Introduction
Acute lung injury (ALI) and acute respiratory
distress syndrome (ARDS) are major clinical syndromes of acute
respiratory failure that can be caused by infectious factors, such
as bacterial and viral infections, and non-infectious factors, such
as inhalation of toxic gas, blood transfusion, drug poisoning and
acute pancreatitis (1,2). ALI may lead to alveolar epithelium or
vascular endothelial cell damage, and an increase in pulmonary
vascular permeability (1). However,
the pathogenesis of ALI is complicated, and among the various
putative mechanistic explanations, an increased production of
pro-inflammatory factors in the lungs is currently recognized as
the most important mechanism (2).
The increased release and activation of inflammatory cytokines can
lead to an uncontrolled inflammatory response, causing damage to
the pulmonary capillary endothelial cells and alveolar epithelial
cells (2). Damaged endothelial
cells release reactive oxygen species, which subsequently leads to
an increase in the permeability of lung endothelial cells, allowing
large quantities of protein-rich substances to enter the bronchial
tubes and alveolar cavities, causing pulmonary edema, thereby
reducing the effectiveness of lung ventilation, and even leading to
respiratory failure (3,4). There are currently no specific drugs
available for the treatment of ALI. Therefore, the development of
effective treatment strategies is of great clinical significance in
treating ALI.
The apelin family comprises a class of endogenous
active peptides with different molecular structures encoded by the
same gene. These peptides function as endogenous ligands for the
seven-transmembrane G-protein-coupled receptor, APJ (5). The precursor of the apelin peptides is
a polypeptide composed of 77 amino acids encoded by the X
chromosome, which is hydrolyzed into active polypeptides of
different lengths by proteases (6).
These active polypeptides can be classified according to their
number of amino acids; peptides produced are typically 36, 17 and
13 amino acids in length, in addition to other subtypes. The longer
the peptide chain, the stronger the binding of the apelin peptide
to the APJ receptor (5). One such
naturally occurring apelin peptide comprising 36 amino acid
residues is termed ‘apelin-36’. Apelin-36 is a long peptide
fragment of the apelin precursor protein that is widely distributed
in the central nervous system and in peripheral tissues (7). Studies have demonstrated that
apelin-36 is closely associated with a variety of diseases,
including nervous system diseases, myocardial ischemia and diabetes
(8–12). A study by Fan et al (5) revealed that the levels of apelin-13
and apelin-36 were increased in the plasma, lung tissues and
bronchoalveolar lavage fluid (BLAF) following lipopolysaccharide
(LPS)-induced lung injury, and the addition of apelin-13 could
reduce LPS-induced lung injury. However, further studies are
required to further elucidate the underlying mechanisms.
Furthermore, a recent study demonstrated that apelin-36 could
regulate oxidative stress, autophagy and apoptosis to exert
neuroprotective effects via inhibiting the apoptosis
signal-regulating kinase 1 (ASK1)/c-Jun N-terminal kinase
(JNK)/caspase-3 apoptotic pathway in a mouse model of Parkinson's
disease (13). In the present
study, the aim was to investigate whether apelin-36 could regulate
LPS-induced ALI both in vivo and in vitro, as well as
exploring the potential mechanisms of apelin-36 in an LPS-induced
ALI rat model.
Materials and methods
Animals and regents
A total of 25 male Sprague-Dawley rats (age, 7–8
weeks) weighing 250–300 g were purchased from the Nanjing Jiancheng
Bioengineering Institute. All animals were kept in a specific
pathogen-free environment at 25°C under a controlled 12/12 h
light/dark cycle, and all rats received food and water ad
libitum. All experimental procedures were carried out in
accordance with the US National Institutes of Health Guidelines for
the Care and Use of Laboratory Animals, and were approved by the
Ethical Committee on Animal Research at People's Hospital of
Ningxia Hui Autonomous Region (approval no. IACUC-20191002-10;
Yinchuan, China).
LPS was also obtained from Nanjing Jiancheng
Bioengineering Institute. Apelin-36 was purchased from Phoenix
Pharmaceuticals, Inc. The apelin-36 kit (cat. no. kt98790) was
provided by MSK Biotechnology Co., Ltd., whereas the ELISA kits and
antibodies were obtained from Abcam.
LPS-induced ALI
Rats were randomly assigned to four treatment
groups, with 5 rats in each group, as follows: i) Control group;
ii) apelin-36 group; iii) LPS group; and iv) LPS + apelin-36 group.
Apelin-36 was intraperitoneally injected into rats at a
concentration of 10 nmol/kg for 24 h, in accordance with a previous
study (5). For induction of ALI,
rats were intratracheally instilled with 5 mg/kg LPS for 24 h,
whereas the control animals were instilled intratracheally with an
equal volume of normal saline, as previously described (14,15).
For the LPS + apelin-36 co-treatment group, apelin-36 was
administered 4 h after LPS instillation. At 24 h after the LPS or
apelin-36 treatment, rats were anesthetized using 1% pentobarbital
(50 mg/kg, intraperitoneal injection) for the subsequent
experiments.
Collection of BALF
After treatment with LPS for 24 h, the rats were
anesthetized using 1% pentobarbital (50 mg/kg, intraperitoneally
injection) and sacrificed by cervical dislocation. After exposing
the chest cavity and intubating the trachea, PBS was slowly dropped
into the lung bronchus, and the BALF was collected for further
analysis, as described previously (16).
Detection of the lung wet/dry
ratio
Following sacrifice, the lungs of the rats were
isolated, and the wet lungs were harvested and subsequently weighed
to obtain the wet weight. The blood on the lungs was then blotted
dry, and the lungs were dried in an 80°C incubator for 48 h, after
which the weight of the lungs was recorded as the dry weight. The
lung wet/dry weight ratio was then calculated (wet weight/dry
weight), and used to assess the degree of pulmonary edema.
