Cancer cachexia is a multifactorial syndrome
characterized by a continuous decline in skeletal muscle mass, with
or without a reduction in adipose tissue, which cannot be reversed
by conventional nutritional treatments and eventually leads to
progressive muscle dysfunction (1). The diagnostic criteria are a weight
loss of >5%, or a weight loss >5%, or a weight loss >2% in
individuals with a body mass index <20 kg/m2 or
sarcopenia (2). Alternative
criteria are a skeletal muscle index of the extremities meetings
the criteria for sarcopenia (males, <7.26 kg/m2;
females, <5.45 kg/m2) and a weight loss of >2%
(3).
The pathophysiological characteristic of cancer
cachexia is a negative protein and energy balance caused by a
combination of factors, such as reduced food intake and metabolic
abnormalities (4–7). Cachexia develops during the
progression of a number of types of malignant tumor, especially
upper gastrointestinal cancer and lung cancer (LC). According to
statistical analyses, >80% of patients with advanced pancreatic
and gastric cancer and ~60% of patients with advanced LC may have
cancer cachexia (8). A previous
survey also revealed that the prevalence of geriatric cancer
cachexia at a geriatric oncology clinic was 65% (9). The clinical symptoms of cancer
cachexia in patients include muscle atrophy and weight loss,
accompanied by various other manifestations, such as loss of
appetite, anorexia, fatigue, anemia, edema and hypoproteinemia,
which significantly impact the quality of life of the patients
(10). In addition, cancer
cachexia has been discovered to reduce the patient sensitivity and
tolerance to treatment and shortens their survival (11).
To the best of our knowledge, the mechanism by which
cancer cachexia causes muscle atrophy is not completely clear.
Skeletal muscle protein undergoes decreased synthesis and increased
degradation during cancer cachexia (1); these changes are attributed to the
upregulation of inflammatory mediators (12–14),
the activation of related transcription factors (15) and signaling pathways (16–18),
abnormalities in the expression of angiotensin II (Ang II)
(19), insulin-like growth
factor-1 (IGF-1) (20) and various
receptors (21,22), proteins and kinases (23), and organelle dysfunction (24). These processes eventually lead to
muscle atrophy during the development of cancer cachexia. To date,
three main pathways of skeletal muscle protein degradation have
been identified: The ubiquitin (Ub)-proteasome, cell
autophagy/lysosomal and Ca2+-activated degradation
pathways (25–27). The most significant of these
pathways is the Ub-proteasome system (UPS) (28). The activation of the above pathways
is often accompanied by the presence of inflammatory mediators,
including IL-1β (14), IL-6
(29) and TNFα (30), and the phosphorylation (17) or abnormal expression of important
molecules (18). The abnormal
catabolism is often related to the dysfunction of organelles, such
as the endoplasmic reticulum (ER) (31) and mitochondria (32). Proteins such as Ang II (19) and IGF-1 (20) are also involved in cancer
cachexia-induced muscle atrophy.
The pathogenesis of cancer cachexia-induced muscle
atrophy is complex and has not been fully elucidated. Currently, no
particular effective treatment method is available; The most
effective treatment includes a multitarget approach including
appetite stimulants, inhibitors of cachectic signaling molecules,
along with nutritional supplementation and physical activity
(33). The present review aimed to
summarize the pathogenesis and comprehensive treatment of muscle
atrophy caused by cancer cachexia and provide novel ideas for the
early detection and timely intervention of cancer cachexia-induced
muscle atrophy.
Protein degradation in cells is a carefully
controlled process, and the UPS serves an important role in the
process of skeletal muscle protein degradation (28). The UPS is composed of Ub and a
series of related enzymes, including Ub-activating enzyme (E1),
Ub-conjugating enzyme (E2), Ub-ligating enzyme (E3) and proteasomes
(34). In this system, proteins
are targeted for degradation by covalent ligation to Ub, a 76 amino
acid residue protein (35). Ub
must first be activated by E1 (36) and then transferred to E2 (37). E2 recognizes E3, which then
specifically recognizes and binds to specific proteins to form a
Ub-protein chain (38,39). A proteasome is a large, 26S,
multi-catalytic protease that degrades polyubiquitinated proteins
to small peptides (40,41). Currently, two E3 protein ligases
have been proven to be very active in the proteolysis of muscle
atrophy, namely, muscle atrophy Fbox-1 protein (MAFbx; also called
atrogin-1) and muscle ring finger protein 1 (MuRF1) (42), which are regulated by a variety of
signaling pathways, such as the NF-κB, IL-6 and p38 MAPK signaling
pathways (12,43–46).
A number of proinflammatory and transfer factors, in
addition to the activation of several pathways have been identified
in skeletal muscle and were illustrated to be involved in cancer
cachexia-induced muscle atrophy; for example, TNF-α (47), Twist1 (48), the NF-κB signaling pathway
(49) and the p38 MAPK signaling
pathway (45), which were all
discovered to be upregulated. The overexpression of proinflammatory
factors, transfer factors or members of signaling pathways in
skeletal muscle in the context of cancer cachexia eventually
converge on the MuRF1 and MAFbx of the UPS, promoting proteasome
hydrolysis in the UPS and leading to skeletal muscle protein
degradation (50–53).
Nonetheless, the majority of the evidence for the
involvement of the UPS in cancer cachexia conditions is currently
derived from animal models of muscle wasting. Further
investigations involving more samples are required to investigate
the regulatory patterns of the UPS in human muscle wasting,
secondary to the above pathologies in cancer cachexia.
Numerous proinflammatory factors have been
discovered to serve important roles in the muscle atrophy caused by
cancer cachexia (13,29,60,61).
IL-6 is produced by macrophages (62) and fibroblasts (63) and was also found to be secreted by
tumor cells (64). Several studies
have reported that severe weight loss due to cancer cachexia was
associated with increased circulating IL-6 levels (65–67).
Clinical studies have also revealed that compared with healthy
controls, patients with non-small cell LC (NSCLC) with cachexia had
smaller muscle fiber cross-sectional areas and significantly
increased plasma IL-6 levels (68). The IL-6/Janus kinase (JAK)/STAT3
signaling pathway was discovered to have an essential role in the
progression of cancer cachexia by regulating the inflammatory
response (13,60). Pin et al (61) intraperitoneally injected ES-2 human
ovarian cancer cells into Nod-SCIDγ mice to establish a cancer
cachexia model; the experimental studies revealed significantly
upregulated IL-6 and phosphorylated STAT3 levels in the plasma and
ascites of model mice compared with control mice. Similarly,
ES-2-conditioned medium directly induced high levels of STAT3
phosphorylation in C2C12 myotubes and caused muscle atrophy in the
mice. Further evidence also suggested that an IL-6/STAT3 signaling
inhibitor (INCB018424) restored myotube size (61). Another previous study also reported
a dose-dependent inhibitory effect of IL-6 on mTOR activity in a
cancer cachexia model and discovered that the suppression of mTOR
activity by IL-6 was dependent on AMP-activated protein kinase
(AMPK) activation and independent of STAT signaling in myotubes
(29). In addition to relieving
the suppression of anabolic signaling, AMPK inhibition also reduced
IL-6-induced MAFbx and ubiquitinated protein expression. Therefore,
on the one hand, IL-6 has been found to inhibit the activity of
mTOR through AMPK activation, thereby inhibiting the synthesis of
muscle protein, while on the other hand, the activation of AMPK
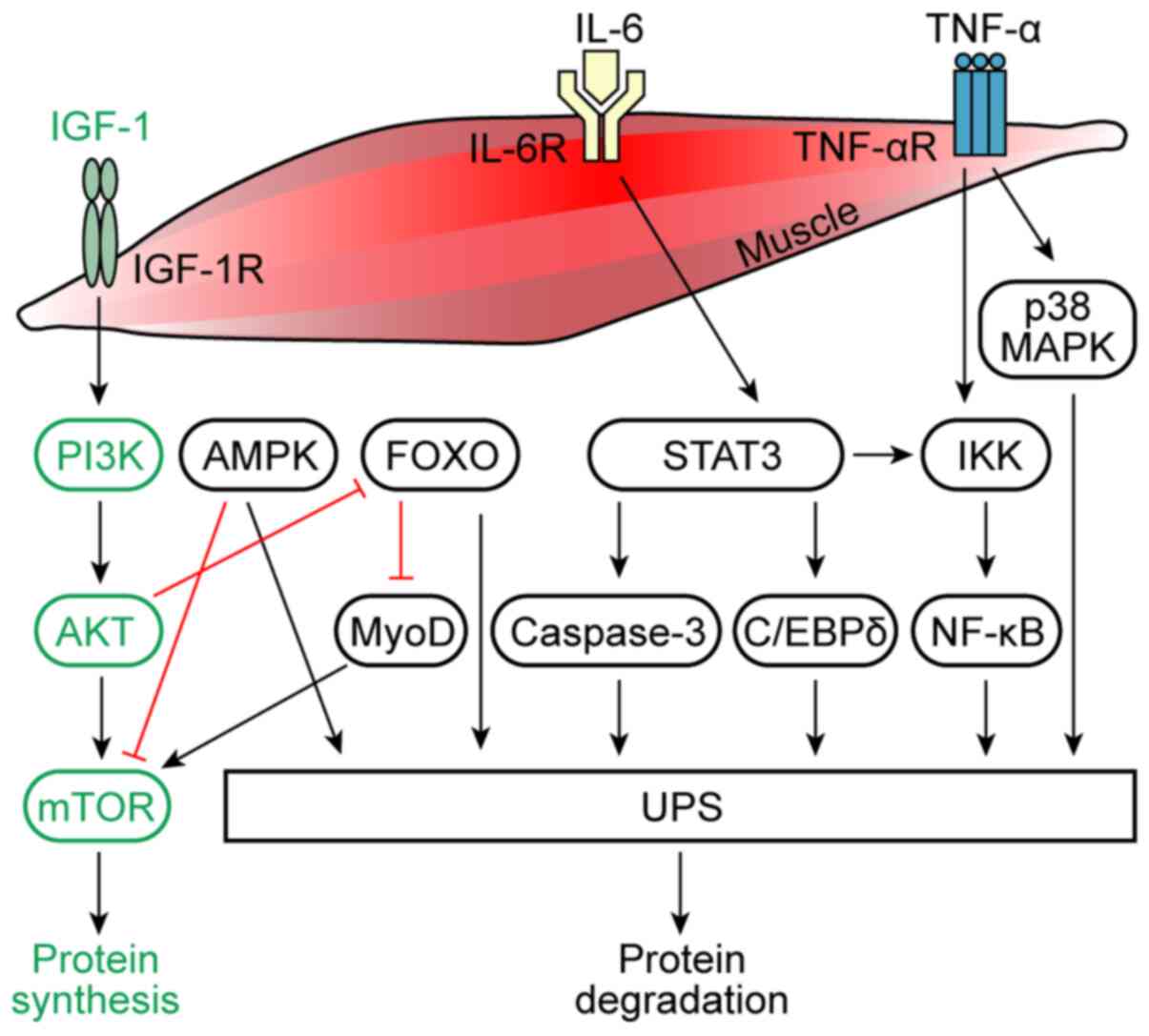
promoted the hydrolysis of muscle protein by the UPS (Fig. 1) (29).
TNF-α is an inflammatory factor secreted by
macrophages and produced by tumor cells, and it has been confirmed
to be a crucial factor associated with cancer cachexia-induced
muscle wasting (69–71). In particular, TNF-α was reported to
have a direct catabolic effect on skeletal muscle, which caused
muscle wasting through the induction of Ub gene expression of the
UPS (Fig. 1) (30,72).
