Introduction
Epilepsy is one of the most common and serious
chronic diseases of the nervous system. According to the World
Health Organization, ~70 million individuals currently suffer from
epilepsy worldwide (1). The
imbalance of excitatory and inhibitory neurotransmission is a
common mechanism of epilepsy. Status epilepticus (SE) is a state in
which failure of the epileptic termination mechanism or an
abnormality of the epileptic initiation mechanism leads to
long-term epileptic seizures and requires emergency administration
of antiepileptic drugs (2).
Failure to terminate SE over time results in neuronal damage and
death, and neural network changes (3). Pilocarpine injection can result in
SE, which triggers a series of molecular and cellular events that
produce neuronal cell death and can culminate in the development of
epilepsy (4,5). In a previous study, the p38 signal
transduction pathway was activated in kainate-induced status
epilepticus and was involved in neuronal damage, moss sprouting,
glial cell proliferation and various other changes in the CA1 and
CA3 areas of the hippocampus following SE (6). Activation of the p38 pathway can
cause damage to hippocampal neurons and increases susceptibility to
seizures (7). In addition, the
mechanism of p38 signaling pathway may also be associated with its
involvement in gliosis following seizures (8). Although there are a number of
antiepileptic drugs that can effectively control epileptic
seizures, certain cases of seizures cannot be controlled over time,
resulting in morbidity and mortality rates ~20% (9). Therefore, exploring the
pathophysiological mechanism of SE and providing novel therapeutic
targets for antiepileptic drugs are urgently required.
Excessive activity of the excitatory
neurotransmitter glutamate results in an imbalance of excitability
and inhibition in vivo, causing excessive neuronal
excitability, leading to a seizure (10). The anticonvulsant effect of the
adenosine system has been confirmed in a number of epilepsy models,
suggesting that the adenosine system serves an important role in
inhibiting neuronal excitation (11,12).
Adenosine has been demonstrated to be an endogenous anticonvulsant
and neuroprotective agent in the brain, and its extracellular
levels are largely dependent on ectonucleotidase-mediated
conversion of ‘activated’ synapse-released ATP and the secretion of
astrocytes (13). However, two-way
regulation of adenosine levels also depends on type 1 equilibrative
nucleoside transporter (ENT1) (13). In addition, the anticonvulsant and
neuroprotective effects of adenosine are largely mediated by
adenosine A1 receptor (A1R) on the presynaptic membrane (11). The activity-dependent release of
adenosine activates A1R to block the release of excitatory
transmitters, such as glutamate (14). Therefore, A1R is an effective
molecular and therapeutic target in the course of epileptic
seizures (12). Moreover,
activated A1R provides a potential endogenous mechanism to inhibit
neuronal excitatory activity and seizure dispersion (15).
MAPKs mediate intracellular signaling cascades in
response to a variety of stimuli by regulating intracellular gene
expression levels, cell division, differentiation, repair and
apoptosis (16). MAPKs are
activated by extracellular stressors, such as UV light, osmotic
pressure, radiation, inflammatory cytokines, growth factors and
shock, and subsequently induce a cascade of three kinases to
transduce signals (17). Upon
stimulation, MAPK kinase kinase is activated by phosphorylation and
then phosphorylates and activates MAPK kinase, which in turn
phosphorylates and activates MAPK (17). p38 is a MAPK primarily activated by
inflammatory factors, stressful stimuli, such as UV light exposure,
hypertonic conditions and heat shock, and glutamate (18). Okamoto et al (19) performed a whole transcriptome
analysis of the hippocampi of epileptic rats and observed that p38
MAPK was overexpressed. Our previous study (20) confirmed that the specific p38 MAPK
inhibitor SB203580 alleviated epileptic seizures in SE rats.
Nitrobenzothioinosine (NBTI), a selective inhibitor of ENT1
(21), blocks the transport of
adenosine into cells and increases extracellular adenosine levels,
thus exhibiting a protective effect against epileptic seizures
(22). A number of studies have
revealed that A1R, a member of the adenosine system, has
significant anticonvulsant effects (13,23).
p38 MAPK affects the levels of ENT1 (24). However, whether the p38
MAPK-specific inhibitor SB203580 regulates the expression levels of
A1R and ENT1, and exerts a protective effect during seizures
remains unclear.
In the present study, SB203580 was used to inhibit
the p38 MAPK signaling pathway in rats. It was observed that
SB203580 could reduce the pathomorphological damage of hippocampal
neurons, prolong the latency of seizure and reduce the degree of
seizure in rats, and reduce the expression of A1R and ENT1.
Materials and methods
Experimental animals
In total, 50 healthy adult male Sprague-Dawley (SD)
(age, 42–49 days) were purchased from the Experimental Animal
Center of Daping Hospital of the Third Military Medical University
[license no. scxk (Chongqing, China) 2012–0005]. The rats were
provided a standard diet and pure drinking water ad libitum in a
room maintained at a constant temperature (24±2°C, and 50–70%
humidity. Animal health and behavior were monitored every day; all
rats were healthy during the experimental period. The experiment
lasted for 2 weeks. All efforts were made to minimize animal
suffering. All procedures were approved by the Animal Care and Use
Committee of Zunyi Medical University (Zunyi, China).
SD rats (weight, ~180–220 g) were randomly divided
into the following 10 groups (5 rats per group): i) Control; ii)
NBTI; iii) NBTI + SB203580; iv) SE, 0 h; v) SE, 1 day; vi) SE, 3
days; vii) SE, 7 days; viii) SE, 14 days; ix) SB203580; and x)
DMSO. A rat model of acute LiCl-pilocarpine SE was established as
described below. Then, randomly selected rats in the SE group
received 127 mg/kg LiCl by intraperitoneal (i.p.) injection,
followed by 1 mg/kg atropine sulfate i.p. 18–20 h later, then 50
mg/kg pilocarpine i.p. after an additional 30 min. A Racine score
of IV–V were recorded (25) to
statistical analysis of rat behavior.
For the control rats, instead of the LiCl and
pilocarpine administered to the SE group, the same amount of saline
was used instead. For rats in the SB20358 group, a solution of the
p38 inhibitor SB203580 (15 mg/kg) and 2% DMSO was injected i.p. 30
min prior to injection of pilocarpine (26–28).
For rats in the NBTI group, an ENT1 inhibitor (15 mg/kg) (29) was injected i.p. 45 min prior to
injection of pilocarpine and 2% DMSO. For rats in the NBTI +
SB20358 group, the same protocol was used as aforementioned. For
rats in the DMSO group, an equal volume of saline and 2% DMSO was
used as a substitution for SB203580.
Euthanasia was accomplished humanely by a trained
laboratory personnel via i.p. injection of saline-diluted
Euthasol® (150 mg/kg) containing pentobarbital sodium
(390 mg/ml) and phenytoin sodium (50 mg/ml), followed by cervical
dislocation at the experimental timepoints (0 h, 1, 3, 7 and 14
days). The death of rats was verified using the following criteria:
Lack of pulse, breathing and corneal reflex, failure to respond to
firm toe pinch, graying of mucus membranes and inability to
auscultate respiratory or heart sounds. Following sacrifice of
rats, the skull was dissected, brain tissue was removed and the
bilateral hippocampus and temporal lobe neocortex were rapidly
dissected on ice. Tissue samples were frozen in liquid nitrogen
immediately and stored until western blot analysis. After
administering complete anesthesia, the control group, SE group and
SB203580 group were injected with saline (~150 ml) rapidly and
continuously. Then, 4% paraformaldehyde (~150 ml) was injected at
room temperature until the limbs, neck and tail became stiff. Then,
rats were decapitated and the brain tissue was removed, incubated
in 4% paraformaldehyde for 12–24 h (4°C) and sent to the Department
of Pathology of Zunyi Medical College to be embedded in paraffin
wax and stored at room temperature prior to
immunohistochemistry.
Tissues for immunofluorescence and
immunohistochemistry were prepared in the same manner. Following
decapitation, the brain tissue was placed in successive 20 and 30%
sucrose solutions for dehydration at 4°C for 24 h each time; after
brain tissue sank, it was sent to the Zunyi Medical College
Affiliated Hospital for the preparation of frozen microtome
sections (thickness, 16 µm) and stored at −20°C.
Western blotting
RIPA buffer and protease inhibitors (Beyotime
Institute of Biotechnology) were used to homogenize all samples.
