Introduction
Non-small cell lung cancer (NSCLC), which represents
~80% of all lung cancers, is the most common type of cancer and the
leading cause of cancer-related deaths worldwide (1,2).
Epidermal growth factor receptor (EGFR)-activating mutations have
been identified in 15–40% of patients with NSCLC and have emerged
as an effective therapeutic target (3). In fact, the first-generation EGFR
tyrosine kinase inhibitors (TKIs), such as gefitinib and erlotinib,
were discovered and recommended as a standard therapy for patients
with advanced NSCLC and EGFR-sensitive mutations (4–6).
However, despite the initial significant response to the treatment
with TKIs, acquired resistance develops in the majority of patients
with NSCLC within a median time of 10–16 months, which limits the
long-term efficacy of TKIs (7). The
acquisition of a secondary T790M mutation in exon 20 of EGFR, which
occurs in ~60% of resistant cases, is the most commonly
characterized mechanism underlying the acquired resistance to the
first-generation of EGFR TKIs (8).
To overcome the resistance induced by the T790M mutation,
osimertinib, as a third-generation EGFR TKI, was developed and
demonstrated a beneficial efficacy in patients with NSCLC carrying
the resistant T790M mutation of EGFR (9,10).
Unfortunately, similar to other EGFR TKIs, the efficacy of
osimertinib is limited due to acquired resistance arising from
multiple mechanisms, including the EGFR C797S mutation and
activation of parallel signaling pathways, amongst others (11,12).
Therefore, there remains an urgent requirement to discover the
underlying mechanisms of resistance and investigate novel
therapeutic strategies for overcoming the recurring resistance to
osimertinib.
Recently, increasing evidence has revealed that
NSCLC cells develop resistance to EGFR TKIs via inhibition of
cellular apoptosis by modulating apoptotic regulators belonging to
the Bcl-2 family (13,14). Among these, Bcl-2 and Bcl-xL, which
are EGFR/AKT downstream antiapoptotic signaling proteins, have been
identified to be responsible for the drug resistance in numerous
types of tumor, such as small-cell lung (15), breast (16) and ovarian cancers (17).
The present study aimed to determine whether the
dysregulation of Bcl-2 and Bcl-xL was involved in the resistance of
NSCLC cells to osimertinib through establishing an
osimertinib-resistant NSCLC cell sub-line, HCC-827/OR. The results
revealed that the expression levels of Bcl-2 and Bcl-xL were
upregulated in HCC-827/OR cells. Moreover, ABT-263, a small
molecular inhibitor of Bcl-2 and Bcl-xL, was discovered to exert
synergetic antitumor effects with osimertinib in HCC-827/OR cells
in vitro and in vivo. Overall, these findings may provide a
potential therapeutic strategy to overcome the resistance of NSCLC
to osimertinib.
Materials and methods
Cell lines and reagents
The NSCLC cell line HCC827 was obtained from The
Cell Bank of Type Culture Collection of the Chinese Academy of
Sciences. Cells were cultured at 60–90% confluence in RPMI-1640
medium (Thermo Fisher Scientific, Inc.), supplemented with 10% FBS
(Thermo Fisher Scientific, Inc.) and 1% penicillin-streptomycin,
and maintained at 37°C in a humidified incubator with 5%
CO2. Osimertinib-resistant HCC827 cell line (HCC-827/OR)
was established beginning with exposing HCC827 parent cells to a
culture medium with 5 nM osimertinib. When the cells were resistant
to 5 nM osimertinib, the concentration was increased to 10 nM.
During over a period of 6 months, the cells were stably grown in a
culture medium containing 100 nM osimertinib. Osimertinib and
ABT-263 were purchased from MedChemExpress and Sigma-Aldrich (Merck
KGaA), respectively. Both drugs were dissolved in DMSO and stored
at −20°C. MTT reagent was purchased from Sigma-Aldrich (Merck
KGaA).
Cell viability assay
The inhibitory effects of osimertinib or ABT-263 on
cell viability were evaluated using an MTT assay. Briefly, cells
were seeded in 96-well plates (5×103 cells per well) and
treated with osimertinib, ABT-263 and osimertinib and ABT-263
together at 1, 2.5, 5 or 10 µM, respectively, or DMSO as the
negative control for 24 h. Then, the medium containing the drugs or
DMSO was replaced with fresh medium containing 0.5 mg/ml MTT and
incubated for 4 h at 37°C. Following the incubation, the
MTT-containing medium was discarded, and 150 µl DMSO was added to
each well to dissolve the formazan and shook for 15 min in the
dark. The absorbance at 570 nm was determined using a microplate
reader (Synergy H4; BioTek Instruments, Inc.).
