for cystic fibrosis patients with rare CFTR
mutations. Am J Physiol Lung Cell Mol Physiol. 314:L529–L543. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pankow S, Bamberger C, Calzolari D,
Martínez-Bartolomé S, Lavallée-Adam M, Balch WE and Yates JR III:
∆F508 CFTR interactome remodelling promotes rescue of cystic
fibrosis. Nature. 528:510–516. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gadsby DC, Vergani P and Csanady L: The
ABC protein turned chloride channel whose failure causes cystic
fibrosis. Nature. 440:477–483. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rommens JM, Iannuzzi MC, Kerem B, Drumm
ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N, et
al: Identification of the cystic fibrosis gene: Chromosome walking
and jumping. Science. 245:1059–1065. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Saint-Criq V and Gray MA: Role of CFTR in
epithelial physiology. Cell Mol Life Sci. 74:93–115. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cuthbert AW: New horizons in the treatment
of cystic fibrosis. Br J Pharmacol. 163:173–183. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang Z and Chen J: Atomic structure of
the cystic fibrosis transmembrane conductance regulator. Cell.
167:1586–1597.e9. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xue X, Mutyam V, Thakerar A, Mobley J,
Bridges RJ, Rowe SM, Keeling KM and Bedwell DM: Identification of
the amino acids inserted during suppression of CFTR nonsense
mutations and determination of their functional consequences. Hum
Mol Genet. 26:3116–3129. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dodge JA: A millennial view of cystic
fibrosis. Dev Period Med. 19:9–13. 2015.PubMed/NCBI
|
|
15
|
Singh M, Rebordosa C, Bernholz J and
Sharma N: Epidemiology and genetics of cystic fibrosis in Asia: In
preparation for the next-generation treatments. Respirology.
20:1172–1181. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Alibakhshi R and Zamani M: Mutation
analysis of CFTR gene in 70 Iranian cystic fibrosis patients. Iran
J Allergy Asthma Immunol. 5:3–8. 2006.PubMed/NCBI
|
|
17
|
Zeitlin PL: Cystic fibrosis and estrogens:
A perfect storm. J Clin Invest. 118:3841–3844. 2008.PubMed/NCBI
|
|
18
|
Lui JK, Kilch J, Fridlyand S, Dheyab A and
Bielick Kotkowski C: Non-classic cystic fibrosis: The value in
family history. Am J Med. 130:e333–e334. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Thomas JM, Durack A, Sterling A, Todd PM
and Tomson N: Aquagenic wrinkling of the palms: A diagnostic clue
to cystic fibrosis carrier status and non-classic disease. Lancet.
389:8462017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Severiche-Bueno D, Gamboa E, Reyes LF and
Chotirmall SH: Hot topics and current controversies in non-cystic
fibrosis bronchiectasis. Breathe (Sheff). 15:286–295. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Andersen DH: Cystic fibrosis of the
pancreas and its relation to celiac disease. Am J Dis Child.
56:1938. View Article : Google Scholar
|
|
22
|
Bell SC, Mall MA, Gutierrez H, Macek M,
Madge S, Davies JC, Burgel PR, Tullis E, Castaños C, Castellani C,
et al: The future of cystic fibrosis care: A global perspective.
Lancet Respir Med. 8:65–124. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gibson LE and Cooke RE: A test for
concentration of electrolytes in sweat in cystic fibrosis of the
pancreas utilizing pilocarpine by iontophoresis. Pediatrics.