The protein concentration of the supernatant was measured with a
BCA kit (Beyotime Institute of Biotechnology) according to the
manufacturer's instructions. A total of 50 µg protein from each
sample was separated via SDS-PAGE (5% separating, 10% stacking
gel), and then proteins were transferred to PVDF membranes (250 mA
for 100 min). After blocking for 1 h at room temperature in skimmed
milk (5%), PVDF membranes were incubated with primary antibodies
[rabbit anti-A1R (1:1,000; cat. no. ab82477; Abcam) and anti-ENT1
(1:500; cat. no. ab223851; Abcam) and mouse anti-β-actin (1:5000;
cat. no. 66009-1-Ig; ProteinTech Group) and anti-β-tubulin
(1:5,000; cat. no. 10094-1-AP; ProteinTech Group)] overnight at
4°C. The blots were washed with PBS with 1% Tween-20 (PBST) three
times and then incubated with horseradish peroxidase
(HRP)-conjugated secondary antibody (1:5,000; cat. no. 35105ES60;
Shanghai Yeasen Biotechnology Co. Ltd) for 1 h at room temperature.
Blots were developed using Super Signal West Pico Chemiluminescent
HRP substrate (Sigma-Aldrich; Merck KGaA) according to the
manufacturer's instructions. Band intensity was analyzed with
Quantity One software (Bio-Rad Laboratories, Inc.).
Immunofluorescence
Slides were incubated in acetone at −20°C for 30 min
washed in PBS three times for 5 min each, then 0.4% Triton X-100
was added to cover the slices. The slices were placed in a 37°C
incubator for 15 min, washed with PBS three times for 5 min each,
immersed in sodium citrate solution, heated at high heat (92-98°C)
in microwave oven for 3 min then at 37°C for 10 min, and washed
with PBS three times. The brain tissue slices were blocked in 20%
goat serum (Beijing Solarbio Science & Technology Co., Ltd.)
for 1–3 h at 37°C, after which the goat serum was discarded. The
slides were incubated with rabbit anti-A1R (1:50; cat. no. ab82477;
Abcam) and anti-ENT1 antibody (1:50; cat. no. ab223851; Abcam), to
which a mixture of mouse anti-glial fibrillary acidic protein
(GFAP; 1:100; cat. no. 3670S; Cell Signaling Technology, Inc.) and
guinea pig anti-microtubule-associated protein 2 (MAP2) antibody
(1:200; cat. no. 188004; Synaptic Systems GmbH) was added. The
sliced brain tissue was incubated at 4°C overnight. Then, the brain
tissue slices were washed with PBS five times for 5 min each. The
slides were incubated with a mixture of DyLight 488-conjugated goat
anti-rabbit (1:50; cat. no. 4412S; Cell Signaling Technology, Inc),
DyLight 594-conjugated goat anti-mouse (1:200; cat. no. ab96881;
Abcam) and DyLight 650-conjugated goat anti-guinea pig antibody
(1:50; cat. no. ab102377; Abcam) in a 37°C incubator in the dark
for 1 h. The slices were washed with PBS three times for 5 min each
in the dark. Following the addition of 50% glycerol solution to
cover the slide for storage, pictures were obtained with a laser
confocal microscope (magnification, ×200) and the slices were
stored in the dark.
Immunohistochemistry
Paraffin sections (thickness, 4 µm) were
deparaffinized, hydrated with an alcohol gradient and incubated in
0.3% H2O2 for 15 min at room temperature. The
sections were incubated for 5 min at 92–98°C in 10 mmol/l sodium
citrate buffer and then blocked with 20% goat serum (Beijing
Solarbio Science & Technology Co., Ltd.) for 30 min at 37°C.
Then, the sections were incubated with rabbit anti-A1R antibody
(1:100; cat. no. ab82477; Abcam) at 37°C for 2 h and washed four
times with PBS. The sections were then incubated with biotinylated
goat anti-rabbit secondary antibody (1:300; cat. no. TA130015;
OriGene Technologies, Inc.) at room temperature for 30 min, washed,
treated with avidin-biotin complex solution for 30 min at 37°C,
washed with PBS and incubated with 3,3′-diaminobenzidine (DAB)
(OriGene Technologies, Inc.) for 3 min. Harris' hematoxylin was
used for counterstaining at room temperature for 5 min. Images were
acquired with an automatic microscope (Nikon Corporation;
magnification, ×200).
Statistical analysis
Each experiment was repeated at least three times.
Data are presented as the mean ± SD and were analyzed using SPSS
software (version 18.0; SPSS, Inc.). The behavioral scores of rats
(latency of seizures and number of seizures) and results of
immunohistochemistry and western blot analysis were analyzed using
one-way ANOVA followed by Tukey's multiple comparison test or
repeated ANOVA. Student's t-test (unpaired) was used to analyze the
immunofluorescence results between two groups. Normality and
homogeneity of variance tests were performed prior to t-test and
ANOVA. P<0.05 was considered to indicate a statistically
significant difference.
Results
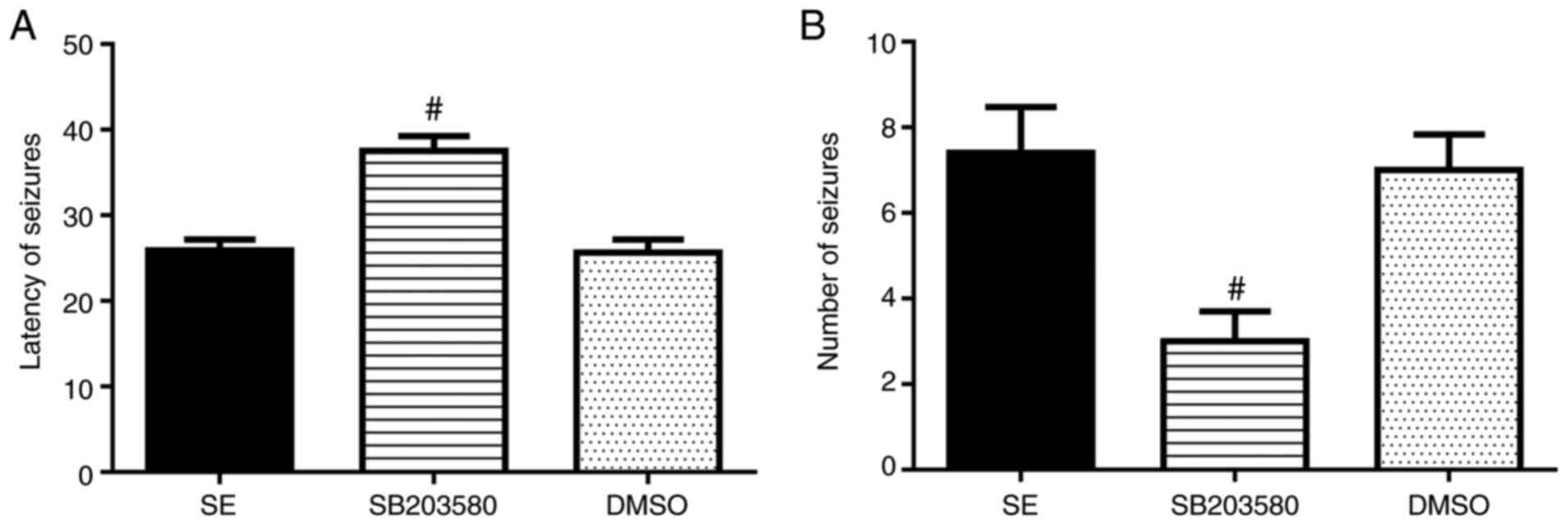
SB203580 affects the latency of the
first seizure and the number of epileptic seizures in rats
Number of seizures was significantly decreased and
latency of seizures was prolonged in the SB203580 group compared
with the SE group (P<0.05). No differences in these parameters
between the DMSO and SE groups were observed (P>0.05) (Fig. 1 and Table I).
 | Table I.SB203580 influences the latency of
the first seizure and the number of epileptic seizures in rats. |
Table I.