Histological staining
To evaluate the histological alterations, lung
tissues were fixed with 4% paraformaldehyde at 4°C for 24 h,
embedded in paraffin, and 4-µm thick sections were cut. After
deparaffinization and dehydration, the sections were stained with
hematoxylin and eosin (Beyotime Institute of Biotechnology) at room
temperature for 2 min using standard histological techniques, and
were subsequently observed under an optical microscope for
pathological examination. Furthermore, some of the tissue sections
were deparaffinized, rehydrated and subjected to TUNEL (Beyotime
Institute of Biotechnology) staining, three fields of view were
examined and the extent of cellular death was determined using a
fluorescence microscope (Olympus Corporation). DAPI was used to
stain the nuclei (blue) at room temperature for 1 min. The TUNEL
positive cells were calculated using the following formula: TUNEL
positive cells=the number of green cells/total (blue) cells ×100%,
this was determined using ImageJ 1.52 software (National Institutes
of Health).
Cell culture and treatment
Human bronchial epithelial (Beas-2B) cells (American
Type Culture Collection) were cultured in RPMI-1640 medium with 10%
FBS (Wisent, Inc.), and 1% streptomycin-penicillin antibiotics
under an atmosphere of 5% CO2 at 37°C. The cells were
pretreated with apelin-36 (0.1, 0.5 or 1 µM) for 1 h, and
subsequently stimulated with LPS (1 µg/ml) for 6 h. Untreated cells
served as a control.
For overexpression of ASK1, recombinant full-length
human ASK1 (2 µg/ml) was cloned into the pcDNA3.0 vector
(Invitrogen; Thermo Fisher Scientific, Inc.), and then transfected
into cells (at a density of 60–70%) using Lipofectamine®
2000 (Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. A pcDNA3.0 empty vector was used as a
negative control (pcDNA-NC). At 48 h post-transfection, cells were
selected for subsequent experiments.
Cell Counting Kit-8 (CCK-8) assay
To determine the cell viability, cells were seeded
into 96-well plates at a density of 4×104, and then
exposed to the aforementioned treatments. Subsequently, cells were
incubated with 50 µl CCK-8 (Beyotime Institute of Biotechnology)
working solution diluted in 0.5 ml cell culture medium for 2 h
under normal cell culture conditions, and then the absorbance at
450 nm was detected with a microplate reader.
ELISA
The concentration of apelin-36 in BALF, and the
activities of inflammatory factors, including interleukin-6 (IL-6),
monocyte chemoattractant protein-1 (MCP-1) and tumor necrosis
factor-α (TNF-α) in BALF and the supernatant of the cells, were
detected using an ELISA assay following previously described
methods (5).
Western blot analysis
In order to obtain the total protein extract, lung
tissues were homogenized using 50 ms pulse ultrasonication at 4°C
for 5–10 min, and total protein of the lung tissues or Beas-2B
cells was extracted using RIPA lysis buffer (Thermo Fisher
Scientific, Inc.). After quantification using a BCA kit (Thermo
Fisher Scientific, Inc.), a total of 10 µg protein samples were
separated via 12% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis, and subsequently transferred to polyvinylidene
fluoride membranes (EMD Millipore). After blocking with 5% non-fat
milk at room temperature for 2 h, the membranes were incubated with
primary antibodies against Bcl-2 (cat. no. sc-7382; 1:1,000), Bax
(cat. no. sc-7480; 1:1,000), caspase-3 (cat. no. sc-271759; 1:200),
cleaved-caspase-3 (cat. no. sc-373730; 1:200), ASK1 (cat. no.
sc-390275; 1:500), p38 (cat. no. sc-7972; 1:500), phosphorylated
(p)-p38 (cat. no. sc-166182; 1:500), JNK (cat. no. sc-7345; 1:500),
p-JNK (cat. no. sc-6254; 1:500), extracellular signal-regulated
kinase (ERK; cat. no. sc-514302; 1:500), p-ERK (cat. no. sc-7383;
1:200) and GAPDH (cat. no. sc-47724; 1:1,000; all purchased from
Santa Cruz Biotechnology, Inc.). The primary antibodies were
detected using horseradish peroxidase-conjugated goat anti-rabbit
(cat. no. ab6721; 1:2,000) and goat anti-mouse (cat. no. ab6789;
1:2,000; both purchased from Abcam) secondary antibodies at room
temperature for 2 h, and visualized by chemiluminescence (Thermo
Fisher Scientific, Inc.). The protein levels were normalized
against GAPDH. ImageJ software (v1.46r; National Institutes of
Health) was used to semi-quantify the intensity of each protein
band.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA from Beas-2B cells was isolated using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) and reverse transcribed to cDNA using the PrimeScript RT
Reagent Kit with gDNA Eraser (Takara Bio, Inc.), according to the
manufacturer's instructions. A total of 50 ng cDNA was subsequently
used for qPCR using TB Green® Fast qPCR Mix (Takara
Biotechnology Co., Ltd.). The following primers were used: ASK1,
forward 5′-AGACCCTGCATTTCGGGAAG-3′ and reverse
5′-GCCAGATCGAAGGTCGGTAG-3′; GAPDH, forward,
5′-AGTGCCAGCCTCGTCTCATA-3′ and reverse 5′-CTCGTGGTTCACACCCATCA-3′.
GAPDH was used as the control. The following thermocycling
conditions were used for qPCR: Initial denaturation at 95°C for 30
sec; and 40 cycles of 95°C for 5 sec and 60°C for 15 sec, followed
by default of melt curve (Applied Biosystems 7500; Thermo Fisher
Scientific, Inc.). Differential expression of mRNA was calculated
using the 2−ΔΔCq method (17).
Flow cytometric analysis
Cells were subjected to flow cytometry, based on
Annexin V/PI staining (Beyotime Institute of Biotechnology).
Briefly, cells at a density of 5×105 cells/well seeded
in 6-well plates were collected after the aforementioned
treatments, washed with PBS, and subsequently 100 µl binding buffer
was added. Next, 5 µl Annexin V-FITC and 5 µl PI were added,
followed by an incubation in the dark at room temperature for 15
min. The apoptotic rate (early and late apoptosis) was subsequently
measured using a BD FACSCelesta™ flow cytometer (BD Biosciences).
Data were analyzed using flow cytometry software (iSort™ Automated
Cell Sorter; Thermo Fisher Scientific, Inc.).