23:545–549. 1959.PubMed/NCBI
|
|
24
|
Di Sant'agnese PA, Darling RC, Perera GA
and Shea E: Abnormal electrolyte composition of sweat in cystic
fibrosis of the pancreas; clinical significance and relationship to
the disease. Pediatrics. 12:549–563. 1953.PubMed/NCBI
|
|
25
|
Davis PB: Cystic fibrosis since 1938. Am J
Respir Crit Care Med. 173:475–482. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yamada A, Komaki Y, Komaki F, Micic D,
Zullow S and Sakuraba A: Risk of gastrointestinal cancers in
patients with cystic fibrosis: A systematic review and
meta-analysis. Lancet Oncol. 19:758–767. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Goetz D and Ren CL: Review of cystic
fibrosis. Pediatr Ann. 48:e154–e161. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fanen P, Wohlhuter-Haddad A and Hinzpeter
A: Genetics of cystic fibrosis: CFTR mutation classifications
toward genotype-based CF therapies. Int J Biochem Cell Biol.
52:94–102. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Linsdell P: Cystic fibrosis transmembrane
conductance regulator (CFTR): Making an ion channel out of an
active transporter structure. Channels (Austin). 12:284–290. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Moran O: The gating of the CFTR channel.
Cell Mol Life Sci. 74:85–92. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mall MA and Galietta LJ: Targeting ion
channels in cystic fibrosis. J Cyst Fibros. 14:561–570. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gentzsch M and Mall MA: Ion channel
modulators in cystic fibrosis. Chest. 154:383–393. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shah VS, Meyerholz DK, Tang XX, Reznikov
L, Abou Alaiwa M, Ernst SE, Karp PH, Wohlford-Lenane CL, Heilmann
KP, Leidinger MR, et al: Airway acidification initiates host
defense abnormalities in cystic fibrosis mice. Science.
351:503–507. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu F, Zhang Z, Csanády L, Gadsby DC and
Chen J: Molecular structure of the human CFTR ion channel. Cell.
169:85–95.e8. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cook DP, Rector MV, Bouzek DC, Michalski
AS, Gansemer ND, Reznikov LR, Li X, Stroik MR, Ostedgaard LS, Abou
Alaiwa MH, et al: Cystic fibrosis transmembrane conductance
regulator in sarcoplasmic reticulum of airway smooth muscle.
Implications for airway contractility. Am J Respir Crit Care Med.
193:417–426. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Iitiä A, Høgdall E, Dahlen P, Hurskainen
P, Vuust J and Siitari H: Detection of mutation delta F508 in the
cystic fibrosis gene using allele-specific PCR primers and
time-resolved fluorometry. PCR Methods Appl. 2:157–162. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xia E, Zhang Y, Cao H, Li J, Duan R and Hu
J: TALEN-mediated gene targeting for cystic fibrosis-gene therapy.
Genes (Basel). 10:392019. View Article : Google Scholar
|
|
38
|
Costa C, Pruliere-Escabasse V, de
Becdelievre A, Gameiro C, Golmard L, Guittard C, Bassinet L,
Bienvenu T, Georges MD, Epaud R, et al: A recurrent deep-intronic
splicing CF mutation emphasizes the importance of mRNA studies in
clinical practice. J Cyst Fibros. 10:479–482. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Brandt C, Roehmel J, Rickerts V, Melichar
V, Niemann N and Schwarz C: Aspergillus bronchitis in patients with
cystic fibrosis. Mycopathologia. 183:61–69. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Strom CM, Ginsberg N, Rechitsky S, Cieslak
J, Ivakhenko V, Wolf G, Lifchez A, Moise J, Valle J, Kaplan B, et
al: Three births after preimplantation genetic diagnosis for cystic
fibrosis with sequential first and second polar body analysis. Am J
Obstet Gynecol. 178:1298–1306. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Girardet A, Viart V, Plaza S, Daina G, De
Rycke M, Des Georges M, Fiorentino F, Harton G, Ishmukhametova A,
Navarro J, et al: The improvement of the best practice guidelines
for preimplantation genetic diagnosis of cystic fibrosis: Toward an
international consensus. Eur J Hum Genet. 24:469–478. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Brennan ML and Schrijver I: Cystic
fibrosis: A review of associated phenotypes, use of molecular
diagnostic approaches, genetic characteristics, progress, and
dilemmas. J Mol Diagn. 18:3–14. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bell CJ, Dinwiddie DL, Miller NA, Hateley
SL, Ganusova EE, Mudge J, Langley RJ, Zhang L, Lee CC, Schilkey FD,
et al: Carrier testing for severe childhood recessive diseases by
next-generation sequencing. Sci Transl Med. 3:65ra42011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Rengaraju B, Thana K, La A, Pavithra K,
Durairaj V, Challapalli SH and Das A: Inquest of the SNP in cystic
fibrosis-A bioinformatic approach. Int J Curr Microbiol Appl Sci.