Research has also discovered that TNF-α exposure upregulated MAFbx
mRNA expression levels within 2 h in C2C12 myotubes, and that
exposing myotubes to TNF-α also promoted the general activation of
p38 MAPK. MAFbx upregulation and the associated increase in
Ub-conjugating activity were both inhibited by p38 inhibitors,
either SB203580 or curcumin. These data indicated that TNF-α may
act via p38 MAPK to increase MAFbx gene expression levels in
skeletal muscle to induce muscle atrophy (Fig. 1) (12).
IL-1 is a cytokine that is produced by monocytes,
endothelial cells, fibroblasts and other cell types in response to
infection and exists in two forms: IL-1α and IL-1β. IL-1 was
identified to be an important factor in cachexia (14,75,76).
For example, Cannon et al (75) established squamous cell carcinoma
cachexia model mice and detected and quantified the levels of 18
cytokines and chemokines, including IL-1β, IL-1α, IL-6, TNF-α and
IFN-γ, among others. The results revealed that only IL-1β levels
were significantly elevated in the tumor-bearing mice compared with
the controls. In addition, MuRF1 levels were significantly
upregulated in the carcinoma cachexia model mice compared with the
controls. Therefore, these findings indicated that IL-1β may
mediate MuRF1 regulation and lead to muscle wasting, and therefore
atrophy in tumor-bearing mice. In another study, mice implanted
with Lewis LC (LLC) cells revealed a robust increase in the
expression of IL-1β in the hypothalamus. Concurrent with the
presence of central inflammation, the atrophy program was activated
in the skeletal muscle as indicated by the upregulation of MAFbx,
MuRF1 and FOXO1 expression levels, which occurred in the context of
muscle wasting in the tumor-bearing animals. The study further
demonstrated that central nervous system (CNS) IL-1β signaling
alone evoked a catabolic program in the muscle, rapidly inducing
atrophy. This effect was dependent on
hypothalamic-pituitary-adrenal axis activation, as CNS
IL-1β-induced atrophy was discovered to be abrogated by
adrenalectomy (14).
As a master cytokine involved in the
pathophysiological characteristics of cancer cachexia, preclinical
studies have demonstrated the role of IL-1 in mediating muscle
wasting in cachexia (14,77), which may reveal new therapeutic
targets for muscle wasting diseases.
Under physiological conditions, growth factors and
nutrients activate AKT through PI3K-dependent processes that
activate mTOR, leading to increased muscle cell proliferation and
muscle protein synthesis (Fig. 1)
(78–80). The serine-threonine kinase AKT, as
a downstream target of PI3K, was discovered to serve an important
role in myogenic differentiation (81). The expression of constitutively
active forms of AKT was discovered to markedly enhance myotube
formation and the expression levels of the muscle-specific proteins
myoblast determination protein 1 (MyoD), creatine kinase, myosin
heavy chain (MyHC) and desmin (81). The activation of the PI3K/AKT
signaling pathway stimulates mTOR signaling cascades, modulating
two master molecules associated with the initiation of mRNA
translation, namely, 70-kDa ribosomal protein S6 kinase (p70S6K)
(82,83) and eukaryotic initiation factor 4E
binding protein 1 (4EBP1) (84,85).
The PI3K/AKT signaling pathway was illustrated to
prevent the induction of the muscle-specific Ub ligases MAFbx and
MuRF1 through a mechanism involving the AKT-mediated inhibition of
the FOXO family of transcription factors (Fig. 1) (86,87).
In addition, a previous study used western blotting to analyze
skeletal muscle and liver tissue extracts from 8 patients with
pancreatic cancer with cachexia and 8 patients with nonmalignant
tumors; compared with the patients without cachexia, the patients
with cachexia had significantly reduced levels of MyHC and actin in
the muscle, a 55% decrease in AKT protein expression levels, a
4-fold decrease in the abundance and/or phosphorylation of the
transcription factors FOXO1 and FOXO3a, and significant reductions
in the expression levels of mTOR (−82%) and p70S6K (−39%) (16). This study demonstrated that the
cachexia-associated loss of AKT-dependent signaling in human
skeletal muscle was associated with the decreased activity of
regulators of protein synthesis (16).
Cachexia was discovered to decrease mTOR
phosphorylation, and the phosphorylation of mTOR substrates, S6
ribosomal protein and 4EBP, independent of AKT activation. These
changes in mTOR-related protein signaling pathways were accompanied
by modest increases in the levels of Beclin-1, which is associated
with autophagy, but not the protein ubiquitination or cardiomyocyte
apoptosis in an ApcMin/+ mouse model of colorectal cancer. The
study suggested that the loss of cardiac mass during cachexia
progression in the ApcMin/+ mice was associated with the
AKT-independent suppression of anabolic signaling and increased
autophagy (88). Furthermore, the
mTOR signaling pathway was demonstrated to control myofiber
formation and myofiber growth during muscle regeneration via
kinase-independent and kinase-dependent mechanisms, respectively
(89). A previous study discovered
that IGF-2 expression during the early phase of regeneration was
sensitive to rapamycin in an mTOR kinase-independent manner,
whereas p70S6K was required for mTOR kinase-dependent myofiber
growth (89). In summary, the
findings of these previous reports indicated that the PI3K/AKT/mTOR
signaling pathway may serve an important role in the process of
muscle protein synthesis, and that the regulation of this pathway
is very complicated, which has undoubtedly influenced the direction
of future PI3K/AKT/mTOR pathway research.
Cachexia phenotypes, such as skeletal muscle
wasting, have been causally linked to the cytokine-activated
transcription factor STAT3. Binding of IL-6 to its receptor induces
STAT3, which was found to lead to proteolysis and muscle wasting
(13,90,91).
STAT3 may be considered as a therapeutic target for patients with
cachexia with gastric, lung and breast cancer. For example, a
previous study identified that IL-6 mediated STAT3 activation in
cachectic patients with gastric and breast cancer (60). STAT3 can also induce the IκB kinase
(IKK)/NF-κB signaling pathway, which was discovered to mediate
apoptosis and muscle atrophy (92). A previous study with C2C12 myotubes
cultured in a simulated cachexia environment revealed that IFN-γ
and TNF-α promoted STAT3 phosphorylation on the myofiber-specific
cytoplasmic Y705 residue by activating JAK kinase (92). Interestingly, pY505-STAT3 and NF-κB
formed a complex that rapidly entered the nucleus and bound the
inducible nitric oxide synthase (iNOS) promoter to activate the
iNOS/nitric oxide (NO) pathway, which induced muscle atrophy
(92). Moreover, another study
identified that cancer cachexia activated STAT3 in the muscle to
stimulate muscle atrophy via two signaling pathways; in one
pathway, phosphorylated (p)-STAT3 stimulated caspase-3
transcription and activity, which induced the activation of the
UPS; while in the second pathway, p-STAT3 stimulated
CCAAT/enhancer-binding protein δ expression and activity, which
increased myostatin and MAFbx and MuRF1 (Fig. 1) (17).
NF-κB resides in the cytosol of cells and is tightly
bound via covalent bonds to IκB, which maintains it in an inactive
state (93). NF-κB activation
occurs by severing the covalent bonds with IκB via the action of
IKK. IKK is a kinase that phosphorylates IκB and initiates IκB
degradation via the Ub proteasome pathway, leaving NF-κB free and
active (94). The canonical
activation of NF-κB due to the degradation of the inhibitor of
IKBα/β by IKK is dependent on TNF (93). The activation of NF-κB was
identified as a key event in the processes that mediate muscle
atrophy (95). NF-κB-inducing
kinase (NIK) serves as a proximal inducer of the IKK complex, which
is an upstream convergence point for numerous signals leading to
NF-κB activation. The overexpression of NIK in primary human
skeletal muscle myotubes increased skeletal muscle atrophy
biomarkers, while NIK knockdown significantly attenuated
glucocorticoid-induced increases in NIK and MAFbx (49). Multiple studies have revealed that
the NF-κB signaling pathway also mediated muscle atrophy by
activating the downstream iNOS/NO pathway (92,96).
In addition, another study investigated the effect of muscle
metabolism on patients with cachexia and advanced NSCLC; compared
with healthy volunteers, patients with NSCLC had significantly
upregulated NF-κB mRNA expression levels (18).
MAPK family proteins, which are evolutionarily
conserved serine/threonine protein kinases, serve a central role in
the p38 signal transduction pathway. ERKs, p38 MAPK, JNKs and ERK5
represent the four MAPK subfamilies (97). The p38 MAPK signaling pathway was
found to serve a critical role in the regulation of E3 ligase
expression and skeletal muscle atrophy (98–100) and is activated by a number of
extra- and intracellular stimuli, including the proinflammatory
factors TNF-α (12,101), endotoxin (102) and reactive oxygen species (ROS),
as well as stressors such as oxidative stress (45).
A previous study identified that oxidative
stress-induced the expression of an autophagy-related gene,
autophagy related 7 (ATG7), in the autophagy-lysosomal proteolytic
(ALP) pathway, and the E3 ligases (MuRF1 and MAFbx) in the UPS were
temporally associated with the activation of the p38 MAPK pathway
independent of NF-κB- and FOXO-dependent transcriptional activation
in cultured muscle cells. These findings provided direct evidence
for the functional role of the p38 MAPK signaling pathway in
mediating oxidative stress through the ALP pathway in cachectic
muscle wasting (45). Based on the
above findings, a model was proposed in which oxidative
stress-induced p38 MAPK activation was suggested to initiate and
participate in cachectic muscle wasting through both the UPS and
ALP mechanisms.
Several transcription factors have been identified
to play important roles in muscle atrophy, particularly FOXO
factors (86,87,103), thus the inhibition of FOXO
factors is an attractive approach to combat muscle wasting. One
study showed that when FOXO1 expression was blocked both in cells
and in mice, the expression levels of MyoD, a myogenic factor, were
upregulated (Fig. 1) (104). Moreover, constitutively active
FOXO3 acted on the MAFbx promoter to cause MAFbx transcription and
enhance the atrophy of myotubes and muscle fibers (87). In animal models of cancer cachexia,
bioinformatics analysis of upregulated gene transcripts that
required FOXO revealed an enrichment of the proteasome, activator
protein 1, and IL-6 pathways, and included several atrophy-related
transcription factors, such as STAT3, Fos and
CCAAT/enhancer-binding protein β (C/EBPβ). Furthermore, the study
validated these findings in limb muscles and the diaphragm through
RT-qPCR and demonstrated that FOXO1 and FOXO3a were sufficient to
increase STAT3, Fos, C/EBPβ and the C/EBPβ target gene,
E3-Ub-protein ligase Ubr2 (15).
Experimental studies have also explored the function of the
transcription factor Twist1 in cancer-driven muscle atrophy; for
example, a previous study demonstrated that Twist1 expression drove
the upregulation of MuRF1 and MAFbx expression levels, leading to
muscle protein degradation (48).
A previous study analyzed the transcriptomes of
cells in atrophied skeletal muscle of cancer cachexia model mice
and revealed that the involved transcription factors and
transcription factor families included Oct1, sex-determining region
Y protein, myogenin, TNF receptor superfamily member 25, zinc
finger protein ZIC 2, T-Box transcription factor 5, sterol
regulatory element-binding protein 1, STAT, PU1, T3R, TAL1BETAITF2,
heat shock factor protein, lymphoid enhancer-binding factor 1, S8,
protein C-ets-1 and SOX9_BP1. In addition, various transcription
factors with specific effects on myogenesis were identified,
including myogenin, FOXO3, NF-κB p65 and paired box 1 (105). Furthermore, Marchildon et
al (106) found that
myoblasts exposed to an in vitro cancer cachexia environment
exhibited upregulated C/EBPβ expression, which led to diminished
myogenin expression and myogenesis.