SB203580 influences the latency of
the first seizure and the number of epileptic seizures in rats.
| Group | Latency of seizures
(min) | Number of epileptic
seizures, n |
|---|
| SE | 25.89±1.27 | 7.40±1.07 |
| SB203580 |
37.53±1.73a |
3.00±0.70a |
| DMSO | 25.65±1.56 | 7.00±0.83 |
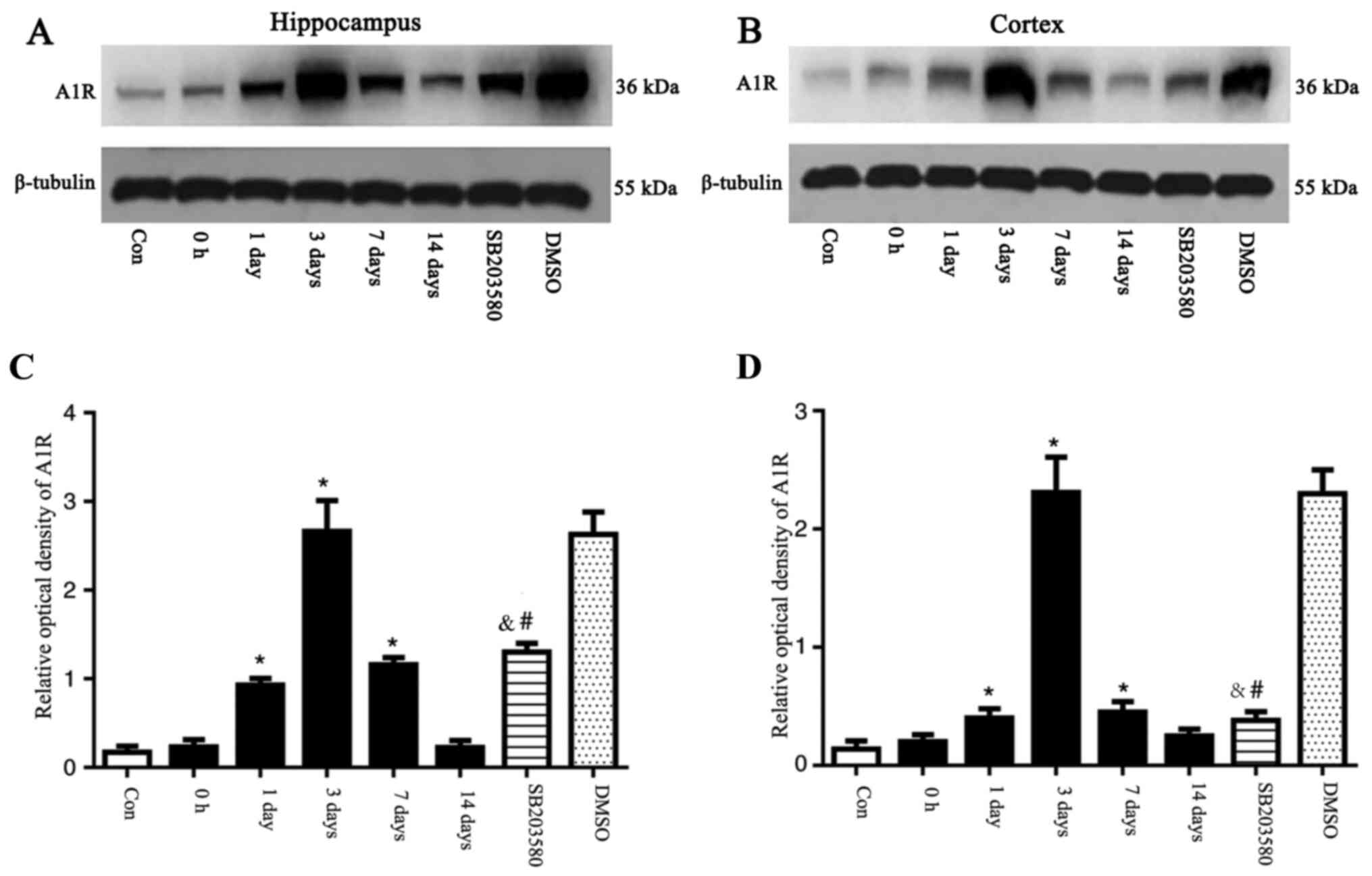
A1R expression levels in the
hippocampus and temporal lobe neocortex of epileptic rats
The expression levels of A1R in the hippocampus and
temporal lobe neocortex of rats were detected by western blotting.
The expression levels of A1R were significantly elevated at 1 and 3
days and 7 days after seizure induction. A1R expression levels
peaked at 3 days, and were significantly different compared with
the control group (P<0.05). No significant differences in the
expression levels of A1R at 0 h and 14 days after seizure induction
were observed between the control and SE groups (P>0.05). An
intervention time point of 3 days was chosen based on changes in
A1R expression. Mice were pre-treated with SB203580, then,
following 3 days of successful establishment of the epileptic
seizure model, the brain tissue was taken for the next experiment,
and DMSO was applied as the parallel solvent control. At day 3, A1R
was expressed at significantly lower levels in the SB203580 group
than the SE groups (P<0.05), although the difference in A1R
expression levels between the DMSO and the SE group was not
statistically significant (P>0.05; Fig. 2). Preliminary experiments
demonstrated that there was no difference in the expression levels
of A1R in the normal, solvent and inhibitor groups at different
time points (data not shown).
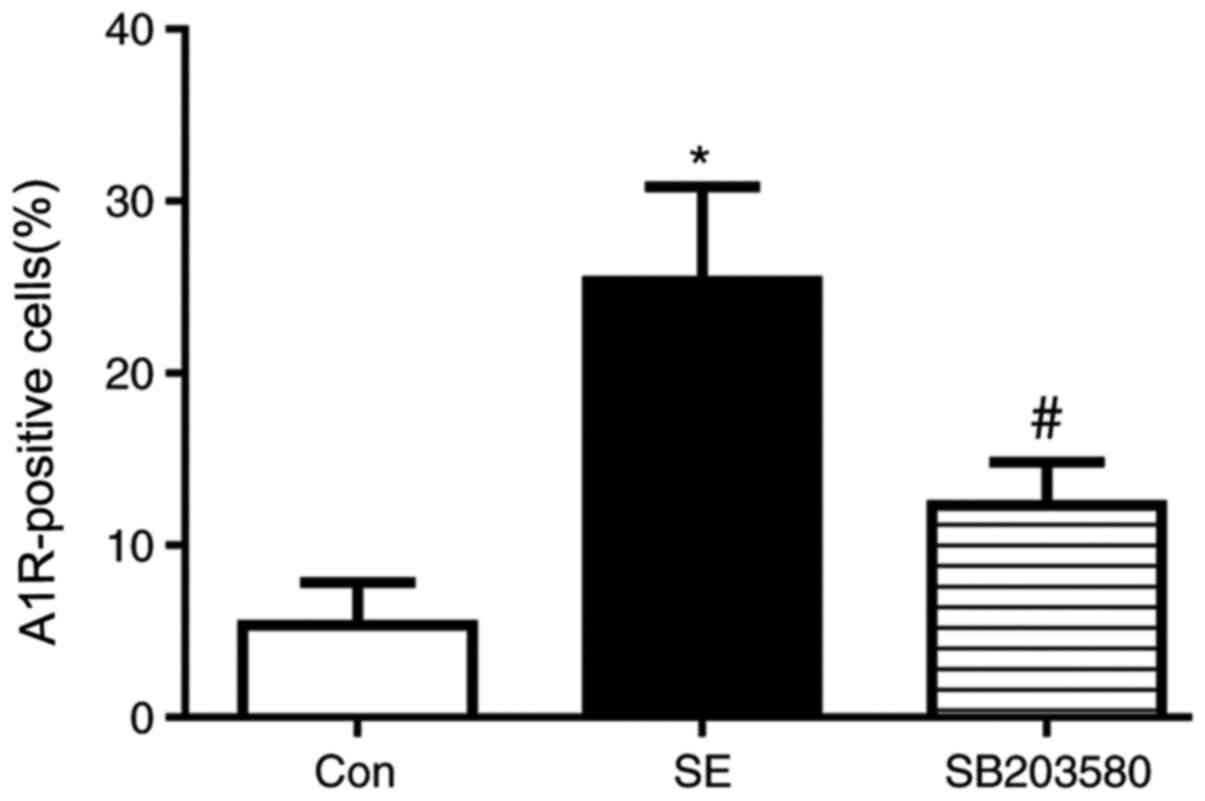
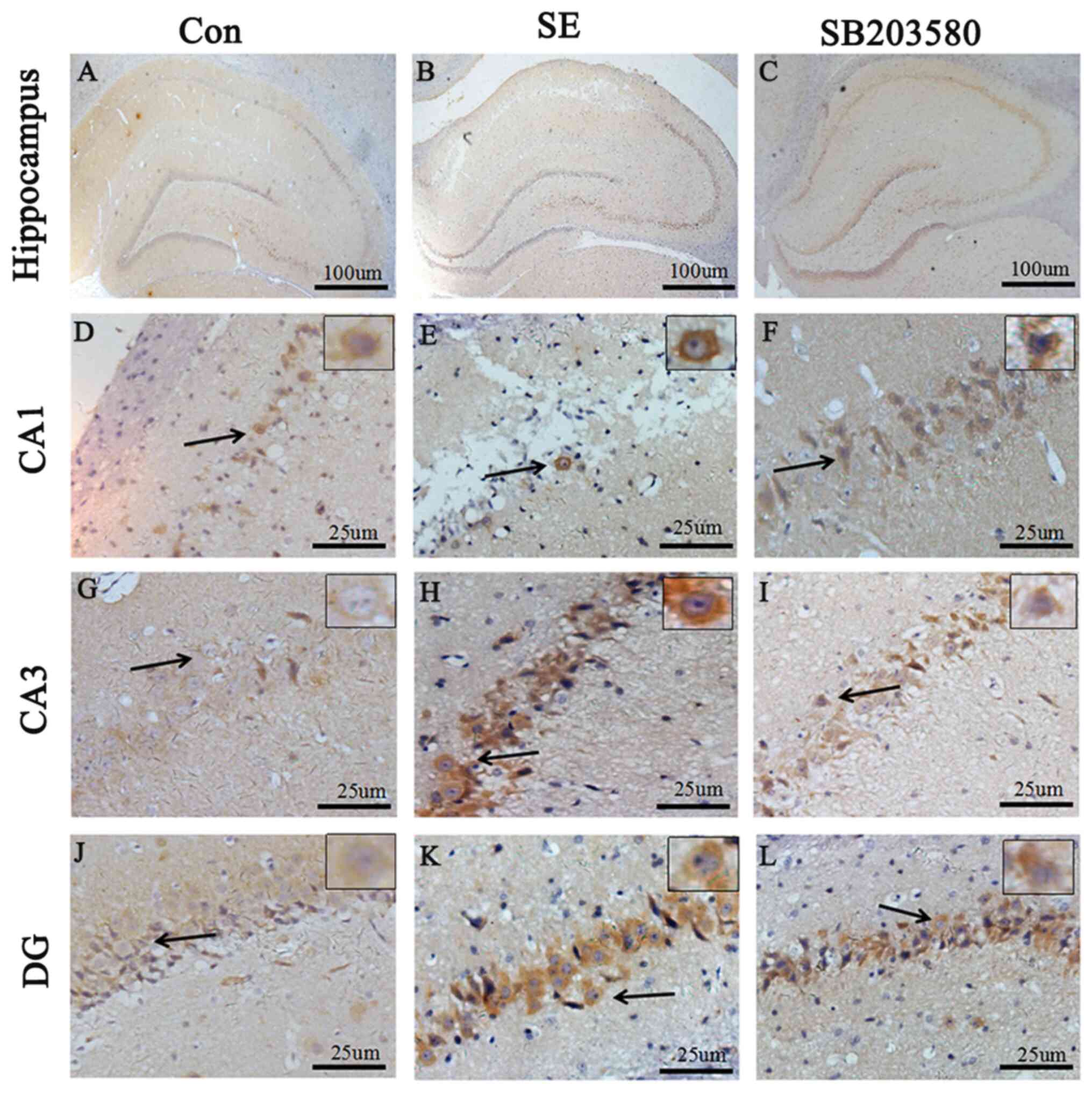
Immunohistochemical detection of A1R
expression levels in the hippocampus and temporal lobe
neocortex
Compared with the control group, A1R protein levels
in the hippocampus and neocortex of SE rats were elevated.
Expression level profiles of A1R in the hippocampus and neocortex
were assessed by immunohistochemistry. Positive staining for A1R
was primarily detected in neurons located in the hippocampus (CA1
and CA3 regions and the dentate gyrus). In addition, Compared with