Statistical analysis
In the present study, GraphPad Prism 6.0 (GraphPad
Software, Inc.) was used for statistical analyses. All data are
expressed as the mean ± standard deviation. The differences between
groups were analyzed using one-way analysis of variance followed by
Tukey's multiple-comparison test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Apelin-36 alleviates lung injury,
inflammation and apoptosis in LPS-treated rats
First, LPS was used to induce ALI in the
experimental rats, and subsequently the normal rats or LPS-induced
rats were treated with or without apelin-36. Lung tissues and BALF
in the different treatment groups were collected, and the
concentration of apelin-36 was found to be significantly increased
in the BALF of LPS-treated rats (Fig.
1A), suggesting its potential role in regulating ALI. As shown
in Fig. 1B, the lung tissues of
rats that underwent LPS administration showed clear pathological
alterations compared with the control rats, including
intra-alveolar hemorrhage, inter-alveolar septum thickening and
inflammatory cell infiltration. However, in the LPS + apelin-36
group, a relatively normal lung structure and an improvement in
inflammatory cell infiltration were observed. Furthermore,
apelin-36 treatment alone did not cause any histological
alterations of the lung tissues compared with those of the control
group. Additionally, rats treated with LPS showed increased wet/dry
ratios compared with the control group, which were significantly
reduced upon co-treatment with apelin-36 (Fig. 1C). These data demonstrated the
protective effect of apelin-36 on LPS-induced lung damage.
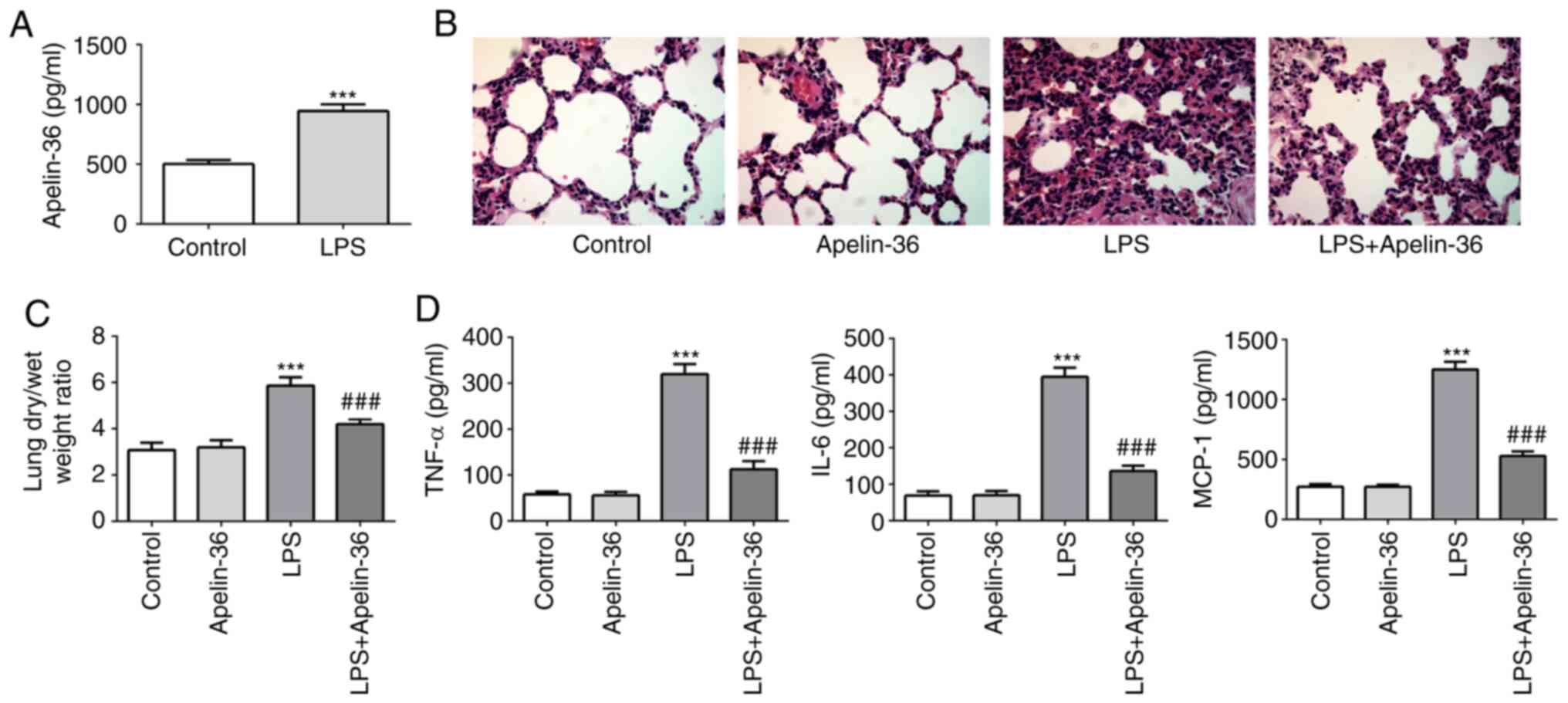 | Figure 1.Apelin-36 alleviates lung injury and
inflammation in LPS-treated rats. (A) The concentration of
apelin-36 in the BALF of normal rats and LPS-treated rats is
presented (n=5). (B) Morphological changes in the lung, as
determined by hematoxylin and eosin staining (magnification, ×400).
The sections are shown after staining with hematoxylin (blue,
nuclei) and eosin (pink, muscle fibers). (C) The lung dry/wet
weight ratio of rats subjected to the respective treatments (n=5).
(D) The concentrations of TNF-α, IL-6 and MCP-1 in the BALF of rats
subjected to the respective treatments (n=5). ***P<0.001 vs.
control; ###P<0.001 vs. LPS treatment. LPS,
lipopolysaccharide; BALF, bronchoalveolar lavage fluid; IL-6,
interleukin-6; MCP-1, monocyte chemoattractant protein-1; TNF-α,
tumor necrosis factor-α. |
Subsequently, the concentrations of pro-inflammatory
cytokines, including IL-6, MCP-1 and TNF-α in BALF, were measured.