Flow cytometric analysis of
apoptosis
Cellular apoptosis was analyzed using an Annexin
V-FITC/PI Apoptosis Detection kit (BD Biosciences), according to
the manufacturer's protocol. Briefly, cells were incubated with the
indicated concentrations of osimertinib, ABT-263, osimertinib and
ABT-263, or DMSO as the negative control for 24 h. The cells were
subsequently trypsinized, washed and resuspended in 200 µl binding
buffer, then stained with 5 µl PE and FITC for 15 min in the dark
at room temperature. Finally, the stained cells were subjected to
analysis by flow cytometry (Cytomics FC 500 MPL; Beckman Coulter,
Inc.). The obtained data were analyzed by CytExpert v2.3 (Beckman
Coulter, Inc.). The early apoptosis was evaluated based on the
percentage of cells with Annexin V+/PI−,
while the late apoptosis was that of cells with Annexin
V+/PI+. The results are presented as the mean
± SD of three independent experiments.
Small interfering RNA (siRNA)-mediated
knockdown of gene expression
Bcl-2, Bcl-xL and Bcl-2-like protein 11 (Bim) siRNA
or control siRNA were purchased from Shanghai GenePharma Co., Ltd.,
and transfected into cells using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. Briefly, cells were seeded in the 6-well
plates (1×105 cells/ml) with growth medium without
antibiotics 1 day before transfection, such that they will be
30–50% confluence at the time of transfection. For each
transfection sample, the transfection complex was prepared by
dilution of 100 pmol siRNA oligomer and 5 µl Lipofectamine 2000
with 250 µl Opti-MEM medium (Thermo Fisher Scientific, Inc.)
without serum, which was added to each well. After incubation at
37°C in a CO2 incubator for 8–10 h, the cultured medium
containing siRNA and transection agent was replaced with fresh
medium. To confirm the downregulation of the expression levels of
the target proteins, cells were collected and lysed 48 h after
transfection, and western blotting or reverse
transcription-quantitative PCR (RT-qPCR) was used to confirm the
transfection efficiency. The following siRNA sequences (Thermo
Fisher Scientific, Inc.) were used: Bcl-2 sense,
5′-GGAUGACUGAGUACCUGAAdTdT-3′ and antisense,
3′-dTdTCCUACUGACUCAUGGACUU-5′ (14); Bcl-xL sense,
5′-GGUAUUGGUGAGUCGGAUCdTdT-3′ and antisense,
3′-dTdTCCAUAACCACUCAGCCUAG-5′ (18); Bim sense,
5′-AAUUGUCUACCUUCUCGGUdTdT-3′ and antisense,
3′-dTdTUUAACAGAUGGAAGAGCCA (19);
and control siRNA sense, 5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense,
5′-ACGUGACACGUUCGGAGAATT-3′.
Western blotting
Cells were harvested and lysed with RIPA lysis
buffer (Beyotime Institute of Biotechnology). Total protein
concentration was determined using a BCA protein assay kit
(Beyotime Institute of Biotechnology) and 30 µg protein/lane was
separated by 12% SDS-PAGE. The separated proteins were transferred
onto PVDF membranes (EMD Millipore) and blocked by 5% skimmed milk
for 1 h at room temperature. The membranes were then incubated with
primary antibodies (diluted 1:1,000 with 1% skimmed milk for Bcl-2,
Bcl-xL, BimEL, cytochrome c, cleaved- and full length PARP-1 and
caspase-3, and diluted 1:10,000 with 1% skimmed milk for β-actin)
overnight at 4°C, followed by incubation with a horseradish
peroxidase (HRP)-conjugated secondary antibody (1:5,000 diluted by
TBST) for 1 h at room temperature. Protein bands were visualized
using an ECL reagent (EMD Millipore). β-actin was used as an
endogenous loading control. ImageJ (version 1.48; National
Institutes of Health) was used to analyze the grey level of the
bands. Representative data from ≥3 independent experiments are
presented. Primary antibodies against Bcl-2 (cat. no. 12789-1-AP),
Bcl-xL (cat. no. 26967-1-AP), BimEL (cat. no. 22037-1-AP),
cytochrome c (cat. no. 10993-1-AP) were purchased from ProteinTech
Group, Inc. Primary antibody against cleaved poly(ADP-ribose)
polymerase-1 (PARP-1; cat. no. sc-56196), PARP-1 (cat. no.