6:1255–1263. 2017. View Article : Google Scholar
|
|
45
|
Beauchamp KA, Johansen Taber KA, Grauman
PV, Spurka L, Lim-Harashima J, Svenson A, Goldberg JD and Muzzey D:
Sequencing as a first-line methodology for cystic fibrosis carrier
screening. Genet Med. 21:2569–2576. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Baker MW, Atkins AE, Cordovado SK, Hendrix
M, Earley MC and Farrell PM: Improving newborn screening for cystic
fibrosis using next-generation sequencing technology: A technical
feasibility study. Genet Med. 18:231–238. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Marangi M and Pistritto G: Innovative
therapeutic strategies for cystic fibrosis: Moving forward to
CRISPR technique. Front Pharmacol. 9:3962018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hodges CA and Conlon RA: Delivering on the
promise of gene editing for cystic fibrosis. Genes Dis. 6:97–108.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Park S and Beal PA: Off-target editing by
CRISPR-guided DNA base editors. Biochemistry. 58:3727–3734. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Schwank G, Koo BK, Sasselli V, Dekkers JF,
Heo I, Demircan T, Sasaki N, Boymans S, Cuppen E, van der Ent CK,
et al: Functional repair of CFTR by CRISPR/Cas9 in intestinal stem
cell organoids of cystic fibrosis patients. Cell Stem Cell.
13:653–658. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Crane AM, Kramer P, Bui JH, Chung WJ, Li
XS, Gonzalez-Garay ML, Hawkins F, Liao W, Mora D, Choi S, et al:
Targeted correction and restored function of the CFTR gene in
cystic fibrosis induced pluripotent stem cells. Stem Cell Reports.
4:569–577. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Liang P, Xu Y, Zhang X, Ding C, Huang R,
Zhang Z, Lv J, Xie X, Chen Y, Li Y, et al: CRISPR/Cas9-mediated
gene editing in human tripronuclear zygotes. Protein Cell.
6:363–372. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Saayman SM, Ackley A, Burdach J, Clemson
M, Gruenert DC, Tachikawa K, Chivukula P, Weinberg MS and Morris
KV: Long non-coding RNA BGas regulates the cystic fibrosis
transmembrane conductance regulator. Mol Ther. 24:1351–1357. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Dimartino D, Colantoni A, Ballarino M,
Martone J, Mariani D, Danner J, Bruckmann A, Meister G, Morlando M
and Bozzoni I: The long non-coding RNA lnc-31 interacts with Rock1
mRNA and mediates its YB-1-dependent translation. Cell Rep.
23:733–740. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kishore S and Stamm S: The snoRNA HBII-52
regulates alternative splicing of the serotonin receptor 2C.
Science. 311:230–232. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Gil N and Ulitsky I: Regulation of gene
expression by cis-acting long non-coding RNAs. Nat Rev Genet.
21:102–117. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Fabbri E, Tamanini A, Jakova T, Gasparello
J, Manicardi A, Corradini R, Sabbioni G, Finotti A, Borgatti M,
Lampronti I, et al: A peptide nucleic acid against MicroRNA
miR-145-5p enhances the expression of the cystic fibrosis
transmembrane conductance regulator (CFTR) in Calu-3 cells.