To further understand the pathogenesis of cancer
cachexia, previous studies have conducted experiments
comprehensively analyzing the muscle atrophy network regulated by
miRNAs. Indeed, researchers have predicted new miRNA/mRNA
interactions, such as miR-27a/FOXO1, miR-27a/myocyte-specific
enhancer factor 2C (MEF2C), miR-27b/stromal cell-derived factor 1
(CXCL12), miR-27b/MEF2C, miR-140/CXCL12, miR-199a/caveolin-1 and
miR-199a/JunB, which may cause muscle atrophy in cancer cachexia
(107). In addition, a previous
study evaluated the miRNA profile of cancer cachexia-induced
skeletal muscle atrophy in a mouse model and identified 9
significantly differentially expressed miRNAs associated with
cancer, intercellular signaling and cell development (108). Overall, these results provided a
basis for future research into genetic targets for reducing muscle
loss in cancer cachexia (108).
Previous studies have noted the important role of
the autophagy-lysosome system in regulating muscle mass, in which
several key components of the autophagy machinery were discovered
to be transcriptionally upregulated during muscle wasting (109–111). A colon-26 (C26) cancer cachexia
mouse model was established to observe the effects of autophagy
inhibition (Beclin-1 knockout) or promotion [tumor protein p53
inducible nuclear protein 2 (TP53INP2/DOR) overexpression] on
cancer-induced muscle loss; the results revealed that Beclin-1
knockout could not prevent muscle atrophy in tumor-bearing mice and
that TP53INP2-mediated autophagy exacerbated the muscle loss.
Furthermore, an increase in autophagy was shown to clearly lead to
a decrease in muscle mitochondrial function in another study
(112). A recent report
identified that activin A served in an autocrine manner to promote
the synthesis and secretion of IL-6 from cancer cells. The
inhibition of activin signaling reduced the production of IL-6 in
cancer cells and the ability of cancer cells to accelerate
autophagy in non-cancerous cells in vivo, which reversed
cachexia and counteracted the loss of all measured muscle groups
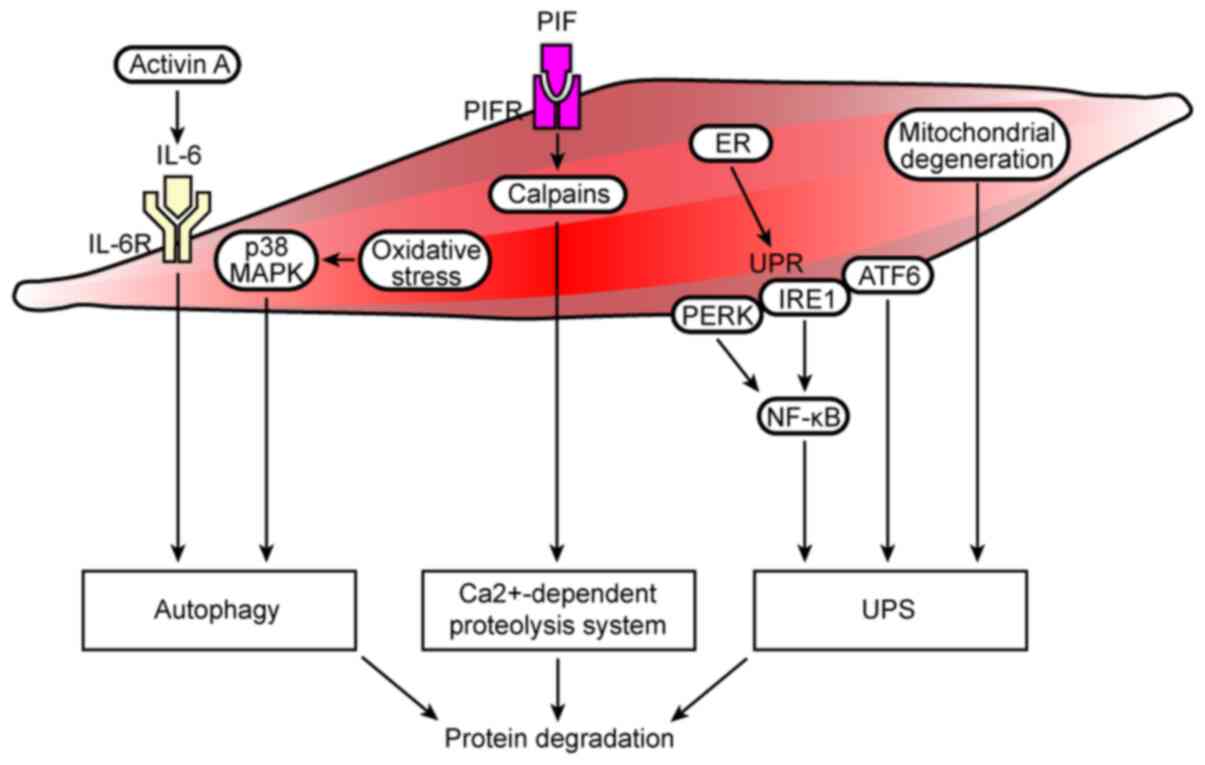
(Fig. 2) (113). In addition, oxidative
stress-induced expression of the autophagy-related gene ATG7 in the
ALP pathway was found to be temporally associated with activation
of the p38 MAPK signaling pathway (Fig. 2) (45).
Currently, to the best of our knowledge, little is
known about the relevance of the Ca2+-dependent
proteolytic system in cancer cachexia. Previous research has
demonstrated that proteolysis-inducing factor (PIF) induced muscle
loss in cancer cachexia through its high-affinity membrane bound
receptor (114). In vitro,
the binding of PIF to its receptor in skeletal muscle triggered an
increase in Ca2+, which initiated the
Ca2+-dependent proteolytic system, leading to an
increase in protein degradation (Fig.
2) (114). Pin et al
(22) established experimental
models of cachexia using Yoshida AH-130 liver cancer cells and C26
colon cancer cells; the results revealed that calpain-1 was
overexpressed in cachexia model rats with AH-130 liver cancer,
while the expression levels of calpastatin (a physiological
Ca2+-dependent protease inhibitor) expression were
downregulated. Interestingly, these data indicated, for the first
time, that the Ca2+-dependent proteolysis system was
also overactivated in the C26 rat model. However, interference with
Ca2+−dependent proteolysis did not alter the course of
muscle wasting in experimental cancer cachexia.
Skeletal muscle contains a plentiful network of ER,
which serves an important role in the regulation of proteostasis
and Ca2+ homeostasis. Protein folding in the ER is
exquisitely sensitive to changes in the environment, which leads to
disrupted protein folding to cause the accumulation of unfolded or
misfolded proteins, a condition termed ER stress (115,116). The ER manages such stress by
initiating the unfolded protein response (UPR), which is controlled
by three transmembrane proteins, namely, RNA-dependent protein
kinase-like ER eukaryotic translation initiation factor 2α kinase
(PERK) (117), inositol-requiring
protein 1 (IRE1) (117,118) and activating transcription factor
6 (ATF6) (119,120), which are activated to alleviate
ER stress. In the absence of stress, the intra-luminal domains of
PERK, IRE1 and ATF6 bind to the ER luminal protein
glucose-regulated protein 78 (GRP78), also known as heat shock
protein A or binding immunoglobulin protein. However, the
accumulation of misfolded and/or unfolded proteins in the ER lumen
leads to the dissociation of PERK, IRE1 and ATF6 from GRP78,
thereby activating downstream signaling cascades (121). The main function of the UPR is to
restore homeostasis (122), but
excessive or prolonged activation of the UPR can lead to
pathological conditions (116).
Previous studies have reported emerging roles of ER
stress and the UPR in cancer cachexia-induced muscle atrophy
(31,123,124). In fact, markers of ER stress and
the UPR were upregulated in the muscles of cachectic patients with
cancer (124). Moreover, studies
have shown that the optimal activation of NF-κB during ER stress
requires inputs from both IRE1 and PERK activities in cancer cells
(Fig. 2) (125). In addition, the mRNA and protein
expression levels of ATF6 were significantly upregulated in the
vastus lateralis (VL) of patients with LC-induced cachexia, and
ATF6 was also discovered to potentially interact with protein
degradation pathways, such as the UPS. The gene expression levels
of MAFbx and MuRF1 were also significantly upregulated in the VL of
LC-induced cachexia patients compared with healthy controls
(124). Evidence from previous
studies has also revealed that multiple markers of ER stress and
the UPR, such as PERK, IRE1a and ATF6, were highly activated in the
skeletal muscle of LLC and Apc(Min/+) mouse models of cancer
cachexia. The inhibition of the UPR reduced the activity of the
AKT/mTOR signaling pathway and upregulated the expression levels of
MuRF1 and MAFbx and autophagy in LLC-bearing mice. Therefore, the
study provided initial evidence to suggest that ER stress and the
UPR pathway may be essential for maintaining skeletal muscle mass
and strength, in addition to the protection against cancer cachexia
(31).
Based on these aforementioned findings, some
divergent views are notably present, and the majority of the
studies regarding the regulation of ER stress and the UPR in cancer
cachexia-induced muscle atrophy were performed using cultured cells
or preclinical animal models. Therefore, additional studies in
patients are required to obtain corroborative clinical data
regarding ER stress, the UPR and cancer cachexia-induced muscle
atrophy.
Numerous studies have illustrated that muscle
atrophy caused by cancer cachexia was related to mitochondrial
dysfunction (126–130). For example, the expression of
mitochondrial quality control (MQC) axis mediators was detected in
rectus abdominis muscle biopsies from 18 elderly patients with
gastric adenocarcinoma (9 with cancer cachexia and 9 without
cachexia) and 9 controls; the expression levels of the mitotic
protein Fis1 were upregulated in the patients with cancer cachexia,
while the fusion index [mitofusin-2 (Mfn2)/Fis1 ratio] was
decreased in the patients. Therefore, these results suggested that
cachexia may be related to mitochondrial dynamics and signal
transduction through the muscle MQC axis (24).
Previous studies have demonstrated that
mitochondrial degeneration precedes muscle atrophy in the
development of cancer cachexia in tumor-bearing mice, providing
novel evidence for mitochondrial damage preceding
cachexia-associated muscle loss (127). Neyroud et al (131) established a C26-induced cancer
cachexia model in CD2F1 mice and observed the mitochondrial
respiratory capacity and content of skeletal muscle. Indeed,
skeletal muscle mitochondrial respiration, mitochondrial coupling
and the mitochondrial content were all reduced.