the control group, the percentage of A1R-positive cells was
significantly increased in the SE group. The difference in the
percentage of A1R-positive cells between the SE and control groups
was statistically significant (P<0.05). In addition, the
percentage of A1R-positive cells was significantly decreased in the
SB203580 group compared with the SE group, and the SB203580 group
showed mild staining for A1R (P<0.05; Figs. 3 and 4). There was no significant difference in
the percentage of A1R-positive cells between the SE and DMSO groups
(P>0.05; Figs. S1 and S2).
Localization of A1R in the hippocampus
and temporal lobe neocortex
Immunohistochemical staining for A1R in the
hippocampus and temporal cortex showed the expression levels of A1R
in the hippocampal neurons of SE rats. The localization of A1R in
the hippocampus and neocortex was compared between the control and
the SE group (3 days after seizure induction) by multiple
immunofluorescence staining. The fluorescence intensity of A1R in
the hippocampus (CA1 and CA3 regions) and neocortex was
significantly higher in the SE group compared with in the control
group (P<0.05). A1R was expressed in cells labeled with
dendrite-specific MAP2 but not in cells labeled with
astrocyte-specific GFAP (Fig.
5).
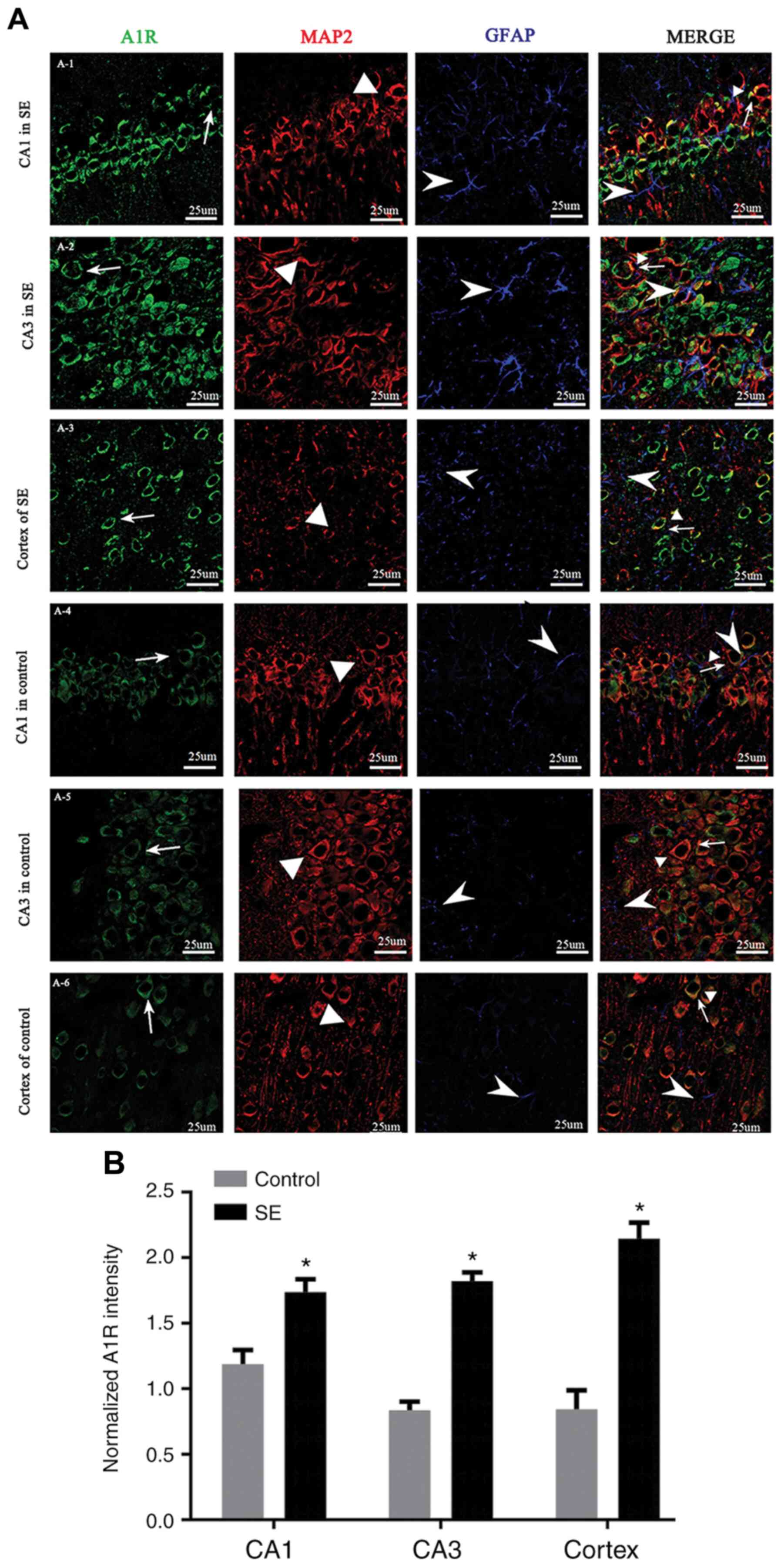 | Figure 5.Localization of A1R in the
hippocampus and temporal lobe neocortex. (A) Immunofluorescence
staining showing A1R in the (A-1) CA1 and (A-2) CA3 regions of the
hippocampus, and (A-3) temporal cortex in SE rats, and the (A-4)
CA1 and (A-5) CA3 region, and (A-6) temporal cortex in control
rats. A1R (green) was expressed in MAP2-positive neurons (red), but
not GFAP-positive astrocytes (blue) in the temporal cortex and
hippocampus (CA1 and CA3 regions). The fluorescence intensity of
A1R was higher in the SE group than the control group, and A1R was
expressed primarily in the neuronal cytoplasm. Line and arrow,
A1R-positive cells; triangle, MAP2-positive cells; arrowhead,
GFAP-positive cells. Scale bar, 25 µm. (B) Immunofluorescence
analysis showed that the fluorescence intensity of A1R in the SE
group was significantly higher than in the control group.