Molecules. 23:712017. View Article : Google Scholar
|
|
58
|
Megiorni F, Cialfi S, Dominici C,
Quattrucci S and Pizzuti A: Synergistic post-transcriptional
regulation of the Cystic Fibrosis Transmembrane conductance
Regulator (CFTR) by miR-101 and miR-494 specific binding. PLoS One.
6:e266012011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Li Z, Yao JN, Huang WT, He RQ, Ma J, Chen
G and Wei QJ: Expression of miR-542-3p in osteosarcoma with miRNA
microarray data, and its potential signaling pathways. Mol Med Rep.
19:974–983. 2019.PubMed/NCBI
|
|
60
|
Hassan F, Nuovo GJ, Crawford M, Boyaka PN,
Kirkby S, Nana-Sinkam SP and Cormet-Boyaka E: MiR-101 and miR-144
regulate the expression of the CFTR chloride channel in the lung.
PLoS One. 7:e508372012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ramachandran S, Karp PH, Jiang P,
Ostedgaard LS, Walz AE, Fisher JT, Keshavjee S, Lennox KA, Jacobi
AM, Rose SD, et al: A microRNA network regulates expression and
biosynthesis of wild-type and DeltaF508 mutant cystic fibrosis
transmembrane conductance regulator. Proc Natl Acad Sci USA.
109:13362–13367. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Fesen K, Silveyra P, Fuentes N, Nicoleau
M, Rivera L, Kitch D, Graff GR and Siddaiah R: The role of
microRNAs in chronic pseudomonas lung infection in Cystic fibrosis.
Respir Med. 151:133–138. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Balloy V, Koshy R, Perra L, Corvol H,
Chignard M, Guillot L and Scaria V: Bronchial epithelial cells from
cystic fibrosis patients express a specific long non-coding RNA
signature upon Pseudomonas aeruginosa infection. Front Cell
Infect Microbiol. 7:2182017. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
McKiernan PJ, Molloy K, Cryan SA,
McElvaney NG and Greene CM: Long noncoding RNA are aberrantly
expressed in vivo in the cystic fibrosis bronchial epithelium. Int
J Biochem Cell Biol. 52:184–191. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kumar P, Sen C, Peters K, Frizzell RA and
Biswas R: Comparative analyses of long non-coding RNA profiles in
vivo in cystic fibrosis lung airway and parenchyma tissues. Respir
Res. 20:2842019. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
McKiernan PJ, Cunningham O, Greene CM and
Cryan SA: Targeting miRNA-based medicines to cystic fibrosis airway
epithelial cells using nanotechnology. Int J Nanomed. 8:3907–3915.
2013.
|
|
67
|
Lu Q, Liu T, Feng H, Yang R, Zhao X, Chen
W, Jiang B, Qin H, Guo X, Liu M, et al: Circular RNA circSLC8A1
acts as a sponge of miR-130b/miR-494 in suppressing bladder cancer
progression via regulating PTEN. Mol Cancer. 18:1112019. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Yu CY, Li TC, Wu YY, Yeh CH, Chiang W,
Chuang CY and Kuo HC: The circular RNA circBIRC6 participates in
the molecular circuitry controlling human pluripotency. Nat Commun.
8:11492017. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Nowacka-Zawisza M and Wiśnik E: DNA
methylation and histone modifications as epigenetic regulation in
prostate cancer (Review). Oncol Rep. 38:2587–2596. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Sirinupong N and Yang Z: Epigenetics in
cystic fibrosis: Epigenetic targeting of a genetic disease. Curr
Drug Targets. 16:976–987. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Morandini AC, Santos CF and Yilmaz Ö: Role
of epigenetics in modulation of immune response at the junction of
host-pathogen interaction and danger molecule signaling. Pathog
Dis. 74:ftw0822016. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Chen Y, Armstrong DA, Salas LA, Hazlett
HF, Nymon AB, Dessaint JA, Aridgides DS, Mellinger DL, Liu X,
Christensen BC and Ashare A: Genome-wide DNA methylation profiling
shows a distinct epigenetic signature associated with lung
macrophages in cystic fibrosis. Clin Epigenetics. 10:1522018.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Magalhães M, Tost J, Pineau F, Rivals I,
Busato F, Alary N, Mely L, Leroy S, Murris M, Caimmi D, et al:
Dynamic changes of DNA methylation and lung disease in cystic
fibrosis: Lessons from a monogenic disease. Epigenomics.