The overexpression of the VDR in tumor-bearing
animals has been reported to impair muscle regeneration and cause
protein degradation. Therefore, caution should be exercised when
considering vitamin D supplementation for patients with chronic
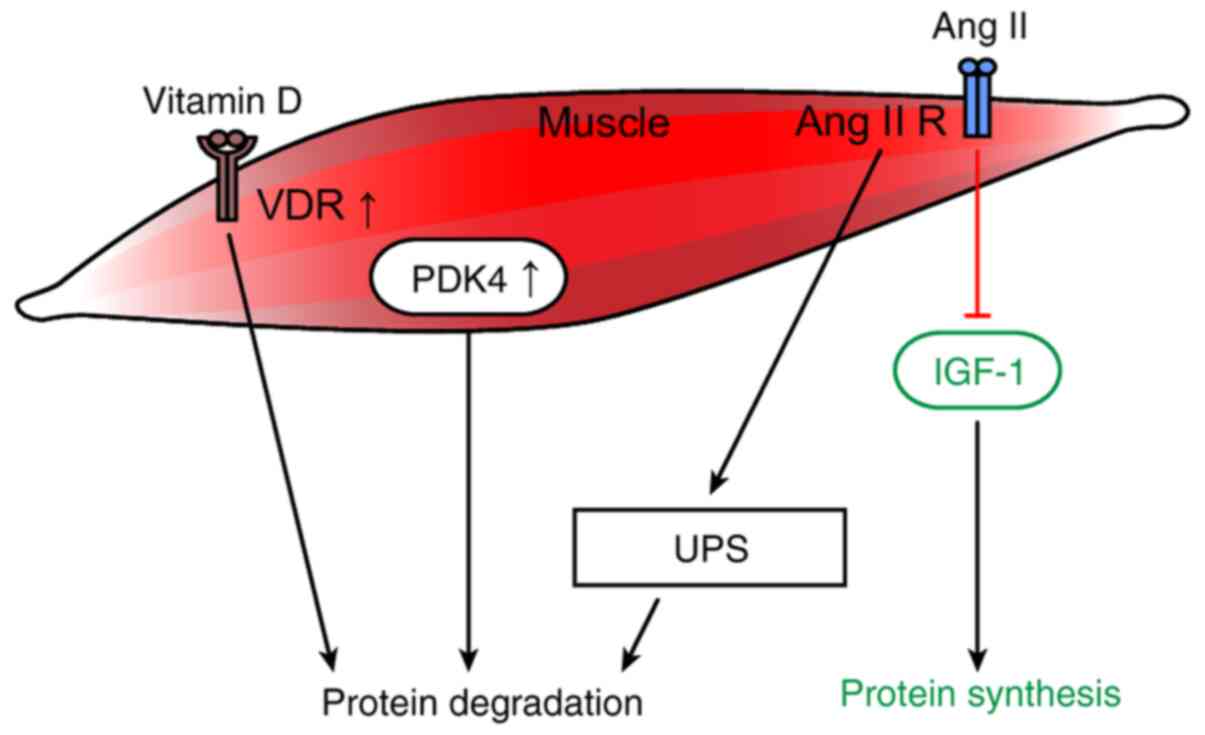
diseases that may involve muscle regeneration (Fig. 3) (23).
A previous study has indicated that in a mouse
pancreatic cancer cachexia model, ZIP4 could stimulate the release
of heat shock protein (HSP)70 and HSP90 from the extracellular
vesicles to stimulate p38 MAPK-mediated abnormal muscle catabolism
(21).
PDK4 is an important regulator of cellular energy
metabolism. High PDK4 and abnormal energetic metabolism were found
in the skeletal muscle of colon-26 tumor hosts, as well as in mice
fed a diet enriched in Pirinixic acid. Viral-mediated PDK4
overexpression in myotube cultures was sufficient to promote
myofiber shrinkage. On the contrary, blockade of PDK4 was
sufficient to restore myotube size in C2C12 cultures exposed to
tumor media. This study by Pin et al (132) was the first to confirm the direct
role of PDK4 in promoting cancer-related changes in muscle
metabolism and skeletal muscle atrophy through in vitro and
in vivo experiments (Fig.
3).
Ang II is the main effector molecule of the
renin-angiotensin system and increasing evidence has revealed that
it also serves an important role in the development of muscle
atrophy (133,134). In addition, Ang II has been
reported to induce myonuclear apoptosis during muscle atrophy
(135).
Ang II was discovered to activate the UPS to induce
muscle atrophy by generating ROS and inhibiting the IGF-1 signaling
pathway (Fig. 3) (136). Muscle atrophy has been suggested
to depend on the impairment of the IGF-1 signaling transduction
pathway. Sugiyama et al (19) reported the effects of ghrelin on
body weight and muscle catabolism in Ang II-induced cachexia mice.
The IGF-1 levels were reduced in the gastrocnemius of the Ang
II-treated mice (Fig. 3).
Consistently, researchers have reported that ghrelin can improve
weight loss and skeletal muscle catabolism in mice treated with Ang
II. These effects are thought to be related to the early recovery
of IGF-1 mRNA and the improved nutritional status in the skeletal
muscle. However, other research has indicated that although the
IGF-1 system is downregulated in cancer cachexia, no simple
relationship linking IGF-1 and/or MAFbx mRNA expression levels and
muscle atrophy could be observed under experimental conditions
(Fig. 3) (20).
Clinical research has revealed that compared with
patients with cancer without cachexia, those with pre-cachexia or
cachexia had upregulated plasma neutrophil-derived protease (NDP)
mRNA expression levels and significantly higher Ang II, TGF-β1 and
C-reactive protein (CRP) levels. These findings suggested that Ang
II, TGFβ1, CRP and NDP may serve as potential circulating
biomarkers for cancer cachexia, which may facilitate the early
diagnosis and prevention of cancer cachexia (137).
The pathogenesis of cancer cachexia-associated
muscle atrophy is complex and not completely understood. Therefore,
multimodal comprehensive treatment should be adopted to delay
muscle atrophy caused by cachexia. Related measures include
activating the PI3K/AKT/mTOR signaling pathway (138,139), inhibiting the UPS (140,141), proinflammatory factors (142), signal transduction pathways
(53,143) and transcription factors (144), and regulating the expression of
certain organelles (145) and
receptors (146) related to
muscle atrophy caused by cancer cachexia.
The results of multiple studies have suggested that
the activation of the PI3K/AKT/mTOR signaling pathway may improve
muscle atrophy caused by cancer cachexia. For example, a C26 cancer
cell cachexia model in mice was established, and skeletal muscle
responses to aerobic exercise and resistance training were
compared. Interestingly, neither aerobic nor resistance training
prevented tumor-induced weight loss. However, aerobic training
maintained the motor function and reduced the inflammatory response
in the spleen; therefore, it may slightly improve muscle atrophy by
activating the mTOR pathway and exert therapeutic value for
patients with cancer cachexia (147). In contrast, resistance training
induced the expression of genes related to muscle damage and
repair, such as myogenin and IGF-IEb, which might be due to the
excessive stress caused by the high resistance load in the
tumor-bearing state (147). In
addition, Tanaka et al (148) discovered that low-intensity
exercise inhibited Yoshida AH130 ascites LC cell-induced cancer
cachexia through the skeletal UPS in male Wistar rats. In addition,
low-intensity exercise increased the levels of hypoxia-inducible
factor-1α and p-AMPK, which suppressed the loss of muscle mass and
the inactivation of mTOR in the soleus muscle. Furthermore, C2C12
myotubes were cultured in C26 conditioned medium in vitro,
and the pharmacological activity of the myostatin (MSTN) pathway
inhibitor IMB0901, which inhibits MSTN promoter activity, MSTN
signal transduction and MSTN positive feedback regulation, was
determined; the results identified that this compound suppressed
muscle atrophy caused by cancer cachexia by inhibiting Ub-mediated
proteolysis and enhancing AKT/mTOR-mediated protein synthesis
(139).
The UPS is the main regulatory mechanism of protein
degradation in cancer cachexia-induced muscle atrophy. Previous
evidence of potential strategies to protect against skeletal muscle
wasting through inhibition of E3 (MuRF-1 and MAFbx) have been
summarized (140,149). For example, Levolger et al
(141) studied the ability of the
ActRIIB and TGF-β receptor type-1 inhibitors, SB431542 and
GW788388, to prevent muscle atrophy in a C26-CD2F1 cachexia model;
it was discovered that the treatment with GW788388 prevented cancer
cachexia and downregulated MAFbx.
Another previous study illustrated that valproic
acid reduced MAFbx expression levels by inhibiting C/EBPβ binding
to the MAFbx promoter, which subsequently decreased skeletal muscle
degradation in cancer cachexia mice (150). The traditional Chinese medicine
(TCM) Zhimu and Huangbai herb pair was shown to not only inhibit
the UPS genes (MuRF1 and FOXO3) associated with muscle atrophy in
C57BL/6 colon cancer cachexia model mice, but also activate the
IGF-1/AKT and autophagy signaling pathways to facilitate protein
synthesis (151). Another study
revealed that Baoyuan Jiedu Decoction and Paeonia lactiflora root
extract inhibited muscle atrophy in cancer cachexia model mice by
downregulating atrogin-1 and MuRF1 expression levels (140,149). Meanwhile, modified Sijunzi
decoctions (Zhen-Qi; ZQ-SJZ) have been extensively used to treat
cachexia and improve the quality of life of patients with cancer
undergoing chemotherapy. The administration of ZQ-SJZ was found to
recover tumor- and/or cisplatin-induced body weight loss, as well
as the forelimb grip strength and myofiber size. ZQ-SJZ also
increased the expression levels of myogenic proteins, such as MyHC
and myogenin, and downregulated the expression levels of the
atrophy-related protein atrogin-1 in cisplatin-treated C2C12
myotubes in vitro (152).
Inflammation is hypothesized to regulate pathways
controlling skeletal muscle wasting. A previous study identified
that IL-6 and its receptor, as well as JAK2 and STAT3 were
significantly attenuated with kimchi. Kimchi was discovered to be a
potential option to ameliorate cancer cachexia through its ability
to suppress IL-6 and decrease muscle atrophy in a C26 mouse model
(142). In addition, in an
experimental model of C26-induced cancer cachexia, 20S,
21-epoxy-resibufogenin-3-acetate (ERBA) markedly inhibited body
weight loss. ERBA is a specific small molecule with IL-6 receptor
antagonist activity (142).
Furthermore, another previous study revealed that aerobic interval
training enhanced the IL-10/TNF-α ratio, an anti-inflammatory
index, and IL-15 expression levels in the skeletal muscle of
tumor-bearing mice (153). In
vivo trials confirmed that combining exercise training with
antioxidant supplements (selenium nanoparticles) may also be a
potential strategy to control tumor volume and prevent cachexia in
patients with breast cancer (153).
Previous research has reported that important
molecules in signaling pathways related to muscle atrophy (NF-κB,
MAPK and FOXO), proteolytic markers (Ub ligases and proteasomes),
autophagy markers (p62, Beclin-1 and microtubule-associated protein
1A/1B light chain 3B) and myostatin levels were upregulated, while
regeneration and metabolic markers (MyoD, mTOR, AKT and peroxisome
proliferator-activated receptor γ coactivator 1-α) were decreased
in cachexia. These changes were attenuated by the administration of
formoterol in cachexia model rats (154). Moreover, coix seed oil
ameliorated cancer cachexia by counteracting muscle loss and fat
lipolysis in an LLC cachexia model in C57BL/6 mice through the
regulation of the NF-κB/MuRF1 and AMPK/hormone sensitive lipase
pathways (143).
Cryptotanshinone prevented muscle wasting in cancer
cachexia through STAT3 inhibition, therefore it was suggested to be
a promising candidate drug for the treatment of cancer cachexia
(53). Sunitinib was able to
alleviate the overactivation of the STAT3 and MuRF1 signaling
pathways, which prevented body weight loss and muscle wasting
during cancer cachexia (155). In
addition, an in vivo study reported that although
pyrrolidine dithiocarbamate (PDTC) did not reduce the tumor volume
in a C26 ×enograft mouse model, it reduced cancer cachexia
symptoms. In addition, in vitro studies demonstrated that
PDTC inhibited muscle atrophy and lipolysis in an in vitro
cell model induced by TNF-α and C26 tumor cell supernatant, and
impeded the atrophy of C2C12-differentiated myotubes by
downregulating MyoD and upregulating MuRF1 expression levels.
Moreover, in addition to inhibiting NF-κB signaling, PDTC inhibited
p38 MAPK signaling and affected skeletal muscle protein synthesis
by activating AKT signaling (156). In another study, the expression
levels of the transcription factor FOXO were upregulated in
85As2-induced cachectic model rats, and the increased FOXO
expression levels were considered to be associated with the
increased expression of atrogin-1 and MuRF1. Notably, the oral
administration of rikkunshito, a traditional Japanese medicine,
substantially ameliorated the presence of cancer cachexia (144).
These findings provided further possible molecular
mechanisms for the targeted suppression of muscle atrophy induced
by cancer cachexia.