*P<0.05 vs. control. A1R, adenosine A1 receptor; SE, status
epilepticus; MAP, microtubule-associated protein; GFAP, glial
fibrillary acidic protein. |
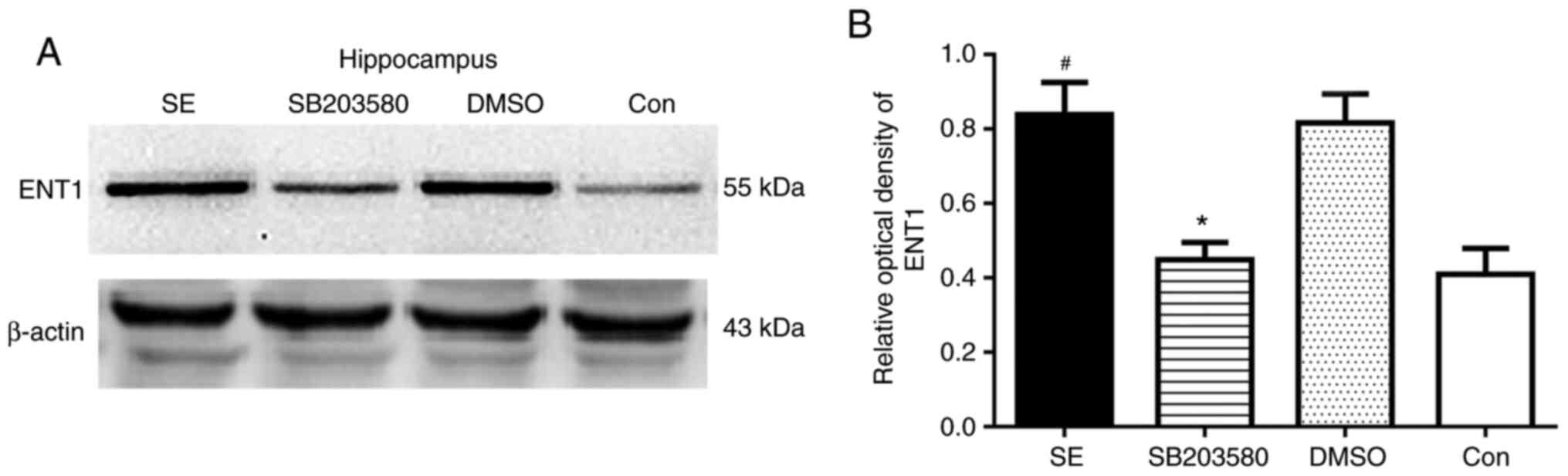
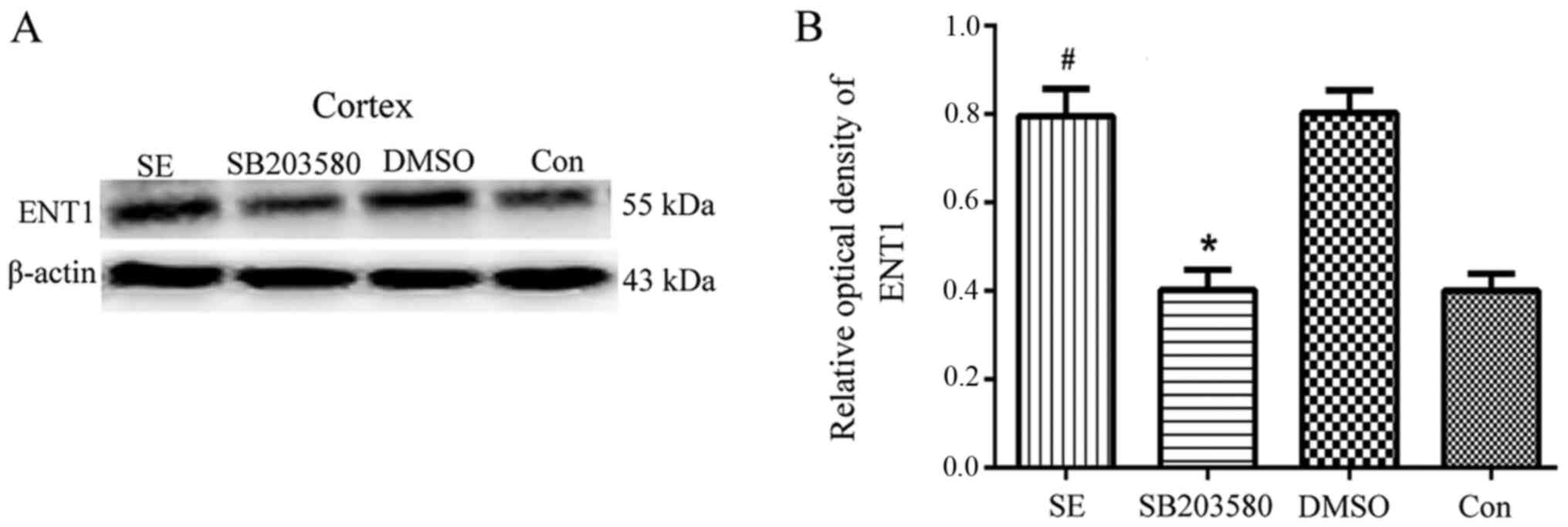
Analysis of ENT1 expression levels in
the hippocampus and temporal lobe neocortex of epileptic rats by
western blotting
Western blotting was used to determine the basal
expression levels of ENT1 protein in the hippocampus and temporal
cortex of control rats. Compared with the control group, the
protein expression levels of ENT1 in the hippocampus and temporal
lobe neocortex were significantly increased in the SE group
(P<0.05). However, compared with the SB203580 group, the protein
expression levels of ENT1 in the hippocampus and temporal lobe
neocortex were significantly increased in the SE group (P<0.05).
No difference in ENT1 protein expression levels in the hippocampus
and temporal lobe neocortex was observed between the DMSO and SE
groups (P>0.05) (Figs. 6 and
7).
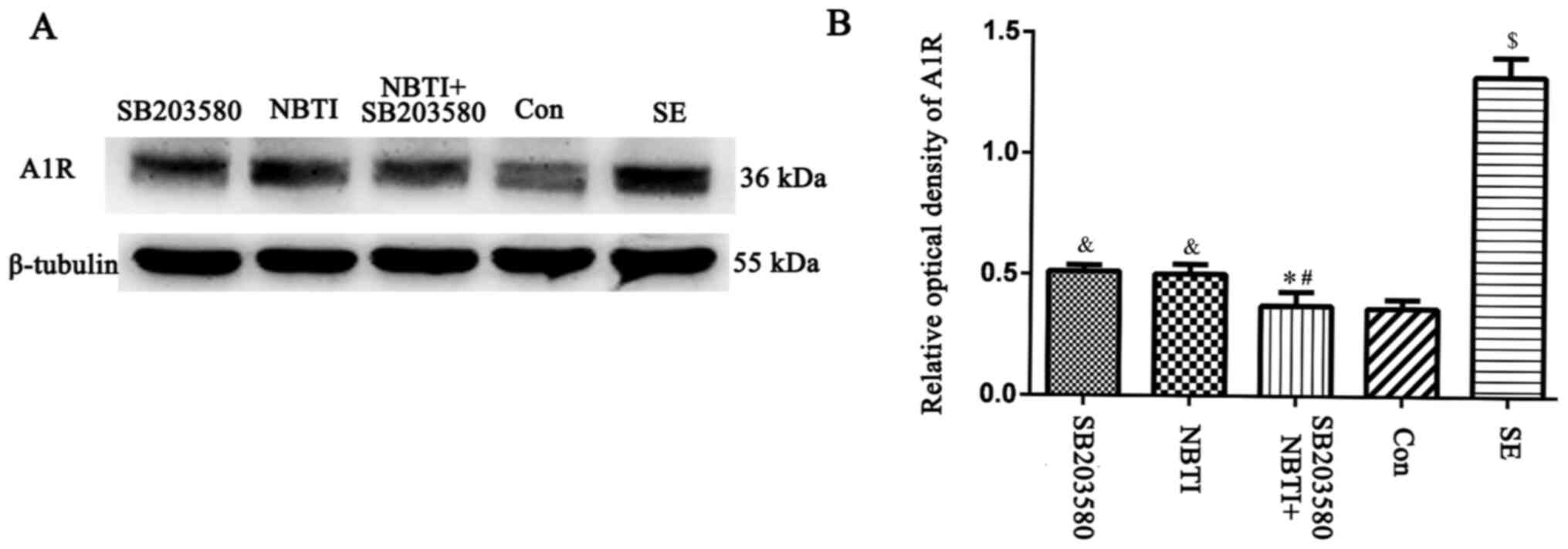
Effect of the p38 MAPK inhibitor
SB203580 on A1R expression levels
The western blotting results showed that A1R
expression levels were upregulated in LiCl-pilocarpine SE model
rats compared with control rats (P<0.05). By contrast, following
administration of the p38 MAPK inhibitor SB203580, A1R expression
levels in the SB203580 group were significantly lower than in the
SE group at 3 days after seizure induction (P<0.05). Following
administration of the ENT1 inhibitor NBTI, the expression levels of
A1R in the NBTI group were significantly lower than in the SE group
at 3 days after seizure induction (P<0.05). By contrast, no
significant difference in A1R expression levels was observed
between the NBTI and SB203580 groups (P>0.05). In addition,
following administration of NBTI + SB203580, A1R expression levels
were downregulated at 3 days compared with NTI alone. In addition,
a significant decrease in A1R expression levels was observed
between the NBTI and SB203580 groups (P<0.05; Fig. 8).