10:1131–1145. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Scott M and De Sario A: DNA methylation
changes in cystic fibrosis: Cause or consequence? Clin Genet.
98:3–9. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Hutt DM, Herman D, Rodrigues AP, Noel S,
Pilewski JM, Matteson J, Hoch B, Kellner W, Kelly JW, Schmidt A, et
al: Reduced histone deacetylase 7 activity restores function to
misfolded CFTR in cystic fibrosis. Nat Chem Biol. 6:25–33. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Bartling TR and Drumm ML: Loss of CFTR
results in reduction of histone deacetylase 2 in airway epithelial
cells. Am J Physiol Lung Cell Mol Physiol. 297:L35–L43. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Rymut SM, Harker A, Corey DA, Burgess JD,
Sun H, Clancy JP and Kelley TJ: Reduced microtubule acetylation in
cystic fibrosis epithelial cells. Am J Physiol Lung Cell Mol
Physiol. 305:L419–L431. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Bergougnoux A, Rivals I, Liquori A, Raynal
C, Varilh J, Magalhães M, Perez MJ, Bigi N, Des Georges M, Chiron
R, et al: A balance between activating and repressive histone
modifications regulates cystic fibrosis transmembrane conductance
regulator (CFTR) expression in vivo. Epigenetics. 9:1007–1017.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Cutting GR: Cystic fibrosis genetics: From
molecular understanding to clinical application. Nat Rev Genet.
16:45–56. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Davis PB, Drumm M and Konstan MW: Cystic
fibrosis. Am J Respir Crit Care Med. 154:1229–1256. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
De Boeck K, Vermeulen F and Dupont L: The
diagnosis of cystic fibrosis. Presse Med. 46:e97–e108. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Schwarzenberg SJ, Hempstead SE, McDonald
CM, Powers SW, Wooldridge J, Blair S, Freedman S, Harrington E,
Murphy PJ, Palmer L, et al: Enteral tube feeding for individuals
with cystic fibrosis: Cystic Fibrosis Foundation evidence-informed
guidelines. J Cyst Fibros. 15:724–735. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Guglani L, Moir D and Jain A: Sweat
chloride concentrations in children with Idiopathic Nephrotic
Syndrome. Pediatr Pulmonol. 51:49–52. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Brown A, Jenkins L, Reid A, Leavy A,
McDowell G, McIlroy C, Thompson A and McNaughten B: How to perform
and interpret the sweat test. Arch Dis Child Educ Pract Ed.
105:230–235. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Solomon GM, Liu B, Sermet-Gaudelus I,
Fajac I, Wilschanski M, Vermeulen F and Rowe SM: A multiple reader
scoring system for Nasal Potential Difference parameters. J Cyst
Fibros. 16:573–578. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Solomon GM, Bronsveld I, Hayes K,
Wilschanski M, Melotti P, Rowe SM and Sermet-Gaudelus I:
Standardized measurement of nasal membrane transepithelial
potential difference (NPD). J Vis Exp. 570062018.
|
|
87
|
Beka M and Leal T: Nasal potential
difference to quantify trans-epithelial ion transport in mice. J
Vis Exp. 579342018.
|
|
88
|
Old RW, Bestwick JP and Wald NJ: Prenatal
maternal plasma DNA screening for cystic fibrosis: A computer
modelling study of screening performance. F1000Res. 6:18962017.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Sugunaraj JP, Brosius HM, Murray MF,
Manickam K, Stamm JA, Carey DJ and Mirshahi UL: Predictive value of
genomic screening: Cross-sectional study of cystic fibrosis in
50,788 electronic health records. NPJ Genom Med. 4:212019.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Ferlin A and Stuppia L: Diagnostics of
CFTR-negative patients with congenital bilateral absence of vas
deferens: Which mutations are of most interest? Expert Rev Mol
Diagn. 20:265–267. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Wagener JS, Sontag MK and Accurso FJ:
Newborn screening for cystic fibrosis. Curr Opin Pediatr.