The results presented in Fig. 1D
revealed that apelin-36 treatment led to a decrease in the levels
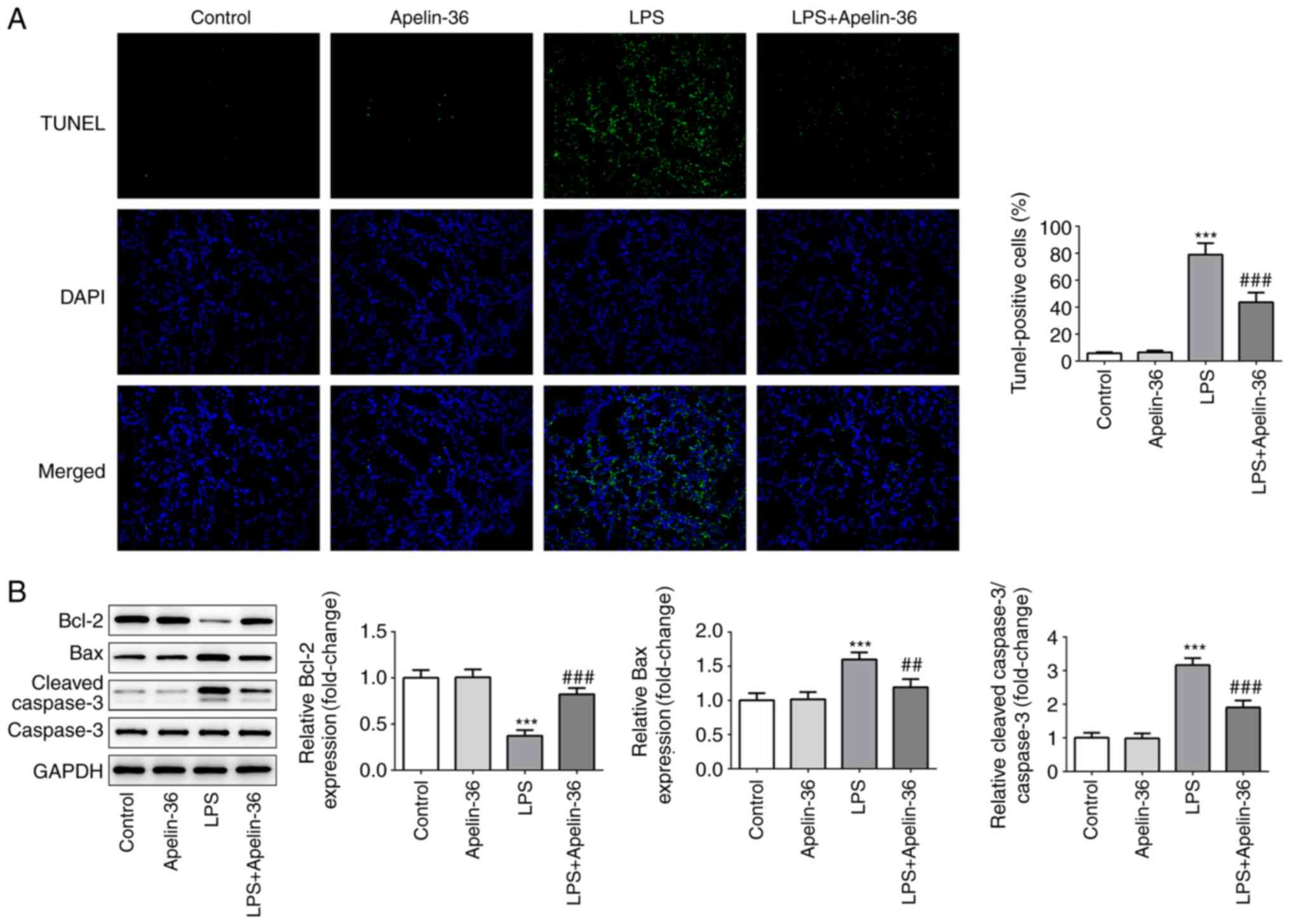
of LPS-induced pro-inflammatory cytokines. As shown in Fig. 2A, lung tissues in each group were
stained with TUNEL to highlight the apoptotic cells, and apelin-36
led to a significant reduction in the number of apoptotic cells
compared with the LPS treatment group. The results shown in
Fig. 2B further confirmed the
aforementioned observations, as LPS reduced the expression of the
anti-apoptotic protein Bcl-2, whereas the expression levels of the
pro-apoptotic proteins Bax and cleaved-caspase-3 were increased.
The presence of apelin-36 partially recovered the expression levels
of these proteins that had been altered by LPS. These findings
suggested that apelin-36 was able to inhibit LPS-induced
inflammation and apoptosis in the lung tissues of rats.
Apelin-36 inhibits the activation of
LPS-induced ASK1/MAPK signaling
To investigate the potential underlying mechanisms
of apelin-36, the expression levels of proteins associated with the
ASK1/MAPK signaling pathway were evaluated. The results presented
in Fig. 3 demonstrated that LPS led
to a significant increase in the expression levels of ASK1, p-p38,
p-JNK and p-ERK, indicating that the ASK1/MAPK pathway was
activated in the lung tissues of rats upon LPS stimulation. On the
other hand, the expression levels of ASK1, p-p38, p-JNK and p-ERK
were significantly reduced in the LPS + apelin-36 co-treatment
groups compared with LPS treatment alone. These results suggested
that apelin-36 could inhibit the LPS-induced activation of the
ASK1/MAPK signaling pathway.