Discussion
SE is defined as the occurrence of sustained
seizures for >5 min (30). Due
to the need for timely termination of SE, delayed intervention of
seizures, particularly tonic-clonic seizures, may lead to nervous
system injury and death (31). SE
is a common, acute and severe neurological disease with high
mortality that is characterized by abnormal discharge of various
stimuli by highly synchronized neurons in the brain, including
neuroinflammation and oxidative stress (32). Seizures are estimated to account
for 0.75% of the global disease burden, based on the research in
predominantly low- and middle-income countries (33). SE needs to be terminated in a
timely manner; the most common and important method is drug therapy
(34,35). Currently used antiepileptic drugs
primarily inhibit nerve excitation, thereby inhibiting epileptic
seizure (36). However, in ~30% of
individuals with epilepsy, existing antiepileptic drugs do not
control repeated epileptic seizures (36). Therefore, studies aiming to
identify novel treatments for patients with epilepsy remain
important (37–39).
Adenosine is present in all cells and is involved in
controlling the function of all types of tissue and organ. Elevated
adenosine levels prevent cell damage and organ dysfunction
(40). In addition, adenosine is
involved in the adjustment of pathophysiological processes, such as
the management of sleep and wakefulness, and affects postsynaptic
receptors involved in the release of neurotransmitters (such as
glutamate, acetylcholine, thyroxine, serotonin and dopamine)
(40). The adenosine system exerts
inhibitory effects on the brain; therefore, adenosine is regarded
as an endogenous anticonvulsant (15). The anticonvulsant effect of
adenosine is primarily mediated by A1R and adenosine A2A receptors,
which are in dynamic equilibrium in normal brain tissue and
maintain the inhibitory effect of the adenosine system (41). A1R inhibits voltage-gated
Ca2+ channels, preventing glutamate release and neuronal
depolarization (42). In addition,
A1R activates K+ channels to enhance potassium ion
outflow, resulting in the hyperpolarization of postsynaptic
membrane and reducing the excitability of nerve conduction
(43). The adenosine A2A receptor
is primarily expressed in the thalamus, where it functions to
promote excitatory synaptic transmission and offset the inhibitory
effect of A1R upon glutamate release (44). Thus, the adenosine system is
regarded as an ‘endogenous antiepilepsy system’ (45).
In the present study, dynamic changes in A1R
expression levels following SE were observed in rats. A1R
expression levels began to increase 1 day after successful
establishment of the SE model and peaked after 3 days before
gradually decreasing over 1–2 weeks after seizure induction. Based
on the immunohistochemistry results, the number of A1R-positive
cells increased and staining for A1R was enhanced in the SE group
at 3 days after seizure induction compared with the control group.
In the normal brain, A1R and adenosine receptor A2A exhibit a
dynamic equilibrium, maintaining the inhibitory effects of the
adenosine system. In previous studies in which changes in the
expression levels of adenosine receptors in a rat model of the
acute phase of epilepsy were examined, A1R was significantly
increased 1 day after seizure induction, thus indicating an
adaptive response to an acute attack (34,38).
However, in patients and animal models of chronic epilepsy, A1R
expression levels in brain tissue are downregulated, whereas A2A
receptor expression levels are notably upregulated, leading to
inhibition of A1R (46). This
decrease in A1R expression levels is considered to reflect nerve
cell degeneration, and increased extracellular adenosine and A1R
expression levels have been revealed to improve the inhibitory
effects of the adenosine system until the end of a seizure
(15).
In the present study, the seizure onset latency was
prolonged and the number of seizures decreased in rats with acute
epilepsy treated with the p38 MAPK inhibitor SB203580. SB203580 has
previously been demonstrated to selectively inhibit the activation
of p38 MAPK, decrease epileptic seizures in rats and protect CA3
neurons, indicating that the p38 MAPK signaling pathway is involved
in an antiepileptic mechanism in the brain (47). In SE, the absence of neurons also
increases the expression levels of phosphorylated-p38 and
inhibition of p38 MAPK decreases neuronal damage (48). Studies have revealed that the
adenosine system activates multiple MAPK signaling pathways via
G-protein coupling (49,50). ENT1 is a subtype of a nucleoside
transporter and its primary function is to regulate the level of
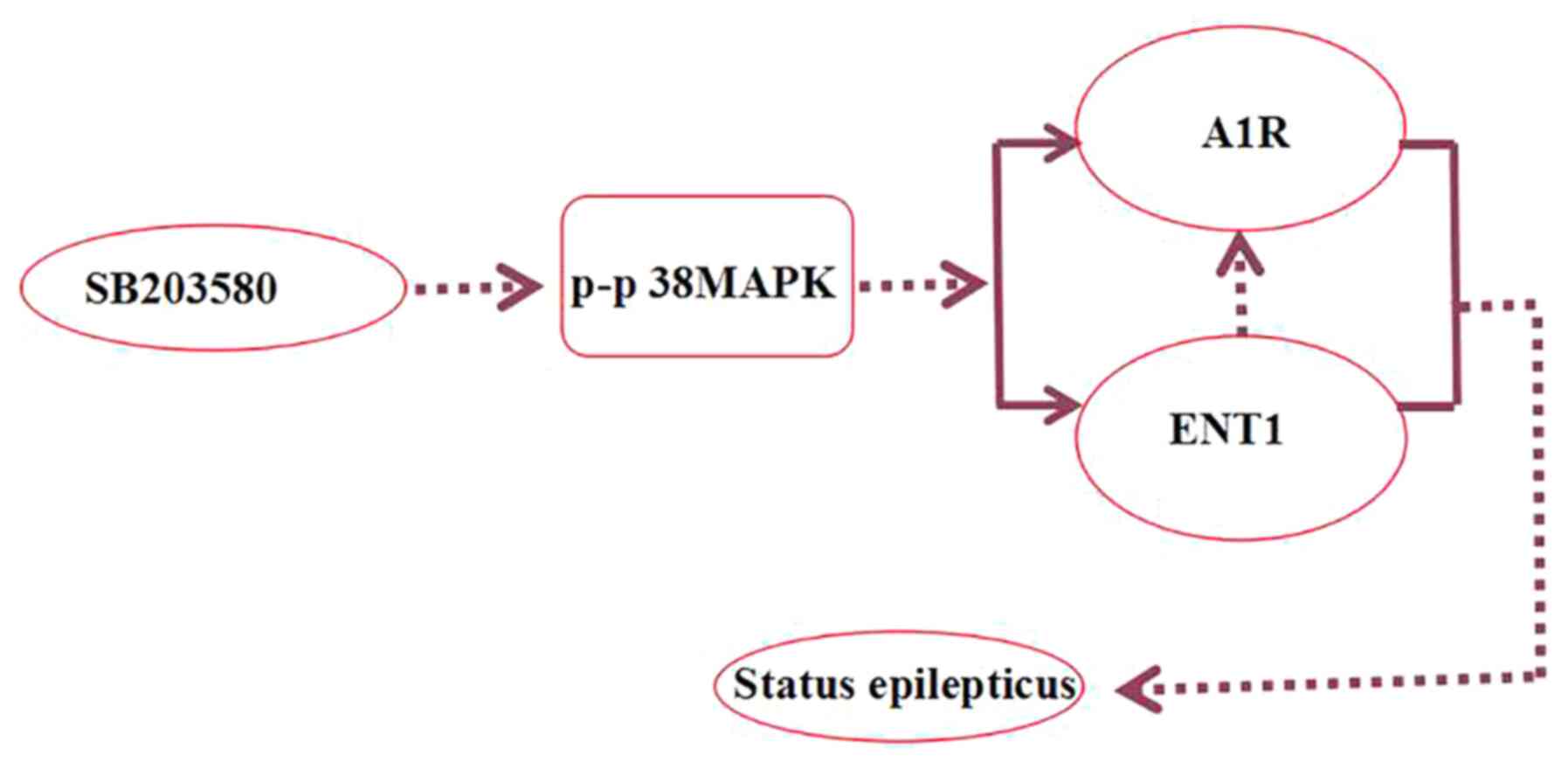
adenosine (51). In the present
study, the p38 MAPK inhibitor SB203580 decreased the number of
seizures and brain tissue damage, as well as stress-induced
endogenous A1R expression levels (Fig.