Mfn2 is highly expressed in muscle cells, and its
function is diminished by disruptions in the mitochondrial network,
which is essential for maintaining normal mitochondrial function
(145). Clinical studies have
reported that Mfn2 expression levels were downregulated in the
rectus abdominis of patients with cancer cachexia (24). Further evidence indicated that Mfn2
overexpression improved TNF-α-induced C2C12 cell muscle atrophy
in vitro. Moreover, in vivo experiments demonstrated
that Mfn2 overexpression in the gastrocnemius muscle partially
reduced gastrocnemius muscle loss caused by cachexia. Overall,
these findings suggested that Mfn2 may be involved in
cachexia-induced muscle loss and may be a potential target for
cachexia treatment (145).
In addition, treatment regimens should include
proper nutritional plans, psychological intervention and to achieve
synergistic effects and change the abnormal cachexia metabolism.
Although current research has demonstrated that reversing the
progression of cachexia with nutritional support alone is
difficult, increasing nutrient intake was found to somewhat delay
the progression of cachexia and improve the quality of life
(158). Previous studies have
investigated the need for nutritional support among patients with
advanced cancer in the palliative care environment; patients with
cancer cachexia had a greater need for nutritional support and
desired additional support from medical staff when the negative
effects of cachexia become apparent, of which adequate nutrition
cannot be guaranteed by oral nutritional supplements (159).
Furthermore, patients with cancer cachexia often
develop complications due to the tumor itself or tumor-related
effects. Several patients with cancer face psychological burdens or
have other medical conditions and experience psychological symptoms
such as fear, anxiety and depression (160). McClement (160) proposed providing psychosocial
support for such patients and their families. Moreover, nurses must
understand the psychological impact of anorexia and cachexia on
affected individuals and suggest interventions that the medical
team can implement to address these issues. Continuous research has
been recommended to obtain a more complete understanding of the
psychological aspects of the patient experience.
The treatment effectiveness of chemical drugs has
not been established by existing cachexia guidelines. In a TCM
study, a large portion of patients with cancer cachexia were
diagnosed with spleen deficiency syndrome and treated with
tonifying TCM, which produced clinical benefits (161). Oral administration of
atractylenolide I (20 mg/kg per day for 30 days) significantly
ameliorated the reduction in body weight and atrophy of the
muscles, spleen and thymus in mice with spleen deficiency and
cachexia (161). Clinical trials
on the efficacy and safety of the Yukgunja-Tang herbal mixture in
the treatment of cancer anorexia are also underway (162).
In conclusion, cancer cachexia is a metabolic
syndrome associated with malignant tumor progression, which
involves multiple mechanisms that cause muscle atrophy. The
pathogenesis is extremely complex and previous studies have
produced inconsistent results. Currently, no single drug that can
effectively reverse cachexia is included in clinical guidelines.
Therefore, the lack of treatment options combined with the
complicated pathogenesis necessitate the development of combination
therapeutics that target multiple pathways and targets.
In the future, it is predicted that cancer cachexia
will become a research hotspot in the field of cancer research;
this will help to further elucidate the pathogenesis of cachexia,
initiate the development of clinical trials and the emergence of
more effective drugs to reduce muscle cachexia associated with
cancer, and thereby improve the quality of life of patients and
extend patient survival.
Not applicable.
No funding was received.
Not applicable.
WY, JH and WG conceptualized and designed the
review. HW, YW, ZD, YL, WW and QW conducted the literature review
and compiled the information. WY and JH wrote the manuscript. WG
revised the manuscript. All authors read and approved the final
manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Fearon K, Strasser F, Anker SD, Bosaeus I,
Bruera E, Fainsinger RL, Jatoi A, Loprinzi C, MacDonald N,
Mantovani G, et al: Definition and classification of cancer
cachexia: An international consensus. Lancet Oncol. 12:489–495.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Blum D, Stene GB, Solheim TS, Fayers P,
Hjermstad MJ, Baracos VE, Fearon K, Strasser F and Kaasa S;
Euro-Impact: Validation of the consensus-definition for cancer
cachexia and evaluation of a classification model-a study based on
data from an international multicentre project (EPCRC-CSA). Ann
Oncol. 25:1635–1642. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Baumgartner RN, Koehler KM, Gallagher D,
Romero L, Heymsfield SB, Ross RR, Garry PJ and Lindeman RD:
Epidemiology of sarcopenia among the elderly in New Mexico. Am J
Epidemiol. 147:755–763. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Emery PW, Edwards RH, Rennie MJ, Souhami
RL and Halliday D: Protein synthesis in muscle measured in vivo in
cachectic patients with cancer. Br Med J (Clin Res Ed).
289:584–586. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Warnold I, Lundholm K and Schersten T:
Energy balance and body composition in cancer patients. Cancer Res.
38:1801–1807. 1978.PubMed/NCBI
|
|
6
|
Chang VT, Xia Q and Kasimis B: The
functional assessment of anorexia/cachexia therapy (FAACT) appetite
scale in veteran cancer patients. J Support Oncol. 3:377–382.
2005.PubMed/NCBI
|
|
7
|
Martin L, Birdsell L, Macdonald N, Reiman
T, Clandinin MT, McCargar LJ, Murphy R, Ghosh S, Sawyer MB and
Baracos VE: Cancer cachexia in the age of obesity: Skeletal muscle
depletion is a powerful prognostic factor, independent of body mass
index. J Clin Oncol. 31:1539–1547. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dewys WD, Begg C, Lavin PT, Band PR,
Bennett JM, Bertino JR, Cohen MH, Douglass HO Jr, Engstrom PF,
Ezdinli EZ, et al: Prognostic effect of weight loss prior to
chemotherapy in cancer patients. Eastern Cooperative Oncology
Group. Am J Med. 69:491–497. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dunne RF, Roussel B, Culakova E, Pandya C,
Fleming FJ, Hensley B, Magnuson AM, Loh KP, Gilles M, Ramsdale E,
et al: Characterizing cancer cachexia in the geriatric oncology
population. J Geriatr Oncol. 10:415–419. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sadeghi M, Keshavarz-Fathi M, Baracos V,
Arends J, Mahmoudi M and Rezaei N: Cancer cachexia: Diagnosis,
assessment, and treatment. Crit Rev Oncol Hematol. 127:91–104.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tazi E and Errihani H: Treatment of
cachexia in oncology. Indian J Palliat Care. 16:129–137. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li YP, Chen Y, John J, Moylan J, Jin B,
Mann DL and Reid MB: TNF-alpha acts via p38 MAPK to stimulate
expression of the ubiquitin ligase atrogin1/MAFbx in skeletal
muscle. FASEB J. 19:362–370. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan
R, Puzis L, Koniaris LG and Zimmers TA: JAK/STAT3 pathway
inhibition blocks skeletal muscle wasting downstream of IL-6 and in
experimental cancer cachexia. Am J Physiol Endocrinol Metab.
303:E410–421. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Braun TP, Zhu X, Szumowski M, Scott GD,
Grossberg AJ, Levasseur PR, Graham K, Khan S, Damaraju S, Colmers
WF, et al: Central nervous system inflammation induces muscle
atrophy via activation of the hypothalamic-pituitary-adrenal axis.
J Exp Med. 208:2449–2463. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Judge SM, Wu CL, Beharry AW, Roberts BM,
Ferreira LF, Kandarian SC and Judge AR: Genome-wide identification
of FoxO-dependent gene networks in skeletal muscle during C26
cancer cachexia. BMC Cancer. 14:9972014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schmitt TL, Martignoni ME, Bachmann J,
Fechtner K, Friess H, Kinscherf R and Hildebrandt W: Activity of
the Akt-dependent anabolic and catabolic pathways in muscle and
liver samples in cancer-related cachexia. J Mol Med (Berl).
85:647–654. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Silva KA, Dong J, Dong Y, Dong Y, Schor N,
Tweardy DJ, Zhang L and Mitch WE: Inhibition of Stat3 activation
suppresses caspase-3 and the ubiquitin-proteasome system, leading
to preservation of muscle mass in cancer cachexia. J Biol Chem.
290:11177–11187. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Murton AJ, Maddocks M, Stephens FB,
Marimuthu K, England R and Wilcock A: Consequences of late-stage
non-small-cell lung cancer cachexia on muscle metabolic processes.
Clin Lung Cancer. 18:e1–e11. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sugiyama M, Yamaki A, Furuya M, Inomata N,
Minamitake Y, Ohsuye K and Kangawa K: Ghrelin improves body weight
loss and skeletal muscle catabolism associated with angiotensin
II-induced cachexia in mice. Regul Pept. 178:21–28. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Costelli P, Muscaritoli M, Bossola M,
Penna F, Reffo P, Bonetto A, Busquets S, Bonelli G, Lopez-Soriano
FJ, Doglietto GB, et al: IGF-1 is downregulated in experimental
cancer cachexia. Am J Physiol Regul Integr Comp Physiol.
291:R674–683. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang J, Zhang Z, Zhang Y, Ni X, Zhang G,
Cui X, Liu M, Xu C, Zhang Q, Zhu H, et al: ZIP4 promotes muscle
wasting and cachexia in mice with orthotopic pancreatic tumors by
stimulating RAB27B-regulated release of extracellular vesicles from
cancer cells. Gastroenterology. 156:722–734.e6. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pin F, Minero VG, Penna F, Muscaritoli M,
De Tullio R, Baccino FM and Costelli P: Interference with
Ca2+-dependent proteolysis does not alter the course of
muscle wasting in experimental cancer cachexia. Front Physiol.
8:2132017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Camperi A, Pin F, Costamagna D, Penna F,
Menduina ML, Aversa Z, Zimmers T, Verzaro R, Fittipaldi R, Caretti
G, et al: Vitamin D and VDR in cancer cachexia and muscle
regeneration. Oncotarget. 8:21778–21793. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Marzetti E, Lorenzi M, Landi F, Picca A,
Rosa F, Tanganelli F, Galli M, Doglietto GB, Pacelli F, Cesari M,
et al: Altered mitochondrial quality control signaling in muscle of
old gastric cancer patients with cachexia. Exp Gerontol. 87:92–99.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Williams A, Sun X, Fischer JE and
Hasselgren PO: The expression of genes in the ubiquitin-proteasome
proteolytic pathway is increased in skeletal muscle from patients
with cancer. Surgery. 126:744–750. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Doyle A, Zhang G, Abdel Fattah EA, Eissa
NT and Li YP: Toll-like receptor 4 mediates
lipopolysaccharide-induced muscle catabolism via coordinate
activation of ubiquitin-proteasome and autophagy-lysosome pathways.
FASEB J. 25:99–110. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Furuno K and Goldberg AL: The activation
of protein degradation in muscle by Ca2+ or muscle injury does not
involve a lysosomal mechanism. Biochem J. 237:859–864. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lecker SH, Solomon V, Mitch WE and
Goldberg AL: Muscle protein breakdown and the critical role of the
ubiquitin-proteasome pathway in normal and disease states. J Nutr.
129 (Suppl 1):S227–S237. 1999. View Article : Google Scholar
|
|
29
|
White JP, Puppa MJ, Gao S, Sato S, Welle
SL and Carson JA: Muscle mTORC1 suppression by IL-6 during cancer
cachexia: A role for AMPK. Am J Physiol Endocrinol Metab.