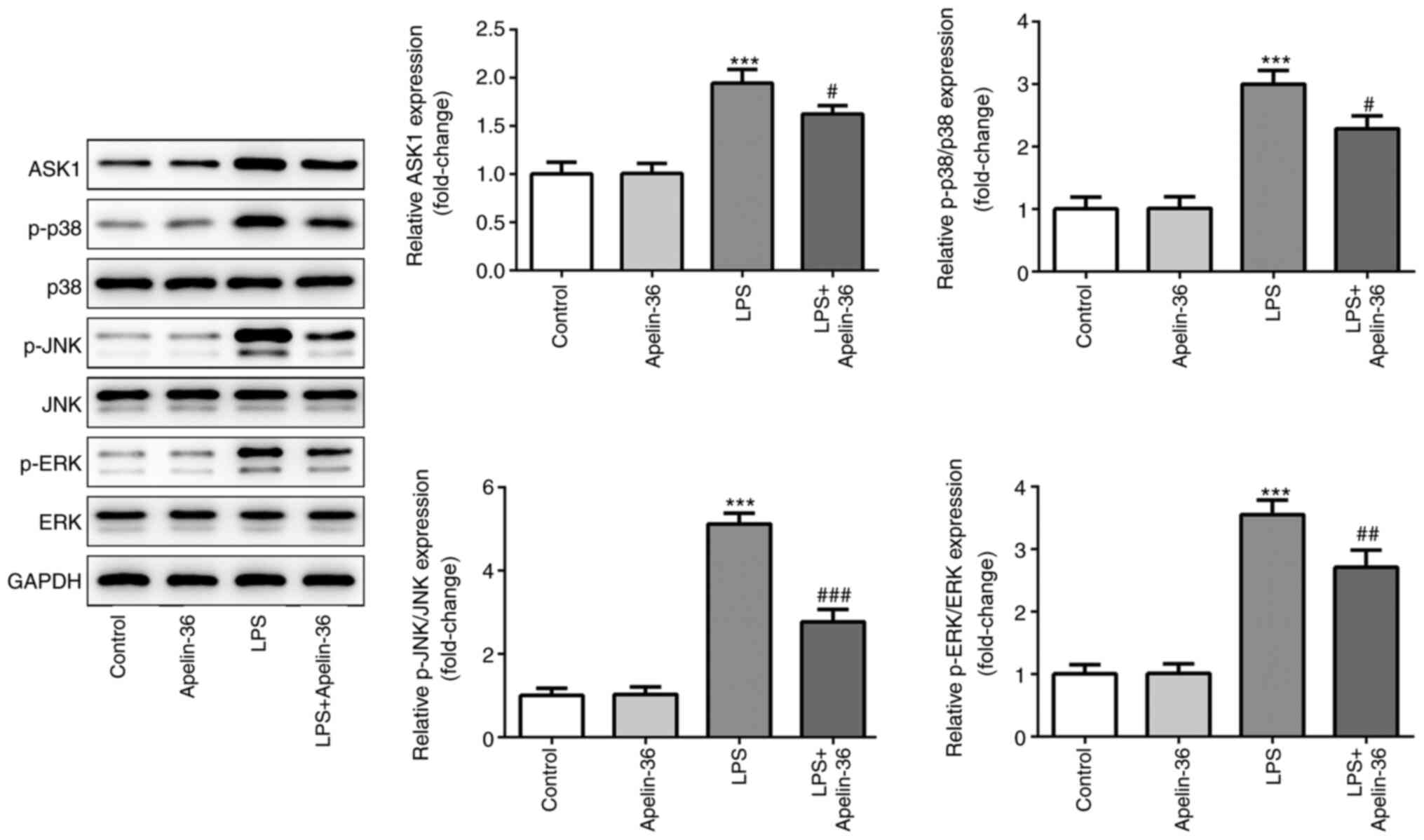 | Figure 3.Apelin-36 suppresses LPS-induced
activation of the ASK1/MAPK signaling pathway. Expression levels of
proteins involved in ASK1/MAPK signaling, including ASK1, p-p38,
p-JNK and p-ERK, in the lung tissues of rats subjected to the
various treatments were detected using western blotting (n=5).
***P<0.001 vs. control; #P<0.05,
##P<0.01 and ###P<0.001 vs. LPS
treatment. LPS, lipopolysaccharide; p-, phosphorylated; ASK1,
apoptosis signal-regulating kinase 1; MAPK, mitogen-activated
protein kinase; JNK, c-Jun N-terminal kinase; ERK, extracellular
signal-regulated kinase. |
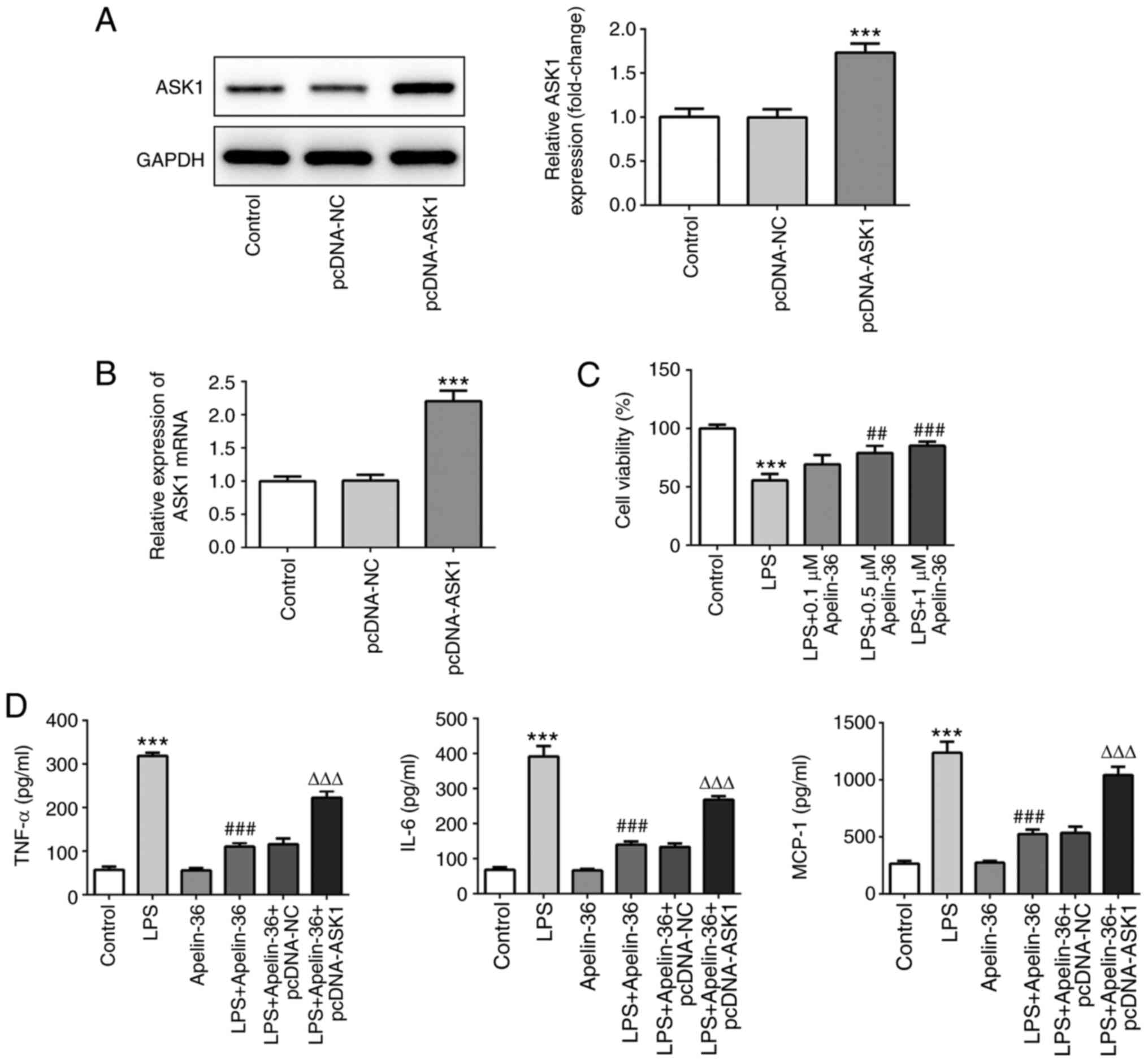
Overexpression of ASK1 suppresses the
inhibitory effect of apelin-36 on inflammation and apoptosis
To further verify the aforementioned observations,
ASK1 was overexpressed in Beas-2B cells. The results presented in
Fig. 4A and B confirmed the
transfection efficiency of ASK1 overexpression. As shown in
Fig. 4C, apelin-36 increased the
cell viability of Beas-2B cells, which had been reduced by LPS, in
a concentration-dependent manner. As 1 µM apelin-36 had the most
significant effects on cell viability, it was chosen as the
concentration for subsequent experiments. Then, cells overexpressed
with or without ASK1 were exposed to LPS, 1 µM apelin-36 or LPS +
apelin-36 (1 µM). LPS treatment led to a significant increase in
the levels of TNF-α, IL-6 and MCP-1, whereas in the apelin-36
co-treatment group, the levels of these inflammatory cytokines were
significantly reduced (Fig. 4D).