15:309–315. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
O'Brien TJ and Welch M: Recapitulation of
polymicrobial communities associated with cystic fibrosis airway
infections: A perspective. Future Microbiol. 14:1437–1450. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Lyczak JB, Cannon CL and Pier GB: Lung
infections associated with cystic fibrosis. Clin Microbiol Rev.
15:194–222. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Savant AP and McColley SA: Cystic fibrosis
year in review 2016. Pediatr Pulmonol. 52:1092–1102. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Wilson J: Treating genes and patients.
Gene Ther. 27:109–110. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Rafeeq MM and Murad HAS: Cystic fibrosis:
Current therapeutic targets and future approaches. J Transl Med.
15:842017. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Moss RB, Flume PA, Elborn JS, Cooke J,
Rowe SM, McColley SA, Rubenstein RC and Higgins M; VX11-770-110
(KONDUCT) Study Group, : Efficacy and safety of ivacaftor in
patients with cystic fibrosis who have an Arg117His-CFTR mutation:
A double-blind, randomised controlled trial. Lancet Respir Med.
3:524–533. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Arjmand B, Larijani B, Sheikh Hosseini M,
Payab M, Gilany K, Goodarzi P, Parhizkar Roudsari P, Amanollahi
Baharvand M and Hoseini Mohammadi NS: The horizon of gene therapy
in modern medicine: Advances and challenges. Adv Exp Med Biol.
1247:33–64. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Yang Q, Soltis AR, Sukumar G, Zhang X,
Caohuy H, Freedy J, Dalgard CL, Wilkerson MD, Pollard HB and
Pollard BS: Gene therapy-emulating small molecule treatments in
cystic fibrosis airway epithelial cells and patients. Respir Res.
20:2902019. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Eymery M, Morfin F, Doleans-Jordheim A,
Perceval M, Ohlmann C, Mainguy C and Reix P: Viral respiratory
tract infections in young children with cystic fibrosis: A
prospective full-year seasonal study. Virol J. 16:1112019.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Tümmler B: Treatment of cystic fibrosis
with CFTR modulators. Pneumologie. 70:301–313. 2016.(In German).
PubMed/NCBI
|
|
102
|
Bessonova L, Volkova N, Higgins M,
Bengtsson L, Tian S, Simard C, Konstan MW, Sawicki GS, Sewall A,
Nyangoma S, et al: Data from the US and UK cystic fibrosis
registries support disease modification by CFTR modulation with
ivacaftor. Thorax. 73:731–740. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Faruqi S, Shiferaw D and Morice AH: Effect
of ivacaftor on objective and subjective measures of cough in
patients with cystic fibrosis. Open Respir Med J. 10:105–108. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Heltshe SL, Mayer-Hamblett N, Burns JL,
Khan U, Baines A, Ramsey BW and Rowe SM; GOAL (the G551D
Observation-AL) Investigators of the Cystic Fibrosis Foundation
Therapeutics Development Network, : Pseudomonas aeruginosa
in cystic fibrosis patients with G551D-CFTR treated with ivacaftor.
Clin Infect Dis. 60:703–712. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Krainer G, Schenkel M, Hartmann A,
Ravamehr-Lake D, Deber CM and Schlierf M: CFTR transmembrane
segments are impaired in their conformational adaptability by a
pathogenic loop mutation and dynamically stabilized by Lumacaftor.