304:E1042–E1052. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li YP, Schwartz RJ, Waddell ID, Holloway
BR and Reid MB: Skeletal muscle myocytes undergo protein loss and
reactive oxygen-mediated NF-kappaB activation in response to tumor
necrosis factor alpha. FASEB J. 12:871–880. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bohnert KR, Gallot YS, Sato S, Xiong G,
Hindi SM and Kumar A: Inhibition of ER stress and unfolding protein
response pathways causes skeletal muscle wasting during cancer
cachexia. FASEB J. 30:3053–3068. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fontes-Oliveira CC, Busquets S, Toledo M,
Penna F, Paz Aylwin M, Sirisi S, Silva AP, Orpí M, García A, Sette
A, et al: Mitochondrial and sarcoplasmic reticulum abnormalities in
cancer cachexia: Altered energetic efficiency? Biochim Biophys
Acta. 1830:2770–2778. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mondello P, Mian M, Aloisi C, Fama F,
Mondello S and Pitini V: Cancer cachexia syndrome: Pathogenesis,
diagnosis, and new therapeutic options. Nutr Cancer. 67:12–26.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hershko A and Ciechanover A: The ubiquitin
system. Annu Rev Biochem. 67:425–479. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hershko A and Ciechanover A: Mechanisms of
intracellular protein breakdown. Annu Rev Biochem. 51:335–364.
1982. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Haas AL and Rose IA: The mechanism of
ubiquitin activating enzyme. A kinetic and equilibrium analysis. J
Biol Chem. 257:10329–10337. 1982.PubMed/NCBI
|
|
37
|
Hershko A, Heller H, Elias S and
Ciechanover A: Components of ubiquitin-protein ligase system.
Resolution, affinity purification, and role in protein breakdown. J
Biol Chem. 258:8206–8214. 1983.PubMed/NCBI
|
|
38
|
Hershko A: The ubiquitin pathway for
protein degradation. Trends Biochem Sci. 16:265–268. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Reiss Y, Heller H and Hershko A: Binding
sites of ubiquitin-protein ligase. Binding of ubiquitin-protein
conjugates and of ubiquitin-carrier protein. J Biol Chem.
264:10378–10383. 1989.PubMed/NCBI
|
|
40
|
Voges D, Zwickl P and Baumeister W: The
26S proteasome: A molecular machine designed for controlled
proteolysis. Annu Rev Biochem. 68:1015–1068. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Glickman MH and Ciechanover A: The
ubiquitin-proteasome proteolytic pathway: Destruction for the sake
of construction. Physiol Rev. 82:373–428. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rom O and Reznick AZ: The role of E3
ubiquitin-ligases MuRF-1 and MAFbx in loss of skeletal muscle mass.
Free Radic Biol Med. 98:218–230. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cai D, Frantz JD, Tawa NE Jr, Melendez PA,
Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ and
Shoelson SE: IKKbeta/NF-kappaB activation causes severe muscle
wasting in mice. Cell. 119:285–298. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li W, Moylan JS, Chambers MA, Smith J and
Reid MB: Interleukin-1 stimulates catabolism in C2C12 myotubes. Am
J Physiol Cell Physiol. 297:C706–C714. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
McClung JM, Judge AR, Powers SK and Yan Z:
p38 MAPK links oxidative stress to autophagy-related gene
expression in cachectic muscle wasting. Am J Physiol Cell Physiol.
298:C542–C549. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kaisari S, Rom O, Aizenbud D and Reznick
AZ: Involvement of NF-κB and muscle specific E3 ubiquitin ligase
MuRF1 in cigarette smoke-induced catabolism in C2 myotubes. Adv Exp
Med Biol. 788:7–17. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Patel HJ and Patel BM: TNF-α and cancer
cachexia: Molecular insights and clinical implications. Life Sci.
170:56–63. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Parajuli P, Kumar S, Loumaye A, Singh P,
Eragamreddy S, Nguyen TL, Ozkan S, Razzaque MS, Prunier C, Thissen
JP and Atfi A: Twist1 activation in muscle progenitor cells causes
muscle loss akin to cancer cachexia. Dev Cell. 45:712–725.e6. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fry CS, Nayeem SZ, Dillon EL, Sarkar PS,
Tumurbaatar B, Urban RJ, Wright TJ, Sheffield-Moore M, Tilton RG
and Choudhary S: Glucocorticoids increase skeletal muscle NF-κB
inducing kinase (NIK): Links to muscle atrophy. Physiol Rep.
4:2016. View Article : Google Scholar
|
|
50
|
Gallot YS, Durieux AC, Castells J,
Desgeorges MM, Vernus B, Plantureux L, Rémond D, Jahnke VE, Lefai
E, Dardevet D, et al: Myostatin gene inactivation prevents skeletal
muscle wasting in cancer. Cancer Res. 74:7344–7356. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bédard N, Jammoul S, Moore T, Wykes L,
Hallauer PL, Hastings KE, Stretch C, Baracos V, Chevalier S,
Plourde M, et al: Inactivation of the ubiquitin-specific protease
19 deubiquitinating enzyme protects against muscle wasting. FASEB
J. 29:3889–3898. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lee H, Lee SJ, Bae GU, Baek NI and Ryu JH:
Canadine from corydalis turtschaninovii stimulates myoblast
differentiation and protects against myotube atrophy. Int J Mol
Sci. 18:27482017. View Article : Google Scholar
|
|
53
|
Chen L, Yang Q, Zhang H, Wan L, Xin B, Cao
Y, Zhang J and Guo C: Cryptotanshinone prevents muscle wasting in
CT26-induced cancer cachexia through inhibiting STAT3 signaling
pathway. J Ethnopharmacol. 260:1130662020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Chong SW, Nguyet LM, Jiang YJ and Korzh V:
The chemokine Sdf-1 and its receptor Cxcr4 are required for
formation of muscle in zebrafish. BMC Dev Biol. 7:542007.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Melchionna R, Di Carlo A, De Mori R,
Cappuzzello C, Barberi L, Musarò A, Cencioni C, Fujii N, Tamamura
H, Crescenzi M, et al: Induction of myogenic differentiation by
SDF-1 via CXCR4 and CXCR7 receptors. Muscle Nerve. 41:828–835.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Bobadilla M, Sainz N, Abizanda G, Orbe J,
Rodriguez JA, Páramo JA, Prósper F and Pérez-Ruiz A: The CXCR4/SDF1
axis improves muscle regeneration through MMP-10 activity. Stem
Cells Dev. 23:1417–1427. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Martinelli GB, Olivari D, Re Cecconi AD,
Talamini L, Ottoboni L, Lecker SH, Stretch C, Baracos VE, Bathe OF,
Resovi A, et al: Activation of the SDF1/CXCR4 pathway retards
muscle atrophy during cancer cachexia. Oncogene. 35:6212–6222.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Winbanks CE, Murphy KT, Bernardo BC, Qian
H, Liu Y, Sepulveda PV, Beyer C, Hagg A, Thomson RE, Chen JL, et
al: Smad7 gene delivery prevents muscle wasting associated with
cancer cachexia in mice. Sci Transl Med. 8:348ra3982016. View Article : Google Scholar
|
|
59
|
Stephens NA, Gallagher IJ, Rooyackers O,
Skipworth RJ, Tan BH, Marstrand T, Ross JA, Guttridge DC, Lundell
L, Fearon KC and Timmons JA: Using transcriptomics to identify and
validate novel biomarkers of human skeletal muscle cancer cachexia.
Genome Med. 2:12010. View
Article : Google Scholar : PubMed/NCBI
|
|
60
|
Eskiler GG, Bezdegumeli E, Ozman Z, Ozkan
AD, Bilir C, Kucukakca BN, Ince MN, Men AY, Aktas O, Horoz YE, et
al: IL-6 mediated JAK/STAT3 signaling pathway in cancer patients
with cachexia. Bratisl Lek Listy. 66:819–826. 2019.PubMed/NCBI
|
|
61
|
Pin F, Barreto R, Kitase Y, Mitra S, Erne
CE, Novinger LJ, Zimmers TA, Couch ME, Bonewald LF and Bonetto A:
Growth of ovarian cancer xenografts causes loss of muscle and bone
mass: A new model for the study of cancer cachexia. J Cachexia
Sarcopenia Muscle. 9:685–700. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Grabiec AM, Korchynskyi O, Tak PP and
Reedquist KA: Histone deacetylase inhibitors suppress rheumatoid
arthritis fibroblast-like synoviocyte and macrophage IL-6
production by accelerating mRNA decay. Ann Rheum Dis. 71:424–431.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ma F, Li Y, Jia L, Han Y, Cheng J, Li H,
Qi Y and Du J: Macrophage-stimulated cardiac fibroblast production
of IL-6 is essential for TGF β/Smad activation and cardiac fibrosis
induced by angiotensin II. PLoS One. 7:e351442012. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Miki S, Iwano M, Miki Y, Yamamoto M, Tang
B, Yokokawa K, Sonoda T, Hirano T and Kishimoto T: Interleukin-6
(IL-6) functions as an in vitro autocrine growth factor in renal
cell carcinomas. FEBS Lett. 250:607–610. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Iwase S, Murakami T, Saito Y and Nakagawa
K: Steep elevation of blood interleukin-6 (IL-6) associated only
with late stages of cachexia in cancer patients. Eur Cytokine Netw.
15:312–316. 2004.PubMed/NCBI
|
|
66
|
Fujimoto-Ouchi K, Onuma E, Shirane M, Mori
K and Tanaka Y: Capecitabine improves cancer cachexia and
normalizes IL-6 and PTHrP levels in mouse cancer cachexia models.
Cancer Chemother Pharmacol. 59:807–815. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
White JP, Baltgalvis KA, Puppa MJ, Sato S,
Baynes JW and Carson JA: Muscle oxidative capacity during
IL-6-dependent cancer cachexia. Am J Physiol Regul Integr Comp
Physiol. 300:R201–R211. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Op den Kamp CM, Gosker HR, Lagarde S, Tan
DY, Snepvangers FJ, Dingemans AM, Langen RC and Schols AM:
Preserved muscle oxidative metabolic phenotype in newly diagnosed
non-small cell lung cancer cachexia. J Cachexia Sarcopenia Muscle.
6:164–173. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Siddiqui RA and Williams JF: Tentative
identification of the toxohormones of cancer cachexia: Roles of
vasopressin, prostaglandin E2 and cachectin-TNF. Biochem Int.
20:787–797. 1990.PubMed/NCBI
|
|
70
|
Llovera M, García-Martínez C,
López-Soriano J, Carbó N, Agell N, López-Soriano FJ and Argiles JM:
Role of TNF receptor 1 in protein turnover during cancer cachexia
using gene knockout mice. Mol Cell Endocrinol. 142:183–189. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Powrozek T, Mlak R, Brzozowska A, Mazurek
M, Golebiowski P and Malecka-Massalska T: Relationship between
TNF-α −1031T/C gene polymorphism, plasma level of TNF-α, and risk
of cachexia in head and neck cancer patients. J Cancer Res Clin
Oncol. 144:1423–1434. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Llovera M, García-Martínez C, Agell N,
López-Soriano FJ and Argilés JM: TNF can directly induce the
expression of ubiquitin-dependent proteolytic system in rat soleus
muscles. Biochem Biophys Res Commun. 230:238–241. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Matsuyama T, Ishikawa T, Okayama T, Oka K,
Adachi S, Mizushima K, Kimura R, Okajima M, Sakai H, Sakamoto N, et
al: Tumor inoculation site affects the development of cancer
cachexia and muscle wasting. Int J Cancer. 137:2558–2565. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Mu X, Agarwal R, March D, Rothenberg A,
Voigt C, Tebbets J, Huard J and Weiss K: Notch signaling mediates
skeletal muscle atrophy in cancer cachexia caused by osteosarcoma.
Sarcoma. 2016:37581622016. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Cannon T, Shores C, Yin X, Dahlman J,
Guttridge D, Lai V, George J, Buzkova P and Couch M:
Immunocompetent murine model of cancer cachexia for head and neck
squamous cell carcinoma. Head Neck. 30:320–326. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Mi L, Lin J, Zheng H, Xu X, Zhang J and
Zhang D: Bacterial translocation contributes to cachexia from
locally advanced gastric cancer. Hepatogastroenterology.