However, by contrast, the overexpression of ASK1 led to an increase
in the levels of these cytokines compared with the LPS + apelin-36
group, suggesting that ASK1 overexpression could suppress the
inhibitory effect of apelin-36 on LPS-induced inflammation in lung
cells.
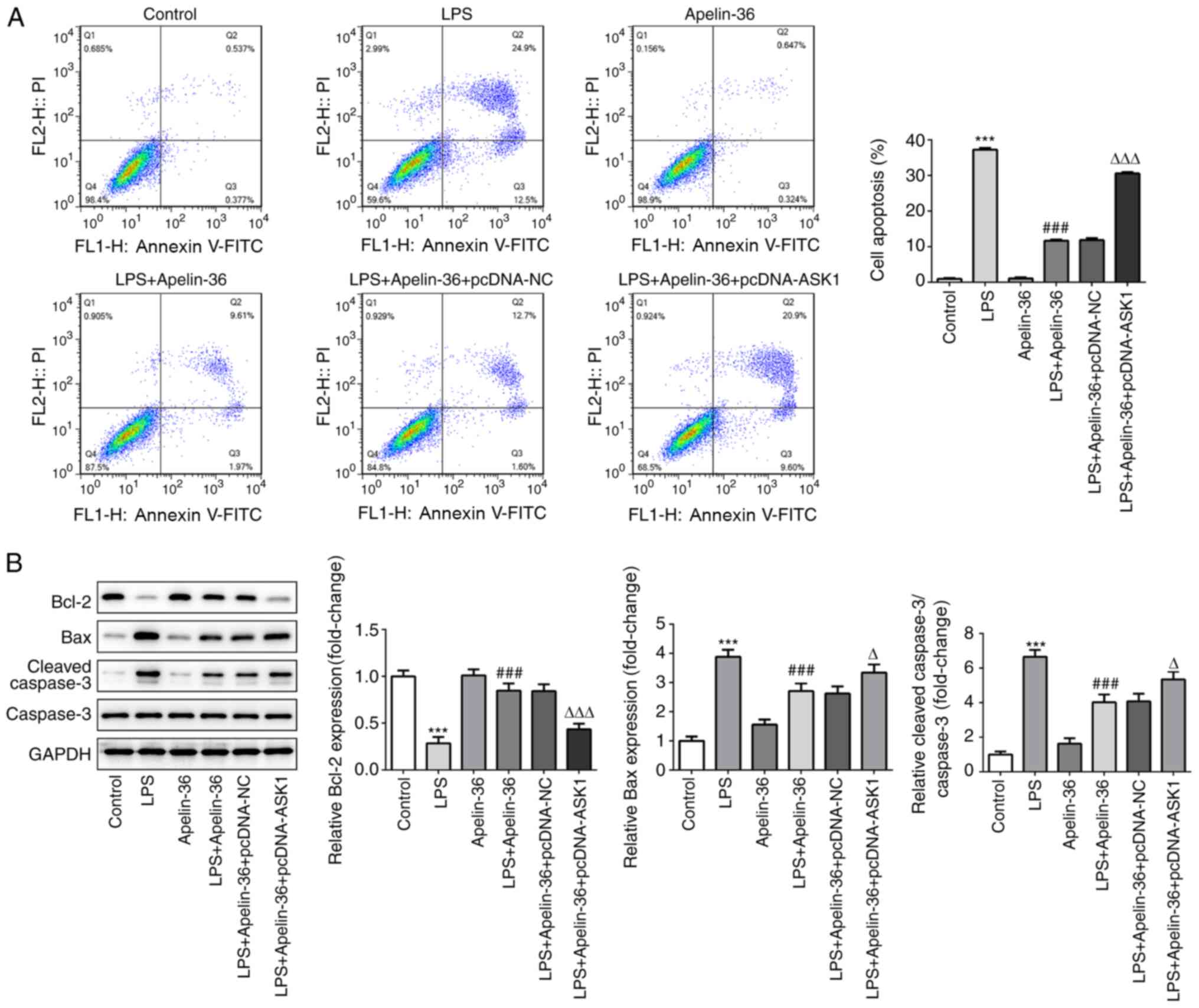
Similarly, as presented in Fig. 5A, LPS treatment resulted in an
increase in cell apoptosis, which was reversed by apelin-36
co-treatment, although this was increased by the overexpression of
ASK1. Furthermore, the expression levels of apoptosis-associated
proteins in the different groups were observed. It was found that
LPS led to a decrease in Bcl-2 expression, whereas the expression
levels of Bax and cleaved-caspase-3 were increased (Fig. 5B), indicating the occurrence of
apoptosis induced by LPS. Apelin-36 co-treatment led to a partial
recovery in the balance of these proteins, although overexpression
of ASK1 reversed the effects of apelin-36 on these
apoptosis-associated proteins. Taken together, the aforementioned
results suggested that overexpression of ASK1 was able to suppress
the inhibitory effects of apelin-36 on LPS-induced inflammation and
apoptosis in Beas-2B cells.