J Biol Chem. 295:1985–1991. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Wainwright CE, Elborn JS and Ramsey BW:
Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous
for Phe508del CFTR. N Engl J Med. 373:1783–1784. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Konstan MW, McKone EF, Moss RB, Marigowda
G, Tian S, Waltz D, Huang X, Lubarsky B, Rubin J, Millar SJ, et al:
Assessment of safety and efficacy of long-term treatment with
combination lumacaftor and ivacaftor therapy in patients with
cystic fibrosis homozygous for the F508del-CFTR mutation
(PROGRESS): A phase 3, extension study. Lancet Respir Med.
5:107–118. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Sala MA and Jain M: Tezacaftor for the
treatment of cystic fibrosis. Expert Rev Respir Med. 12:725–732.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Rowe SM, Daines C, Ringshausen FC, Kerem
E, Wilson J, Tullis E, Nair N, Simard C, Han L, Ingenito EP, et al:
Tezacaftor-ivacaftor in residual-function heterozygotes with cystic
fibrosis. N Engl J Med. 377:2024–2035. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Donaldson SH, Pilewski JM, Griese M, Cooke
J, Viswanathan L, Tullis E, Davies JC, Lekstrom-Himes JA and Wang
LT; VX11-661-101 Study Group, : Tezacaftor/ivacaftor in subjects
with cystic fibrosis and F508del/F508del-CFTR or
F508del/G551D-CFTR. Am J Respir Crit Care Med. 197:214–224. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Taylor-Cousar JL, Munck A, McKone EF, van
der Ent CK, Moeller A, Simard C, Wang LT, Ingenito EP, McKee C, Lu
Y, et al: Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis
Homozygous for Phe508del. N Engl J Med. 377:2013–2023. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Giuliano KA, Wachi S, Drew L, Dukovski D,
Green O, Bastos C, Cullen MD, Hauck S, Tait BD, Munoz B, et al: Use
of a high-throughput phenotypic screening strategy to identify
amplifiers, a novel pharmacological class of small molecules that
exhibit functional synergy with potentiators and correctors. SLAS
Discov. 23:111–121. 2018.PubMed/NCBI
|
|
113
|
Gambari R, Breveglieri G, Salvatori F,
Finotti A and Borgatti M: Therapy for cystic fibrosis caused by
nonsense mutations. Cystic Fibrosis in the Light of New Research
Ch. 13:2015. View
Article : Google Scholar
|
|
114
|
Wang G: Interplay between inhibitory
ferric and stimulatory curcumin regulates phosphorylation-dependent
human cystic fibrosis transmembrane conductance regulator and
DeltaF508 activity. Biochemistry. 54:1558–1566. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Chaudary N: Triplet CFTR modulators:
Future prospects for treatment of cystic fibrosis. Ther Clin Risk
Manag. 14:2375–2383. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Raynal C, Baux D, Theze C, Bareil C,
Taulan M, Roux AF, Claustres M, Tuffery-Giraud S and des Georges M:
A classification model relative to splicing for variants of unknown
clinical significance: Application to the CFTR gene. Hum Mutat.
34:774–784. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Mention K, Santos L and Harrison PT: Gene
and base editing as a therapeutic option for cystic
fibrosis-learning from other diseases. Genes (Basel). 10:3872019.
View Article : Google Scholar
|
|
118
|
Osman G, Rodriguez J, Chan SY, Chisholm J,
Duncan G, Kim N, Tatler AL, Shakesheff KM, Hanes J, Suk JS and
Dixon JE: PEGylated enhanced cell penetrating peptide nanoparticles
for lung gene therapy. J Control Release. 285:35–45. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Condren ME and Bradshaw MD: Ivacaftor: A
novel gene-based therapeutic approach for cystic fibrosis. J
Pediatr Pharmacol Ther. 18:8–13. 2013.PubMed/NCBI
|