sc-74470) and caspase-3 (cat. no. sc-7272) were purchased from
Santa Cruz Biotechnology, Inc. Primary antibodies targeting β-actin
(cat. no. 4970) and cleaved caspase-3 (cat. no. 9664) and
HRP-conjugated secondary antibody (cat. nos. 7074 and 7076) were
purchased from Cell Signaling Technology, Inc.
Colony formation assay
A total of 1×103 cells/well were seeded
into 6-well plates at a single cell density and treated with
osimertinib or/and ABT-263 or DMSO (negative control) at the
indicated concentrations for 48 h. The drug-containing medium was
replaced with fresh medium and cells were cultured for 2 weeks.
Then the cells were fixed with methanol for 15 min at room
temperature, followed by staining with gentian violet (Beijing
Solarbio Science & Technology Co., Ltd.) for 1 h at room
temperature. The colonies with >50 cells were counted under an
inverted microscope (IX71; Olympus Corporation; magnification,
×10).
Xenograft study
The animal experimental protocol was approved by the
Animal Ethics Committee of Fudan University Shanghai Medical School
(Shanghai, China). A total of 24 female BALB/c nude mice (age, 6–7
weeks; weight 20±2 g) were purchased from Shanghai SLAC Laboratory
Animal Co., Ltd., and housed in a specific pathogen-free
environment under a 12-h light-dark cycle at 23±1°C and 50±5%
humidity atmosphere, with free access to standard food and water. A
total of 5×106 HCC827/OR cells/mouse resuspended in 200
µl PBS were subcutaneously implanted into the right flank of nude
mice. The tumor volume was measured using a digital caliper and
calculated using the following formula: Volume = L × S2
× 0.52, where L represents the longest tumor diameter, and S is the
shortest tumor diameter. The tumor volume and mouse body weight
were recorded every 3 days. After the tumors reached a mean volume
of 100 mm3, the animals were randomly assigned into one
of four groups (n=6) and treated with vehicle control, 5 mg/kg
osimertinib, 100 mg/kg ABT-263 alone or in combination; both agents
were administered by oral gavage daily. At the end of experiment
(32 days after implantation of tumor cells), the mice were
sacrificed by carbon dioxide asphyxiation with a flow rate of 15%
chamber volume/min and the tumors were harvested and weighed.
Immunohistochemistry
The xenograft tumor tissues were immersed in 4%
paraformaldehyde for >24 h at room temperature, and transferred
to 70% ethanol. Individual lobes of tumor tissues material were
placed in processing cassettes, dehydrated through a serial alcohol
gradient, and embedded in paraffin wax blocks. Before
immunostaining, 5 µm thick xenograft tumor tissue sections were
dewaxed in xylene, rehydrated through decreasing concentrations of
ethanol and washed in PBS. Then, antigens were unmasked by
microwaving sections in 10 mmol/l citrate buffer, pH 6.0 (15 min),
and blocked with 5% goat serum (cat. no. SL038; Beijing Solarbio
Science & Technology Co., Ltd.) at room temperature for 15 min.
Then, immunostaining was undertaken using the avidinbiotinylated
enzyme complex method with antibodies against Bcl-2 (1:50 dilution,
cat. no. 12789-1-AP; ProteinTech Group, Inc.), Bcl-xL (1:50
dilution, cat. no. 26967-1-AP; ProteinTech Group, Inc.), BimEL
(1:50 dilution, cat. no. 22037-1-AP; ProteinTech Group, Inc.)
overnight at 4°C, and equivalent concentrations of polyclonal
nonimmune IgG controls. After incubation with the biotin-conjugated
secondary antibody (cat. no. GK600705, Gene Tech, Inc.) for 2 h at
room temperature and subsequently with streptavidin solution, color
development was performed using 3,3-diaminobenzidine
tetrahydrochloride (cat. no. P0203; Beyotime Institute of
Biotechnology) as a chromogen at room temperature for 1 min.
Sections were counterstained using Gill-2 hematoxylin (cat. no.