59:2348–2351. 2012.PubMed/NCBI
|
|
77
|
Kumar S, Kishimoto H, Chua HL, Badve S,
Miller KD, Bigsby RM and Nakshatri H: Interleukin-1 alpha promotes
tumor growth and cachexia in MCF-7 ×enograft model of breast
cancer. Am J Pathol. 163:2531–2541. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Zheng R, Huang S, Zhu J, Lin W, Xu H and
Zheng X: Leucine attenuates muscle atrophy and autophagosome
formation by activating PI3K/AKT/mTOR signaling pathway in rotator
cuff tears. Cell Tissue Res. 378:113–125. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Lee S, Kim MB, Kim C and Hwang JK: Whole
grain cereal attenuates obesity-induced muscle atrophy by
activating the PI3K/Akt pathway in obese C57BL/6N mice. Food Sci
Biotechnol. 27:159–168. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Ma XM and Blenis J: Molecular mechanisms
of mTOR-mediated translational control. Nat Rev Mol Cell Biol.
10:307–318. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Jiang BH, Aoki M, Zheng JZ, Li J and Vogt
PK: Myogenic signaling of phosphatidylinositol 3-kinase requires
the serine-threonine kinase Akt/protein kinase B. Proc Natl Acad
Sci USA. 96:2077–2081. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Price DJ, Grove JR, Calvo V, Avruch J and
Bierer BE: Rapamycin-induced inhibition of the 70-kilodalton S6
protein kinase. Science. 257:973–977. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Chung J, Kuo CJ, Crabtree GR and Blenis J:
Rapamycin-FKBP specifically blocks growth-dependent activation of
and signaling by the 70 kd S6 protein kinases. Cell. 69:1227–1236.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Lin TA, Kong X, Saltiel AR, Blackshear PJ
and Lawrence JC Jr: Control of PHAS-I by insulin in 3T3-L1
adipocytes. Synthesis, degradation, and phosphorylation by a
rapamycin-sensitive and mitogen-activated protein
kinase-independent pathway. J Biol Chem. 270:18531–18538. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Graves LM, Bornfeldt KE, Argast GM, Krebs
EG, Kong X, Lin TA and Lawrence JC Jr: cAMP- and
rapamycin-sensitive regulation of the association of eukaryotic
initiation factor 4E and the translational regulator PHAS-I in
aortic smooth muscle cells. Proc Natl Acad Sci USA. 92:7222–7226.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Stitt TN, Drujan D, Clarke BA, Panaro F,
Timofeyva Y, Kline WO, Gonzalez M, Yancopoulos GD and Glass D: The
IGF-1/PI3K/Akt pathway prevents expression of muscle
atrophy-induced ubiquitin ligases by inhibiting FOXO transcription
factors. Mol Cell. 14:395–403. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Sandri M, Sandri C, Gilbert A, Skurk C,
Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH and Goldberg
AL: Foxo transcription factors induce the atrophy-related ubiquitin
ligase atrogin-1 and cause skeletal muscle atrophy. Cell.
117:399–412. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Manne ND, Lima M, Enos RT, Wehner P,
Carson JA and Blough E: Altered cardiac muscle mTOR regulation
during the progression of cancer cachexia in the ApcMin/+ mouse.
Int J Oncol. 42:2134–2140. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Ge Y, Wu AL, Warnes C, Liu J, Zhang C,
Kawasome H, Terada N, Boppart MD, Schoenherr CJ and Chen J: mTOR
regulates skeletal muscle regeneration in vivo through
kinase-dependent and kinase-independent mechanisms. Am J Physiol
Cell Physiol. 297:C1434–C1444. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Zimmers TA, Fishel ML and Bonetto A: STAT3
in the systemic inflammation of cancer cachexia. Semin Cell Dev
Biol. 54:28–41. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Bonetto A, Aydogdu T, Kunzevitzky N,
Guttridge DC, Khuri S, Koniaris LG and Zimmers TA: STAT3 activation
in skeletal muscle links muscle wasting and the acute phase
response in cancer cachexia. PLoS One. 6:e225382011. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Ma JF, Sanchez BJ, Hall DT, Tremblay AK,
Di Marco S and Gallouzi IE: STAT3 promotes IFNγ/TNFα-induced muscle
wasting in an NF-κB-dependent and IL-6-independent manner. EMBO Mol
Med. 9:622–637. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Ghosh S and Karin M: Missing pieces in the
NF-kappaB puzzle. Cell. 109 (Suppl 1):S81–S96. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Hayden MS and Ghosh S: NF-kappaB in
immunobiology. Cell Res. 21:223–244. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Thoma A and Lightfoot AP: NF-kB and
inflammatory cytokine signalling: Role in skeletal muscle atrophy.
Adv Exp Med Biol. 1088:267–279. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Di Marco S, Mazroui R, Dallaire P, Chittur
S, Tenenbaum SA, Radzioch D, Marette A and Gallouzi IE: NF-kappa
B-mediated MyoD decay during muscle wasting requires nitric oxide
synthase mRNA stabilization, HuR protein, and nitric oxide release.
Mol Cell Biol. 25:6533–6545. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Cuenda A and Rousseau S: p38 MAP-kinases
pathway regulation, function and role in human diseases. Biochim
Biophys Acta. 1773:1358–1375. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Kim J, Won KJ, Lee HM, Hwang BY, Bae YM,
Choi WS, Song H, Lim KW, Lee CK and Kim B: p38 MAPK participates in
muscle-specific RING finger 1-mediated atrophy in cast-immobilized
rat gastrocnemius muscle. Korean J Physiol Pharmacol. 13:491–496.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Ryu Y, Lee D, Jung SH, Lee KJ, Jin H, Kim
SJ, Lee HM, Kim B and Won KJ: Sabinene prevents skeletal muscle
atrophy by inhibiting the MAPK-MuRF-1 pathway in rats. Int J Mol
Sci. 20:49552019. View Article : Google Scholar
|
|
100
|
Belova SP, Mochalova EP, Kostrominova TY,
Shenkman BS and Nemirovskaya TL: P38α-MAPK signaling inhibition
attenuates soleus atrophy during early stages of muscle unloading.
Int J Mol Sci. 21:27562020. View Article : Google Scholar
|
|
101
|
Girven M, Dugdale HF, Owens DJ, Hughes DC,
Stewart CE and Sharples AP: l-glutamine improves skeletal muscle
cell differentiation and prevents myotube atrophy after cytokine
(TNF-α) stress via reduced p38 MAPK signal transduction. J Cell
Physiol. 231:2720–2732. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Morales MG, Olguin H, Di Capua G, Brandan
E, Simon F and Cabello-Verrugio C: Endotoxin-induced skeletal
muscle wasting is prevented by angiotensin-(1–7) through a p38
MAPK-dependent mechanism. Clin Sci (Lond). 129:461–476. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
O'Neill BT, Bhardwaj G, Penniman CM,
Krumpoch MT, Suarez Beltran PA, Klaus K, Poro K, Li M, Pan H,
Dreyfuss JM, et al: FoxO transcription factors are critical
regulators of diabetes-related muscle atrophy. Diabetes.
68:556–570. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Liu CM, Yang Z, Liu CW, Wang R, Tien P,
Dale R and Sun LQ: Effect of RNA oligonucleotide targeting Foxo-1
on muscle growth in normal and cancer cachexia mice. Cancer Gene
Ther. 14:945–952. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Blackwell TA, Cervenka I, Khatri B, Brown
JL, Rosa-Caldwell ME, Lee DE, Perry RA Jr, Brown LA, Haynie WS,
Wiggs MP, et al: Transcriptomic analysis of the development of
skeletal muscle atrophy in cancer-cachexia in tumor-bearing mice.
Physiol Genomics. 50:1071–1082. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Marchildon F, Lamarche E, Lala-Tabbert N,
St-Louis C and Wiper-Bergeron N: Expression of CCAAT/enhancer
binding protein beta in muscle satellite cells inhibits myogenesis
in cancer cachexia. PLoS One. 10:e01455832015. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Freire PP, Fernandez GJ, Cury SS, de
Moraes D, Oliveira JS, de Oliveira G, Dal-Pai-Silva M, Dos Reis PP
and Carvalho RF: The pathway to cancer cachexia: MicroRNA-regulated
networks in muscle wasting based on integrative meta-analysis. Int
J Mol Sci. 20:2019. View Article : Google Scholar
|
|
108
|
Lee DE, Brown JL, Rosa-Caldwell ME,
Blackwell TA, Perry RA Jr, Brown LA, Khatri B, Seo D, Bottje WG,
Washington TA, et al: Cancer cachexia-induced muscle atrophy:
Evidence for alterations in microRNAs important for muscle size.
Physiol Genomics. 49:253–260. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Sandri M: Autophagy in health and disease.
3. Involvement of autophagy in muscle atrophy. Am J Physiol Cell
Physiol. 298:C1291–C1297. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Bargiela A, Cerro-Herreros E,
Fernandez-Costa JM, Vilchez JJ, Llamusi B and Artero R: Increased
autophagy and apoptosis contribute to muscle atrophy in a myotonic
dystrophy type 1 Drosophila model. Dis Model Mech. 8:679–690. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Pettersen K, Andersen S, Degen S, Tadini
V, Grosjean J, Hatakeyama S, Tesfahun AN, Moestue S, Kim J, Nonstad
U, et al: Cancer cachexia associates with a systemic
autophagy-inducing activity mimicked by cancer cell-derived IL-6
trans-signaling. Sci Rep. 7:20462017. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Penna F, Ballarò R, Martinez-Cristobal P,
Sala D, Sebastian D, Busquets S, Muscaritoli M, Argilés JM,
Costelli P and Zorzano A: Autophagy exacerbates muscle wasting in
cancer cachexia and impairs mitochondrial function. J Mol Biol.
431:2674–2686. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Pettersen K, Andersen S, van der Veen A,
Nonstad U, Hatakeyama S, Lambert C, Lach-Trifilieff E, Moestue S,
Kim J, Grønberg BH, et al: Autocrine activin A signalling in
ovarian cancer cells regulates secretion of interleukin 6,
autophagy, and cachexia. J Cachexia Sarcopenia Muscle. 11:195–207.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Mirza KA and Tisdale MJ: Role of
Ca2+ in proteolysis-inducing factor (PIF)-induced
atrophy of skeletal muscle. Cell Signal. 24:2118–2122. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Kozutsumi Y, Segal M, Normington K,
Gething MJ and Sambrook J: The presence of malfolded proteins in
the endoplasmic reticulum signals the induction of
glucose-regulated proteins. Nature. 332:462–464. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Bohnert KR, McMillan JD and Kumar A:
Emerging roles of ER stress and unfolded protein response pathways
in skeletal muscle health and disease. J Cell Physiol. 233:67–78.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Liu CY, Schröder M and Kaufman RJ:
Ligand-independent dimerization activates the stress response
kinases IRE1 and PERK in the lumen of the endoplasmic reticulum. J
Biol Chem. 275:24881–24885. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Bertolotti A, Zhang Y, Hendershot LM,
Harding HP and Ron D: Dynamic interaction of BiP and ER stress
transducers in the unfolded-protein response. Nat Cell Biol.
2:326–332. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Yoshida H, Matsui T, Yamamoto A, Okada T
and Mori K: XBP1 mRNA is induced by ATF6 and spliced by IRE1 in
response to ER stress to produce a highly active transcription
factor. Cell. 107:881–891. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Shen J, Chen X, Hendershot L and Prywes R:
ER stress regulation of ATF6 localization by dissociation of
BiP/GRP78 binding and unmasking of Golgi localization signals. Dev
Cell. 3:99–111. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Roy A and Kumar A: ER stress and unfolded
protein response in cancer cachexia. Cancers (Basel). 11:19292019.