Discussion
The involvement of apelin proteins in various
diseases, including cardiovascular diseases, diabetes, obesity and
neurological disorders (11,12,18),
has been extensively reported. To the best of our knowledge, the
present study was the first to investigate the modulatory effects
of apelin-36 on LPS-induced ALI, and demonstrate that apelin-36
could protect against LPS-induced lung injury both in vivo
and in vitro. Additionally, the associated mechanism may
involve the inhibition of ASK1/MAPK signaling.
There is an increasing body of evidence that
supports the role of inflammation and apoptosis in the initiation
and progression of ALI. ALI is a prevailing inflammatory lung
disease characterized by the increased production of
pro-inflammatory mediators, infiltration of inflammatory cells, and
apoptosis of alveolar epithelial cells (19,20).
The role of endotoxins, especially LPS, has been well recognized in
the pathogenesis of ALI. LPS is a potent activator of Toll-like
receptor 4, triggering the NF-κB signaling pathway, thereby
producing pro-inflammatory molecules, such as IL-6, IL-1β and TNF-α
(15). Consistent with this
previous study, the results of the present study showed that LPS
could clearly cause pathological manifestations, including
intra-alveolar hemorrhage, inter-alveolar septum thickening,
inflammatory cell infiltration, pulmonary edema and cell apoptosis,
in both the lung tissues of rats and lung bronchial epithelial
cells. Therefore, controlling aberrant inflammation and apoptosis
is considered to be an effective strategy to attenuate lung
injury.
The APJ receptor and its endogenous ligand apelin
protein may be detected in endothelial cells, cardiomyocytes and
vascular smooth muscle cells, and their interaction helps to
maintain the normal function of the body; dysregulation of this
process is associated with the pathophysiological processes of
various diseases (7,12,21).
For example, apelin-36 can be produced and released by the vascular
endothelium, subsequently binding to neighboring APJ receptors. It
can activate endothelial nitric oxide synthase to stimulate nitric
oxide production, the release of which helps to maintain vascular
tone and regulate blood pressure stability (22). The role of apelin-36 in the
cardiovascular system has become a research hotspot. For instance,
apelin-36 can improve myocardial ischemia, and its mechanism of
action may be associated with the reduction of mitochondrial damage
of myocardial cells under hypoxic conditions and protection of the
energy supply of myocardial cells (18,23).
In brain injury, apelin-36 can significantly reduce the expression
levels of apoptosis markers, including caspase-3 and Bax, and it
may also inhibit activation of the phosphoinositide 3-kinase
signaling pathway, reduce cell apoptosis, and reduce cerebral
cortex damage in hypoxic-ischemic neonatal rats (24). In the present study, it was found
that apelin-36 was upregulated in the BALF of rats subjected to LPS
treatment. This finding was in accordance with a previous study, in
which elevated levels of apelin-36 in the plasma, BALF and lung
tissue were confirmed in rats with ARDS, indicating the important
pathophysiological function of apelin-36 in lung injury (5). To further confirm the role of
apelin-36, it was administered to normal rats or LPS-induced rats.
Apelin-36 administration was shown to have no obvious influence on
the structure and function of the lungs of normal rats, although it
could markedly alleviate the pathological alterations in lung
tissues, lung injury, pulmonary edema, generation of inflammatory
cytokines and cell apoptosis induced by LPS both in rats and in
Beas-2B cells. These results suggested that apelin-36 could protect
against LPS-induced lung injury without producing any notable side
effects.
ASK1 is a member of the MAPK family. MAPKs induce
cell apoptosis and activate downstream signaling pathways of MAPKs,
JNKs and p38 MAPKs. ASK1 is involved in numerous types of stress
response, including apoptosis, and ASK1 has been reported to be
associated with numerous diseases, including lung injury (25,26).
In the present study, it was shown that apelin-36 could
downregulate the LPS-induced expression levels of ASK1, p-p38,
p-JNK and p-ERK, these proteins all being members of the MAPK
family that can be activated in response to LPS. These results
indicated that the actions of apelin-36 may be dependent on the
inhibition of ASK1/MAPK signaling. To verify this hypothesis, ASK1
was overexpressed in cells that were subsequently co-treated with
LPS and apelin-36. The results demonstrated that the overexpression
of ASK1 could suppress the inhibitory effects of apelin-36 on
LPS-induced inflammation and apoptosis to a significant extent,
confirming that apelin-36 exerted anti-inflammatory and
anti-apoptotic effects in LPS-induced lung injury via inhibiting
ASK1. However, the overexpression of ASK1 did not completely
eliminate the effects of apelin-36, suggesting that other targets
are involved in the actions of apelin-36. The MAPK family consists
of a large number of members, including p-38, JNK and ERK, and
whether apelin-36 is able to also directly target these proteins,
or whether the targeting of ASK1 thereby affects p-38, JNK and ERK
downstream in the signaling pathway, remains to be elucidated.
Taken together, the present study demonstrated the
potential pharmacological effects of apelin-36 in ALI, and the
possible underlying mechanism. The results highlighted that
apelin-36 may be a potential novel candidate for the treatment of
injurious inflammatory responses and apoptosis in ALI via targeting
ASK1, thereby inhibiting ASK1/MAPK signaling. However, further
studies are needed to investigate the specific relationship between
apelin-36 and ASK1.
Acknowledgements
Not applicable.
Funding
This study was funded by the National Natural
Science Foundation of China (grant no. 81860056), the Key
Scientific Research Project of Health Department of Ningxia Hui
Autonomous Region (grant no. 2019-NW-018), and The Scientific
Research Project of Ningxia Hui Autonomous Region People's Hospital
(grant no. 201918).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
QH, YW and DL conceived and designed the study. QH,
YW, HY, JW and JZ acquired and analyzed the data. DL prepared the
draft of the manuscript, including the figures. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
All experimental procedures were approved by the
Ethical Committee on Animal Research at the People's Hospital of
Ningxia Hui Autonomous Region (Yinchuan, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Butt Y, Kurdowska A and Allen TC: Acute
lung injury: A clinical and molecular review. Arch Pathol Lab Med.