C0107, Beyotime Institute of Biotechnology) at room temperature for
30 sec. After staining, sections were dehydrated through increasing
concentrations of ethanol and xylene. The sections were observed
and analyzed under a routine light microscope (BX43; Olympus
Corporation; magnification, ×100 and ×200).
Statistical analysis
Statistical analysis was performed using GraphPad
Prism software (v5.0; GraphPad Software, Inc.). The IC50
values were fitted using a non-linear regression model with a
sigmoidal dose response. Results are representative from ≥3
independent experiments and presented as the mean ± SD. Comparisons
between the control and treatment groups were determined using a
paired Student's t-test or a one-way ANOVA, followed by Tukey's
multiple comparison test. P<0.05 was considered to indicate a
statistically significant difference. The combination index was
calculated using CompuSyn software (v1.0.1; ComboSyn, Inc.) by Chou
(20).
Results
Establishment of osimertinib-resistant
NSCLC cells
To investigate the molecular mechanism underlying
the resistance of NSCLC cells to osimertinib, osimertinib-resistant
HCC827 cells (HCC-827/OR) were established through the
dosage-escalation of osimertinib over 6 months. The effects of
osimertinib on cell viability were analyzed using an MTT assay in
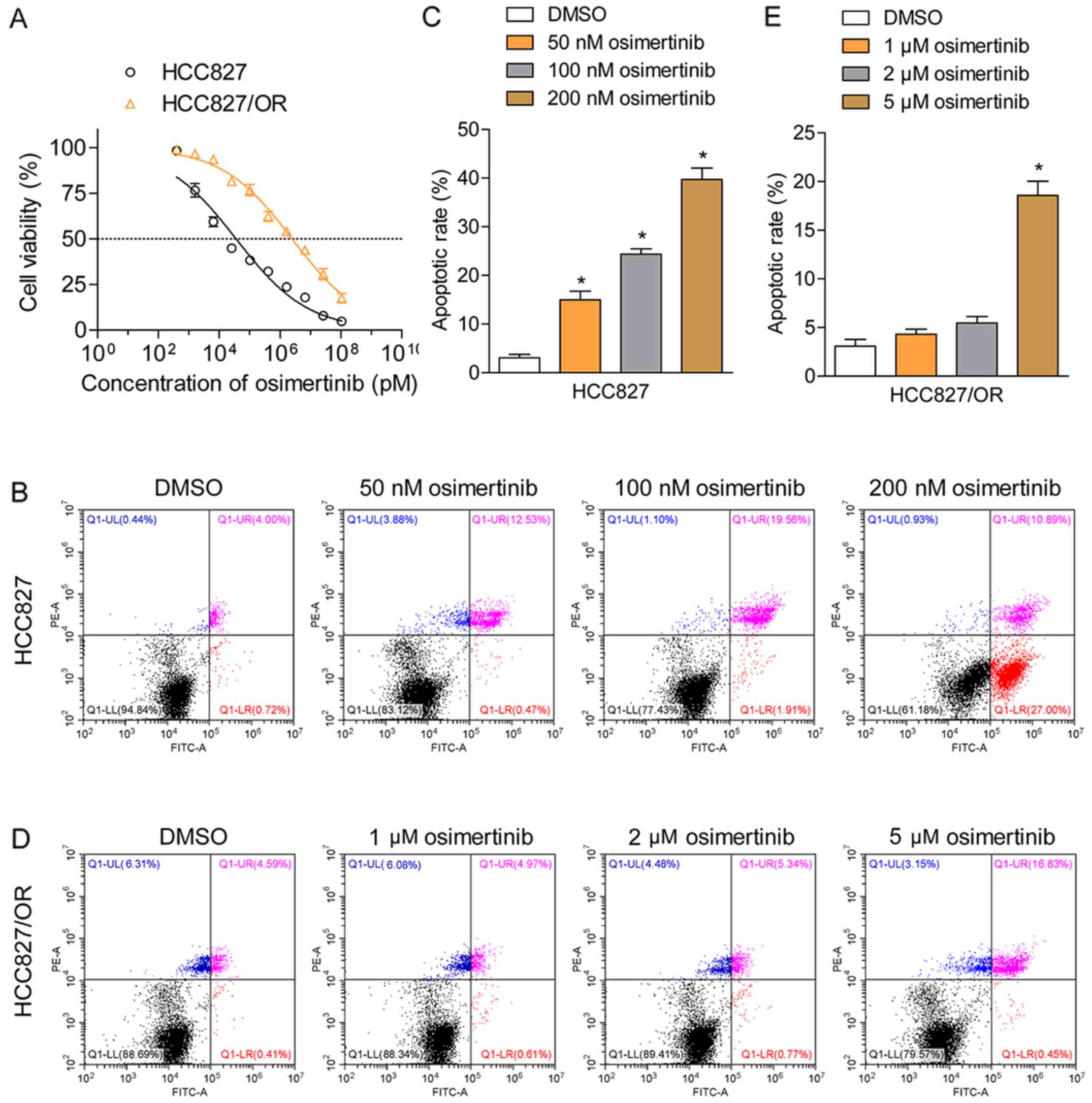
HCC827 and HCC-827/OR cells. HCC827 cells were sensitive to
osimertinib, demonstrating an IC50 value of 35.2 nM,
whereas HCC827/OR cells were resistant to osimertinib, exhibiting
IC50 values of 2.45 µM, ~70-fold larger than sensitive
cells (Fig. 1A). The rate of
cellular apoptosis induced by osimertinib was determined using
Annexin V/PI double staining followed by flow cytometric analysis;
the results revealed that osimertinib effectively induced the
apoptosis of HCC827 cells in a dose-dependent manner (Fig. 1B and C). Upon treatment with 50, 100
or 200 nM, the total proportion of apoptotic cells (Annexin
V-positive staining) increased from 4.7 to 13.0, 21.5 and 37.8%,
respectively. In comparison, the treatment of HCC827/OR cells with
1 or 2 µM osimertinib failed to induce apoptosis (Fig. 1D and E). When HCC827/OR cells were
treated with 5 µM osimertinib, the total proportion of apoptotic
cells (Annexin V-positive staining) was only 18.3%, indicating the