View Article : Google Scholar
|
|
122
|
Wu J and Kaufman RJ: From acute ER stress
to physiological roles of the Unfolded Protein Response. Cell Death
Differ. 13:374–384. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Isaac ST, Tan TC and Polly P: Endoplasmic
reticulum stress, calcium dysregulation and altered protein
translation: Intersection of processes that contribute to cancer
cachexia induced skeletal muscle wasting. Curr Drug Targets.
17:1140–1146. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Barreiro E, Salazar-Degracia A,
Sancho-Muñoz A and Gea J: Endoplasmic reticulum stress and unfolded
protein response profile in quadriceps of sarcopenic patients with
respiratory diseases. J Cell Physiol. 234:11315–11329. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Tam AB, Mercado EL, Hoffmann A and Niwa M:
ER stress activates NF-κB by integrating functions of basal IKK
activity, IRE1 and PERK. PLoS One. 7:e450782012. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
White JP, Puppa MJ, Sato S, Gao S, Price
RL, Baynes JW, Kostek MC, Matesic LE and Carson JA: IL-6 regulation
on skeletal muscle mitochondrial remodeling during cancer cachexia
in the ApcMin/+ mouse. Skelet Muscle. 2:142012. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Brown JL, Rosa-Caldwell ME, Lee DE,
Blackwell TA, Brown LA, Perry RA, Haynie WS, Hardee JP, Carson JA,
Wiggs MP, et al: Mitochondrial degeneration precedes the
development of muscle atrophy in progression of cancer cachexia in
tumour-bearing mice. J Cachexia Sarcopenia Muscle. 8:926–938. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
128
|
VanderVeen BN, Fix DK and Carson JA:
Disrupted skeletal muscle mitochondrial dynamics, mitophagy, and
biogenesis during cancer cachexia: A role for inflammation. Oxid
Med Cell Longev. 2017:32920872017. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
van der Ende M, Grefte S, Plas R,
Meijerink J, Witkamp RF, Keijer J and van Norren K: Mitochondrial
dynamics in cancer-induced cachexia. Biochim Biophys Acta Rev
Cancer. 1870:137–150. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Dave DT and Patel BM: Mitochondrial
metabolism in cancer cachexia: Novel drug target. Curr Drug Metab.
20:1141–1153. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Neyroud D, Nosacka RL, Judge AR and Hepple
RT: Colon 26 adenocarcinoma (C26)-induced cancer cachexia impairs
skeletal muscle mitochondrial function and content. J Muscle Res
Cell Motil. 40:59–65. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Pin F, Novinger LJ, Huot JR, Harris RA,
Couch ME, O'Connell TM and Bonetto A: PDK4 drives metabolic
alterations and muscle atrophy in cancer cachexia. FASEB J.
33:7778–7790. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Du Bois P, Pablo Tortola C, Lodka D, Kny
M, Schmidt F, Song K, Schmidt S, Bassel-Duby R, Olson EN and
Fielitz J: Angiotensin II induces skeletal muscle atrophy by
activating TFEB-mediated MuRF1 expression. Circ Res. 117:424–436.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Kadoguchi T, Kinugawa S, Takada S,
Fukushima A, Furihata T, Homma T, Masaki Y, Mizushima W, Nishikawa
M, Takahashi M, et al: Angiotensin II can directly induce
mitochondrial dysfunction, decrease oxidative fibre number and
induce atrophy in mouse hindlimb skeletal muscle. Exp Physiol.
100:312–322. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Cisternas F, Morales MG, Meneses C, Simon
F, Brandan E, Abrigo J, Vazquez Y and Cabello-Verrugio C:
Angiotensin-(1–7) decreases skeletal muscle atrophy induced by
angiotensin II through a Mas receptor-dependent mechanism. Clin Sci
(Lond). 128:307–319. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Yoshida T, Tabony AM, Galvez S, Mitch WE,
Higashi Y, Sukhanov S and Delafontaine P: Molecular mechanisms and
signaling pathways of angiotensin II-induced muscle wasting:
Potential therapeutic targets for cardiac cachexia. Int J Biochem
Cell Biol. 45:2322–2332. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Penafuerte CA, Gagnon B, Sirois J, Murphy
J, MacDonald N and Tremblay ML: Identification of
neutrophil-derived proteases and angiotensin II as biomarkers of
cancer cachexia. Br J Cancer. 114:680–687. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Cai X, Zhu C, Xu Y, Jing Y, Yuan Y, Wang
L, Wang S, Zhu X, Gao P, Zhang Y, et al: Alpha-ketoglutarate
promotes skeletal muscle hypertrophy and protein synthesis through
Akt/mTOR signaling pathways. Sci Rep. 6:268022016. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Liu D, Qiao X, Ge Z, Shang Y, Li Y, Wang
W, Chen M, Si S and Chen SZ: IMB0901 inhibits muscle atrophy
induced by cancer cachexia through MSTN signaling pathway. Skelet
Muscle. 9:82019. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Bae T, Jang J, Lee H, Song J, Chae S, Park
M, Son CG, Yoon S and Yoon Y: Paeonia lactiflora root extract
suppresses cancer cachexia by down-regulating muscular NF-κB
signalling and muscle-specific E3 ubiquitin ligases in
cancer-bearing mice. J Ethnopharmacol. 246:1122222020. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Levolger S, Wiemer EAC, van Vugt JLA,
Huisman SA, van Vledder MG, van Damme-van Engel S, Ambagtsheer G,
IJzermans JNM and de Bruin RWF: Inhibition of activin-like kinase
4/5 attenuates cancer cachexia associated muscle wasting. Sci Rep.
9:98262019. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
An JM, Kang EA, Han YM, Oh JY, Lee DY,
Choi SH, Kim DH and Hahm KB: Dietary intake of probiotic kimchi
ameliorated IL-6-driven cancer cachexia. J Clin Biochem Nutr.
65:109–117. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Liu H, Li L, Zou J, Zhou T, Wang B, Sun H
and Yu S: Coix seed oil ameliorates cancer cachexia by
counteracting muscle loss and fat lipolysis. BMC Complement Altern
Med. 19:2672019. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Terawaki K, Sawada Y, Kashiwase Y,
Hashimoto H, Yoshimura M, Suzuki M, Miyano K, Sudo Y, Shiraishi S,
Higami Y, et al: New cancer cachexia rat model generated by
implantation of a peritoneal dissemination-derived human stomach
cancer cell line. Am J Physiol Endocrinol Metab. 306:E373–E387.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Xi QL, Zhang B, Jiang Y, Zhang HS, Meng
QY, Chen Y, Han YS, Zhuang QL, Han J, Wang HY, et al: Mitofusin-2
prevents skeletal muscle wasting in cancer cachexia. Oncol Lett.
12:4013–4020. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Toledo M, Busquets S, Penna F, Zhou X,
Marmonti E, Betancourt A, Massa D, López-Soriano FJ, Han HQ and
Argilés JM: Complete reversal of muscle wasting in experimental
cancer cachexia: Additive effects of activin type II receptor
inhibition and beta-2 agonist. Int J Cancer. 138:2021–2029. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Khamoui AV, Park BS, Kim DH, Yeh MC, Oh
SL, Elam ML, Jo E, Arjmandi BH, Salazar G, Grant SC, et al: Aerobic
and resistance training dependent skeletal muscle plasticity in the
colon-26 murine model of cancer cachexia. Metabolism. 65:685–698.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Tanaka M, Sugimoto K, Fujimoto T, Xie K,
Takahashi T, Akasaka H, Kurinami H, Yasunobe Y, Matsumoto T, Fujino
H and Rakugi H: Preventive effects of low-intensity exercise on
cancer cachexia-induced muscle atrophy. FASEB J. 33:7852–7862.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Zhang Y, Han X, Ouyang B, Wu Z, Yu H, Wang
Y, Liu G and Ji X: Chinese herbal medicine Baoyuan Jiedu decoction
inhibited muscle atrophy of cancer cachexia through Atrogin-l and
MuRF-1. Evid Based Complement Alternat Med.
2017:62683782017.PubMed/NCBI
|
|
150
|
Sun R, Zhang S, Hu W, Lu X, Lou N, Yang Z,
Chen S, Zhang X and Yang H: Valproic acid attenuates skeletal
muscle wasting by inhibiting C/EBPbeta-regulated atrogin1
expression in cancer cachexia. Am J Physiol Cell Physiol.
311:C101–C115. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Zhuang P, Zhang J, Wang Y, Zhang M, Song
L, Lu Z, Zhang L, Zhang F, Wang J, Zhang Y, et al: Reversal of
muscle atrophy by Zhimu and Huangbai herb pair via activation of
IGF-1/Akt and autophagy signal in cancer cachexia. Support Care
Cancer. 24:1189–1198. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Chen JM, Yang TT, Cheng TS, Hsiao TF,
Chang PM, Leu JY, Wang FS, Hsu SL, Huang CF and Lai JM: Modified
Sijunzi decoction can alleviate cisplatin-induced toxicity and
prolong the survival time of cachectic mice by recovering muscle
atrophy. J Ethnopharmacol. 233:47–55. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Molanouri Shamsi M, Chekachak S, Soudi S,
Quinn LS, Ranjbar K, Chenari J, Yazdi MH and Mahdavi M: Combined
effect of aerobic interval training and selenium nanoparticles on
expression of IL-15 and IL-10/TNF-α ratio in skeletal muscle of 4T1
breast cancer mice with cachexia. Cytokine. 90:100–108. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Salazar-Degracia A, Busquets S, Argilés
JM, Bargalló-Gispert N, López-Soriano FJ and Barreiro E: Effects of
the beta2 agonist formoterol on atrophy signaling, autophagy, and
muscle phenotype in respiratory and limb muscles of rats with
cancer-induced cachexia. Biochimie. 149:79–91. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Pretto F, Ghilardi C, Moschetta M, Bassi
A, Rovida A, Scarlato V, Talamini L, Fiordaliso F, Bisighini C,
Damia G, et al: Sunitinib prevents cachexia and prolongs survival
of mice bearing renal cancer by restraining STAT3 and MuRF-1
activation in muscle. Oncotarget. 6:3043–3054. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Miao C, Lv Y, Zhang W, Chai X, Feng L,
Fang Y, Liu X and Zhang X: Pyrrolidine dithiocarbamate (PDTC)
attenuates cancer cachexia by affecting muscle atrophy and fat
lipolysis. Front Pharmacol. 8:9152017. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Kadoguchi T, Takada S, Yokota T, Furihata
T, Matsumoto J, Tsuda M, Mizushima W, Fukushima A, Okita K and
Kinugawa S: Deletion of NAD(P)H oxidase 2 prevents angiotensin
II-induced skeletal muscle atrophy. Biomed Res Int.
2018:31949172018. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Bosaeus I: Nutritional support in
multimodal therapy for cancer cachexia. Support Care Cancer.
16:447–451. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Amano K, Morita T, Miyamoto J, Uno T,
Katayama H and Tatara R: Perception of need for nutritional support
in advanced cancer patients with cachexia: A survey in palliative
care settings. Support Care Cancer. 26:2793–2799. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
McClement S: Cancer anorexia-cachexia
syndrome: Psychological effect on the patient and family. J Wound
Ostomy Continence Nurs. 32:264–268. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Zhang WL, Li N, Shen Q, Fan M, Guo XD,
Zhang XW, Zhang Z and Liu X: Establishment of a mouse model of
cancer cachexia with spleen deficiency syndrome and the effects of
atractylenolide I. Acta Pharmacol Sin. 41:237–248. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Kang HJ, Jeong MK, Park SJ, Jun HJ and Yoo
HS: Efficacy and safety of Yukgunja-Tang for treating anorexia in
patients with cancer: The protocol for a pilot, randomized,
controlled trial. Medicine (Baltimore). 98:e169502019. View Article : Google Scholar : PubMed/NCBI
|