9).
Numerous types of nervous system disease (including
epilepsy, neurodegenerative disease, mental disorders and cerebral
blood disease) may involve nucleosides; nucleoside drugs have been
designed to treat a range of types of tumors and viral infections
(52,53). However, the intra- and
extracellular availability of nucleosides and their derivatives
depends on the presence of specific nucleoside transporters
(54). Importantly, our previous
study found that i.p. and hippocampal injections of NBTI, a
selective ENT1 inhibitor, decreased the number of seizures and
prolonged the latency of the first seizure in rats, indicating that
ENT1 plays an important role in the development of epilepsy
(29). Therefore, ENT1 may be a
novel therapeutic target for the control of epileptic seizures in
the clinic (22,28). Based on the results of western blot
analysis in the present study, ENT1 expression levels 3 days after
seizure induction were significantly lower in the SB203580 group
than in the epilepsy group, showing that inhibition of the p38 MAPK
signaling pathway modified ENT1 expression levels in epileptic
rats.
There were certain limitations of the present study.
Following SE, the distribution of A1R inside and outside of cells
was different from that of normal rats; increased extracellular A1R
can protect neurons (55).
Secondly, SB203580 was used to inhibit p38 MAPK, but changes in p38
levels following SB203580 application were not detected.
Immunohistochemistry demonstrated that the SB203580 group had fewer
A1R-positive cells than the SE group; future immunofluorescence
experiments should compare the fluorescence intensity of the SE and
normal control groups. Moreover, changes in fluorescence intensity
of A1R in the SB203580 group require further investigation.
In summary, the results of the present study
demonstrated that the p38 MAPK inhibitor SB203580 decreased
pathological damage to hippocampal neurons, prolonged the seizure
latency and decreased A1R and ENT1 protein expression levels in
rats. The proposed antiepileptic effect and protective effects of
SB203580 in neurons may be due to decreased ENT1 expression levels,
although the specific mechanism requires further investigation.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81560224
and 81660227) and the Guizhou Provincial Science and Technology
Foundation [grant nos. LH (2015) 7520 and LH (2015) 7471].
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZX, XZ and QC conceived and designed the study. XZ,
QC, YC, HZ and HH performed the experiments. JZh, JW, YP, JZe and
ZF conducted statistical analysis, XZ and QC wrote the paper. XZ,
QC, HH, JZh, JW, YP, JZe and ZF reviewed and edited the manuscript.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All procedures were performed in accordance with the
Guide for and Use of Medical Laboratory Animals (Ministry of Health
of China, 1998) and approved by Animal experiment ethics committee
of Zunyi Medical College [approval no. (2016) 2–044].
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mallok A, Vaillant JD, Soto MT,
Viebahn-Hänsler R, Viart ML, Pérez AF, Cedeño RI and Fernández OS:
Ozone protective effects against PTZ-induced generalized seizures
are mediated by reestablishment of cellular redox balance and A1
adenosine receptors. Neurol Res. 37:204–210. 2015.PubMed/NCBI
|
|
2
|
Lawson T and Yeager S: Status epilepticus
in adults: A review of diagnosis and treatment. Crit Care Nurse.
36:62–73. 2016.PubMed/NCBI
|
|
3
|
Sutter R, Semmlack S and Kaplan PW:
Nonconvulsive status epilepticus in adults - insights into the
invisible. Nat Rev Neurol. 12:281–293. 2016.PubMed/NCBI
|
|
4
|
Córdova-Dávalos L, Carrera-Calvo D,
Solís-Navarrete J, Mercado-Gómez OF, Arriaga-Ávila V,
Agredano-Moreno LT, Jiménez-García LF and Guevara-Guzmán R: Status
epilepticus triggers early mitochondrial fusion in the rat
hippocampus in a lithium-pilocarpine model. Epilepsy Res.
123:11–19. 2016.PubMed/NCBI
|
|
5
|
Cui Y, Liu J, Luo Y, He S, Xia Y, Zhang Y,
Yao D and Guo D: Aberrant connectivity during pilocarpine-induced
status epilepticus. Int J Neural Syst. 30:19500292020.PubMed/NCBI
|
|
6
|
Xie L, Li T, Song X, Sun H, Liu J, Yang J,
Zhao W, Cheng L, Chen H, Liu B, et al: Dynamic alteration of
dendrites and dendritic spines in the hippocampus and microglia in
mouse brain tissues after kainate-induced status epilepticus. Int J
Neurosci. Jun 1–2020.(Epub ahead of print).
|
|
7
|
Hu JH, Malloy C and Hoffman DA: P38
Regulates kainic acid-induced seizure and neuronal firing via Kv4.2
phosphorylation. Int J Mol Sci. 21:E59212020.PubMed/NCBI
|
|
8
|
Che Y, Yu YM, Han PL and Lee JK: Delayed
induction of p38 MAPKs in reactive astrocytes in the brain of mice
after KA-induced seizure. Brain Res Mol Brain Res. 94:157–165.
2001.PubMed/NCBI
|
|
9
|
Neligan A and Shorvon SD: Frequency and
prognosis of convulsive status epilepticus of different causes: A
systematic review. Arch Neurol. 67:931–940. 2010.PubMed/NCBI
|
|
10
|
Matute C and Cavaliere F: Neuroglial
interactions mediated by purinergic signalling in the
pathophysiology of CNS disorder. Semin Cell Dev Biol. 22:252–259.
2011.PubMed/NCBI
|
|
11
|
Li T, Quan Lan J, Fredholm BB, Simon RP
and Boison D: Adenosine dysfunction in astrogliosis: Cause for
seizure generation? Neuron Glia Biol. 3:353–366. 2007.PubMed/NCBI
|
|
12
|
Masino SA, Kawamura M Jr and Ruskin DN:
Adenosine receptors and epilepsy: Current evidence and future
potential. Int Rev Neurobiol. 119:233–255. 2014.PubMed/NCBI
|
|
13
|
Boison D: Adenosine dysfunction in
epilepsy. Glia. 60:1234–1243. 2012.PubMed/NCBI
|
|
14
|
Cunha RA: How does adenosine control
neuronal dysfunction and neurodegeneration. J Neurochem.
2016.139(6). 1019–1055. PubMed/NCBI
|
|
15
|
Hargus NJ, Jennings C, Perez-Reyes E,
Bertram EH and Patel MK: Enhanced actions of adenosine in medial
entorhinal cortex layer II stellate neurons in temporal lobe
epilepsy are mediated via A(1)-receptor activation. Epilepsia.
53:168–176. 2012.PubMed/NCBI
|
|
16
|
Ono K and Han J: The p38 signal
transduction pathway: Activation and function. Cell Signal.
12:1–13. 2000.PubMed/NCBI
|
|
17
|
Downey JS and Han J: Cellular activation
mechanisms in septic shock. Front Biosci. 3:d468–d476.
1998.PubMed/NCBI
|
|
18
|
Robinson MJ and Cobb MH: Mitogen-activated
protein kinase pathways. Curr Opin Cell Biol. 9:180–186.
1997.PubMed/NCBI
|
|
19
|
Okamoto OK, Janjoppi L, Bonone FM, Pansani
AP, da Silva AV, Scorza FA and Cavalheiro EA: Whole transcriptome
analysis of the hippocampus: Toward a molecular portrait of
epileptogenesis. BMC Genomics. 11:2302010.PubMed/NCBI
|
|
20
|
Yang Z, Wang J, Yu C, Xu P, Zhang J, Peng
Y, Luo Z, Huang H, Zeng J and Xu Z: Inhibition of p38 MAPK
signaling regulates the expression of EAAT2 in the brains of
epileptic rats. Front Neurol. 9:9252018.PubMed/NCBI
|
|
21
|
Minor TR, Rowe M, Cullen PK and Furst S:
Enhancing brain adenosine signaling with the nucleoside transport
blocker NBTI (S-(4-nitrobenzyl)-6-theoinosine) mimics the effects
of inescapable shock on later shuttle-escape performance in rats.
Behav Neurosci. 122:1236–1247. 2008.PubMed/NCBI
|
|
22
|
Huang H, Wang J, Zhang J, Luo Z, Li D, Qiu
X, Peng Y and Xu Z, Xu P and Xu Z: Nitrobenzylthioinosine mimics
adenosine to attenuate the epileptiform discharge of hippocampal
neurons from epileptic rats. Oncotarget. 8:35573–35582.