resistance of HCC827/OR cells to osimertinib-induced apoptosis.
Bcl-2 and Bcl-xL mediate the
resistance of HCC827/OR cells to osimertinib
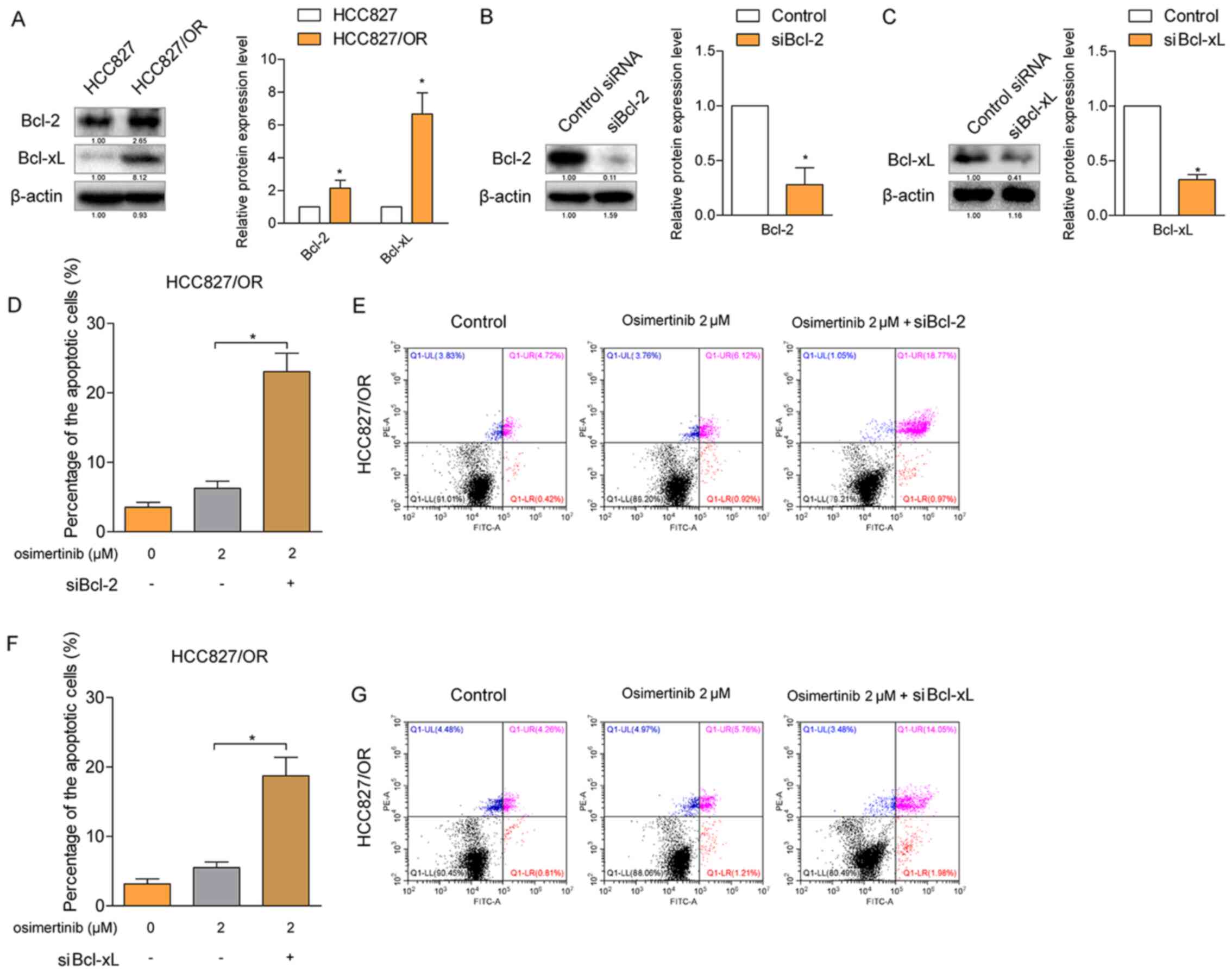
Since the Bcl-2 protein family is a major regulator
of cell apoptosis, the present study investigated whether Bcl-2 and
Bcl-xL were involved in the resistance of HCC827/OR cells to
osimertinib-induced apoptosis. The results from the western
blotting analysis revealed that both the expression levels of Bcl-2
and Bcl-xL were significantly upregulated in HCC827/OR cells
(Fig. 2A) compared with HCC827
cells. Subsequently, the expression levels of Bcl-2 and Bcl-xL were
knocked down by specific siRNAs in HCC827/OR cells (Fig. 2B and C), respectively, and the rate
of osimertinib-induced cellular apoptosis was determined. The
osimertinib-induced rate of cellular apoptosis was significantly
increased following the knockdown of either Bcl-2 or Bcl-xL
(Fig. 2D-G), indicating that both
Bcl-2 and Bcl-xL contributed to the resistance of HCC827/OR cells
to osimertinib-induced apoptosis.
Bcl-2/Bcl-xL inhibitor ABT-263 acts in
synergy with osimertinib in NSCLS cells