140:345–350. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thompson BT, Chambers RC and Liu KD: Acute
respiratory distress syndrome. N Engl J Med. 377:562–572. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hughes KT and Beasley MB: Pulmonary
manifestations of acute lung injury: More than just diffuse
alveolar damage. Arch Pathol Lab Med. 141:916–922. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Monsel A, Zhu YG, Gudapati V, Lim H and
Lee JW: Mesenchymal stem cell derived secretome and extracellular
vesicles for acute lung injury and other inflammatory lung
diseases. Expert Opin Biol Ther. 16:859–871. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fan XF, Xue F, Zhang YQ, Xing XP, Liu H,
Mao SZ, Kong XX, Gao YQ, Liu SF and Gong YS: The Apelin-APJ axis is
an endogenous counterinjury mechanism in experimental acute lung
injury. Chest. 147:969–978. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vinel C, Lukjanenko L, Batut A,
Deleruyelle S, Pradère JP, Le Gonidec S, Dortignac A, Geoffre N,
Pereira O, Karaz S, et al: The exerkine apelin reverses
age-associated sarcopenia. Nat Med. 24:1360–1371. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cheng J, Luo X, Huang Z and Chen L:
Apelin/APJ system: A potential therapeutic target for endothelial
dysfunction-related diseases. J Cell Physiol. 234:12149–12160.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhu J, Dou S, Jiang Y, Bai B, Chen J, Wang
C and Cheng B: Apelin-36 exerts the cytoprotective effect against
MPP+-induced cytotoxicity in SH-SY5Y cells through
PI3K/Akt/mTOR autophagy pathway. Life Sci. 224:95–108. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhu J, Dou S, Wang C, Jiang Y, Wang C and
Cheng B: Apelin-36 mitigates MPTP/MPP+-induced
neurotoxicity: Involvement of α-synuclein and endoplasmic reticulum
stress. Brain Res. 1721:1463342019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Folino A, Montarolo PG, Samaja M and
Rastaldo R: Effects of apelin on the cardiovascular system. Heart
Fail Rev. 20:505–518. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Castan-Laurell I, Dray C, Attané C, Duparc
T, Knauf C and Valet P: Apelin, diabetes, and obesity. Endocrine.
40:1–9. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Antushevich H and Wójcik M: Review: Apelin
in disease. Clin Chim Acta. 483:241–248. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhu J, Gao W, Shan X, Wang C, Wang H, Shao
Z, Dou S, Jiang Y, Wang C and Cheng B: Apelin-36 mediates
neuroprotective effects by regulating oxidative stress, autophagy
and apoptosis in MPTP-induced Parkinson's disease model mice. Brain
Res. 1726:1464932020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ko IG, Hwang JJ, Chang BS, Kim SH, Jin JJ,
Hwang L, Kim CJ and Choi CW: Polydeoxyribonucleotide ameliorates
lipopolysaccharide-induced acute lung injury via modulation of the
MAPK/NF-kappaB signaling pathway in rats. Int Immunopharmacol.
83:1064442020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jin Y, Qian J, Ju X, Bao X, Li L, Zheng S,
Chen X, Xiao Z, Chen X, Zhu W, et al: Osthole protects against
acute lung injury by suppressing NF-κB-Dependent Inflammation.
Mediators Inflamm. 2018:49345922018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhou J, Hu R, Jing S, Xue X and Tang W:
Activated protein C inhibits lung injury induced by LPS via
downregulating MAPK signaling. Exp Ther Med. 16:931–936.
2018.PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yu XH, Tang ZB, Liu LJ, Qian H, Tang SL,
Zhang DW, Tian GP and Tang CK: Apelin and its receptor APJ in
cardiovascular diseases. Clin Chim Acta. 428:1–8. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu ZF, Zheng D, Fan GC, Peng T and Su L:
Heat stress prevents lipopolysaccharide-induced apoptosis in
pulmonary microvascular endothelial cells by blocking calpain/p38
MAPK signalling. Apoptosis. 21:896–904. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cox R Jr, Phillips O, Fukumoto J, Fukumoto
I, Parthasarathy PT, Arias S, Cho Y, Lockey RF and Kolliputi N:
Enhanced resolution of hyperoxic acute lung injury as a result of
aspirin triggered resolvin D1 treatment. Am J Respir Cell Mol Biol.
53:422–435. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hu H, He L, Li L and Chen L: Apelin/APJ
system as a therapeutic target in diabetes and its complications.
Mol Genet Metab. 119:20–27. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mughal A and O'Rourke ST: Vascular effects
of apelin: Mechanisms and therapeutic potential. Pharmacol Ther.
190:139–147. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pisarenko O, Shulzhenko V, Studneva I,
Pelogeykina Y, Timoshin A, Anesia R, Valet P, Parini A and
Kunduzova O: Structural apelin analogues: Mitochondrial ROS
inhibition and cardiometabolic protection in myocardial ischaemia
reperfusion injury. Br J Pharmacol. 172:2933–2945. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gu Q, Zhai L, Feng X, Chen J, Miao Z, Ren
L, Qian X, Yu J, Li Y, Xu X and Liu CF: Apelin-36, a potent
peptide, protects against ischemic brain injury by activating the
PI3K/Akt pathway. Neurochem Int. 63:535–540. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Noguchi T, Ishii K, Fukutomi H, Naguro I,
Matsuzawa A, Takeda K and Ichijo H: Requirement of reactive oxygen
species-dependent activation of ASK1-p38 MAPK pathway for
extracellular ATP-induced apoptosis in macrophage. J Biol Chem.
283:7657–7665. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Han J, Lv W, Sheng H, Wang Y, Cao L, Huang
S, Zhu L and Hu J: Ecliptasaponin A induces apoptosis through the
activation of ASK1/JNK pathway and autophagy in human lung cancer
cells. Ann Transl Med. 7:5392019. View Article : Google Scholar : PubMed/NCBI
|