2017.PubMed/NCBI
|
|
23
|
Boison D: Adenosine as a modulator of
brain activity. Drug News Perspect. 20:607–611. 2007.PubMed/NCBI
|
|
24
|
Huang M, Wang Y, Collins M, Gu JJ,
Mitchell BS and Graves LM: Inhibition of nucleoside transport by
p38 MAPK inhibitors. J Biol Chem. 277:28364–28367. 2002.PubMed/NCBI
|
|
25
|
Rashid K, Van der Zee CE, Ross GM, Chapman
CA, Stanisz J, Riopelle RJ, Racine RJ and Fahnestock M: A nerve
growth factor peptide retards seizure development and inhibits
neuronal sprouting in a rat model of epilepsy. Proc Natl Acad Sci
USA. 92:9495–9499. 1995.PubMed/NCBI
|
|
26
|
Bulavin DV, Phillips C, Nannenga B,
Timofeev O, Donehower LA, Anderson CW, Appella E and Fornace AJ Jr:
Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis
through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf)
pathway. Nat Genet. 36:343–350. 2004.PubMed/NCBI
|
|
27
|
Li Q, Liu H, Du J, Chen B, Li Q, Guo X,
Huang X and Huang Q: Advanced glycation end products induce moesin
phosphorylation in murine brain endothelium. Brain Res. 1373:1–10.
2011.PubMed/NCBI
|
|
28
|
Mannangatti P, NarasimhaNaidu K, Damaj MI,
Ramamoorthy S and Jayanthi LD: A Role for p38 Mitogen-activated
Protein Kinase-mediated Threonine 30-dependent Norepinephrine
Transporter Regulation in Cocaine Sensitization and Conditioned
Place Preference. J Biol Chem. 290:10814–10827. 2015.PubMed/NCBI
|
|
29
|
Xu Z, Xu P, Chen Y, Liu J, Zhang Y, Lv Y,
Luo J, Fang M, Zhang J, Wang J, et al: ENT1 inhibition attenuates
epileptic seizure severity via regulation of glutamatergic
neurotransmission. Neuromolecular Med. 17:1–11. 2015.PubMed/NCBI
|
|
30
|
Marawar R, Basha M, Mahulikar A, Desai A,
Suchdev K and Shah A: Updates in refractory status epilepticus.
Crit Care Res Pract. 2018:97689492018.PubMed/NCBI
|
|
31
|
Smith PE: Management of Established Status
Epilepticus. N Engl J Med. 381:2171–2172. 2019.PubMed/NCBI
|
|
32
|
Terrone G, Frigerio F, Balosso S, Ravizza
T and Vezzani A: Inflammation and reactive oxygen species in status
epilepticus: Biomarkers and implications for therapy. Epilepsy
Behav. 101((Pt B)): 1062752019.PubMed/NCBI
|
|
33
|
Thijs RD, Surges R, O'Brien TJ and Sander
JW: Epilepsy in adults. Lancet. 393:689–701. 2019.PubMed/NCBI
|
|
34
|
Goodrich GS, Kabakov AY, Hameed MQ, Dhamne
SC, Rosenberg PA and Rotenberg A: Ceftriaxone treatment after
traumatic brain injury restores expression of the glutamate
transporter, GLT-1, reduces regional gliosis, and reduces
post-traumatic seizures in the rat. J Neurotrauma. 30:1434–1441.
2013.PubMed/NCBI
|
|
35
|
Wagner RG, Bottomley C, Ngugi AK, Ibinda
F, Gómez-Olivé FX, Kahn K, Tollman S, Newton CR, Wagner R, Twine R,
et al SEEDS Writing Group, : Incidence, remission and mortality of
convulsive epilepsy in rural northeast South Africa. PLoS One.
10:e01290972015.PubMed/NCBI
|
|
36
|
Arnold Cenobamate S.: new hope for
treatment-resistant epilepsy. Lancet Neurol. 19:23–24.
2020.PubMed/NCBI
|
|
37
|
Boison D: The Biochemistry and Epigenetics
of Epilepsy: Focus on Adenosine and Glycine. Front Mol Neurosci.
9:262016.PubMed/NCBI
|
|
38
|
Kwan P and Brodie MJ: Early identification
of refractory epilepsy. N Engl J Med. 342:314–319. 2000.PubMed/NCBI
|
|
39
|
Shao Y, Wang C, Hong Z and Chen Y:
Inhibition of p38 mitogen-activated protein kinase signaling
reduces multidrug transporter activity and anti-epileptic drug
resistance in refractory epileptic rats. J Neurochem.
136:1096–1105. 2016.PubMed/NCBI
|
|
40
|
Świąder MJ, Kotowski J and Łuszczki JJ:
Modulation of adenosinergic system and its application for the
treatment of epilepsy. Pharmacol Rep. 66:335–342. 2014.PubMed/NCBI
|
|
41
|
Sebastião AM and Ribeiro JA: Adenosine
receptors and the central nervous system. Handb Exp Pharmacol.
193:471–534. 2009.
|
|
42
|
Fredholm BB, Chen JF, Cunha RA,
Svenningsson P and Vaugeois JM: Adenosine and brain function. Int
Rev Neurobiol. 63:191–270. 2005.PubMed/NCBI
|
|
43
|
Liu YJ, Chen J, Li X, Zhou X, Hu YM, Chu
SF, Peng Y and Chen NH: Research progress on adenosine in central
nervous system diseases. CNS Neurosci Ther. 25:899–910.
2019.PubMed/NCBI
|
|
44
|
Boison D, Chen JF and Fredholm BB:
Adenosine signaling and function in glial cells. Cell Death Differ.
17:1071–1082. 2020.
|
|
45
|
Boison D: Adenosine as a modulator of
brain activity. Drug News Perspect. 20:607–611. 2007.PubMed/NCBI
|
|
46
|
Huicong K, Zheng X, Furong W, Zhouping T,
Feng X, Qi H, Xiaoyan L, Xiaojiang H, Na Z, Ke X, et al: The
imbalanced expression of adenosine receptors in an epilepsy model
corrected using targeted mesenchymal stem cell transplantation. Mol
Neurobiol. 48:921–930. 2013.PubMed/NCBI
|
|
47
|
Jiang W, Van Cleemput J, Sheerin AH, Ji
SP, Zhang Y, Saucier DM, Corcoran ME and Zhang X: Involvement of
extracellular regulated kinase and p38 kinase in hippocampal
seizure tolerance. J Neurosci Res. 81:581–588. 2005.PubMed/NCBI
|
|
48
|
Cieślak M, Wojtczak A and Komoszyński M:
Role of the purinergic signaling in epilepsy. Pharmacol Rep.
69:130–138. 2017.PubMed/NCBI
|
|
49
|
Kim DS, Min SJ, Kim MJ, Kim JE and Kang
TC: Leptomycin B ameliorates vasogenic edema formation induced by
status epilepticus via inhibiting p38 MAPK/VEGF pathway. Brain Res.
1651:27–35. 2016.PubMed/NCBI
|
|
50
|
Akhtar S, Yousif MH, Chandrasekhar B and
Benter IF: Activation of EGFR/ERBB2 via pathways involving ERK1/2,
P38 MAPK, AKT and FOXO enhances recovery of diabetic hearts from
ischemia-reperfusion injury. PLoS One. 7:e390662012.PubMed/NCBI
|
|
51
|
Zhang YN, Dong HT, Yang FB, Wang ZQ, Ma
ZH, Ma SZ, Ma XD and Duan L: Nrf2-ARE signaling pathway regulates
the expressions of A1R and ENT1 in the brain of epileptic rats. Eur
Rev Med Pharmacol Sci. 22:6896–6904. 2018.PubMed/NCBI
|
|
52
|
Boison D: Adenosine as a neuromodulator in
neurological diseases. Curr Opin Pharmacol. 8:2–7. 2008.PubMed/NCBI
|
|
53
|
Zhang J, Visser F, King KM, Baldwin SA,
Young JD and Cass CE: The role of nucleoside transporters in cancer
chemotherapy with nucleoside drugs. Cancer Metastasis Rev.
26:85–110. 2007.PubMed/NCBI
|
|
54
|
Parkinson FE, Damaraju VL, Graham K, Yao
SY, Baldwin SA, Cass CE and Young JD: Molecular biology of
nucleoside transporters and their distributions and functions in
the brain. Curr Top Med Chem. 11:948–972. 2011.PubMed/NCBI
|
|
55
|
Rombo DM, Dias RB, Duarte ST, Ribeiro JA,
Lamsa KP and Sebastião AM: Adenosine A1 receptor suppresses tonic
GABAA receptor currents in hippocampal pyramidal cells and in a
defined subpopulation of interneurons. Cereb Cortex. 26:1081–1095.
2016.PubMed/NCBI
|