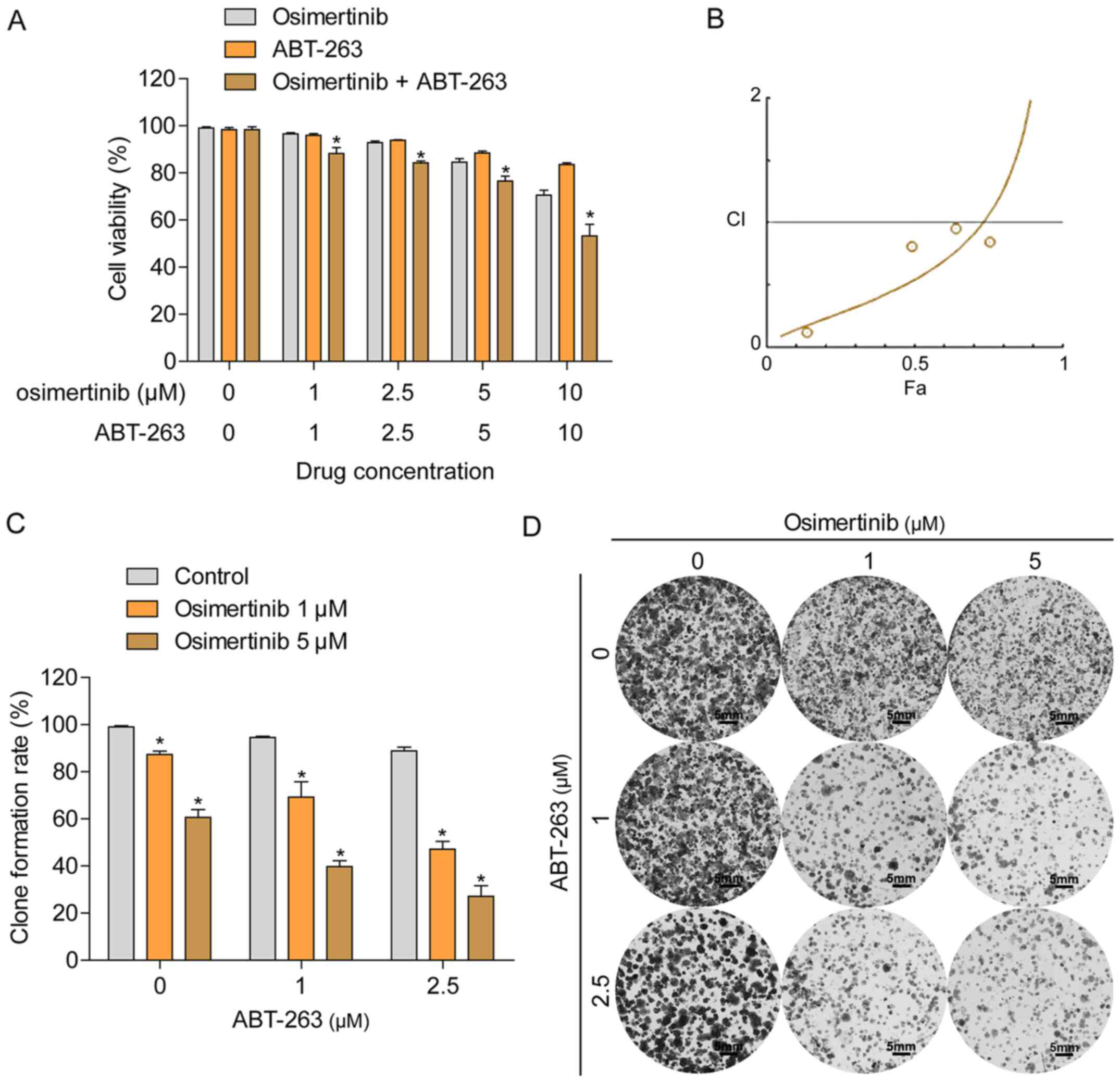
Since the overexpression of Bcl-2 and Bcl-xL were
demonstrated to contribute to the resistance of HCC827/OR cells to
osimertinib, the present study subsequently investigated whether
ABT-263, an inhibitor of Bcl-2 and Bcl-xL, improved the sensitivity
of HCC827/OR cells to osimertinib treatment. HCC827/OR cells were
treated with a combination of osimertinib and ABT-263 or each
individual agent, and cell viability was determined using an MTT
assay. The combined treatment with osimertinib and ABT-263 enhanced
the inhibition of cell viability compared with the treatment with
each single agent alone (Fig. 3A).
The combination index (CI) was calculated using the Chou-Talalay
method (Fig. 3B), indicating the
synergistic effect (CI <1) of osimertinib and ABT-263 in
osimertinib-resistant NSCLC cells. In addition, the synergistic
effect between osimertinib and ABT-263 in cells was further
confirmed using a colony formation assay; the concurrent treatment
of HCC827/OR cells with osimertinib and ABT-263 resulted in the
enhanced inhibition of colony formation compared with the treatment
of cells with either agent alone (Fig.
3C and D).
ABT-263 enhances the rate of cellular
apoptosis induced by osimertinib
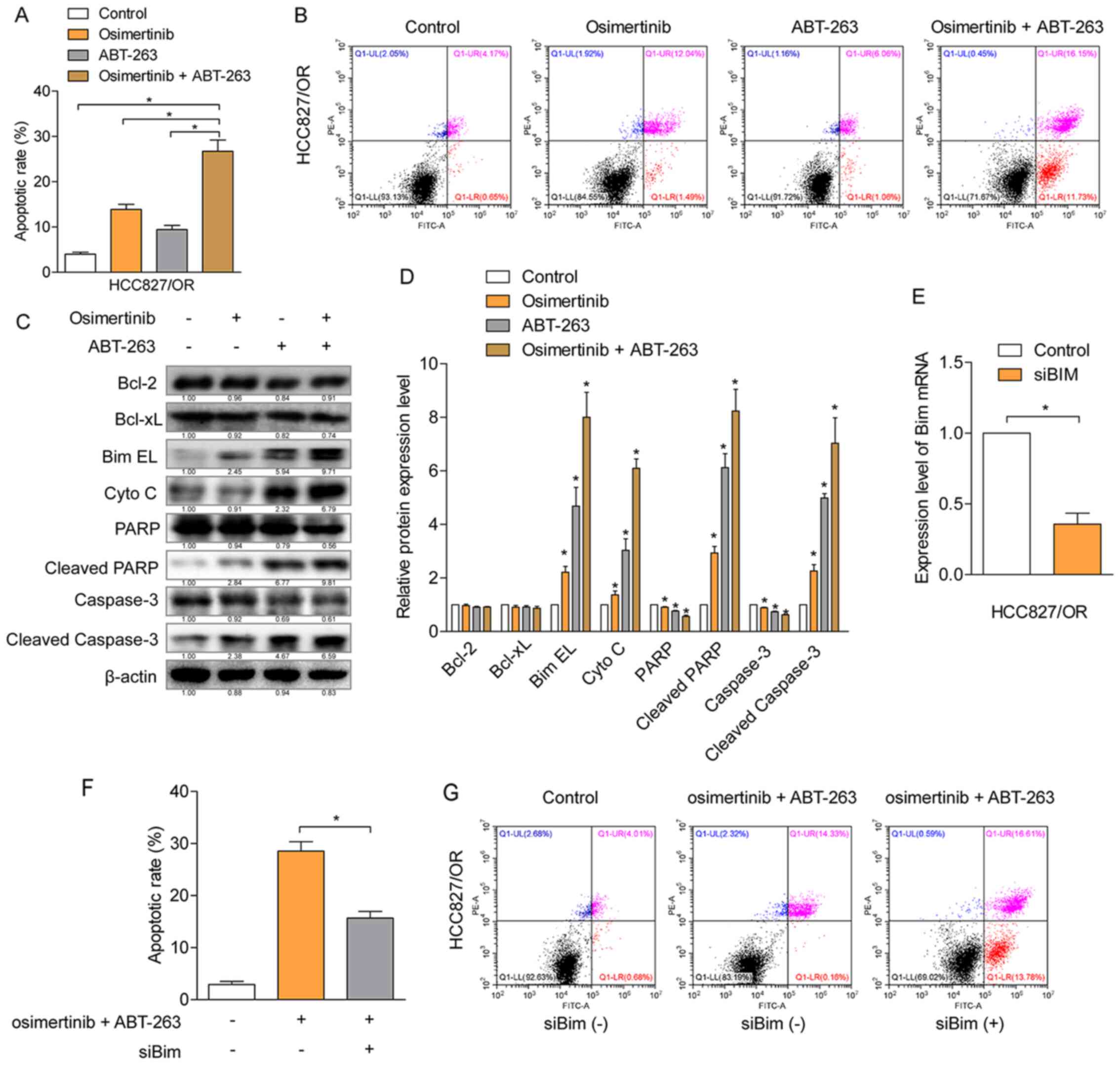
Cellular apoptosis induced by the combination of
osimertinib and ABT-263 or each individual agent were determined
using Annexin V-PI double staining. The total number of apoptotic
cells (Annexin V-positive staining) induced by the combined
treatment of osimertinib and ABT-263 was significantly increased
compared with the number of apoptotic cells induced by each
individual agent alone (Fig. 4A and
B). To further determine the mechanism of apoptosis induced by
osimertinib with ABT-263, protein expression levels were
investigated using western blotting in HCC827/OR cells (Fig. 4C and D). The results revealed that,
compared with the single agent treatment group, the combined
treatment of osimertinib and ABT-263 significantly upregulated the
expression levels of BimEL and cytochrome c, in addition to
promoting the cleavage of caspase-3 and PARP-1. However, the
expression levels of Bcl-2 and Bcl-xL were not significantly
affected. To further verify whether the function of BimEL was
involved in the induction of cellular apoptosis, BimEL was knocked
down using siRNA and the knockdown efficiency was confirmed by
RT-qPCR (Fig. 4E). The knockdown of
BimEL partially attenuated the rate of cellular apoptosis induced
by the combined treatment of osimertinib and ABT-263 (Fig. 4F and G), suggesting the contribution
of BimEL to the induction of cellular apoptosis. These results
indicated that the combined treatment of osimertinib with ABT-263
may enhance the rate of cellular apoptosis through the
mitochondria-initiated apoptotic pathway.
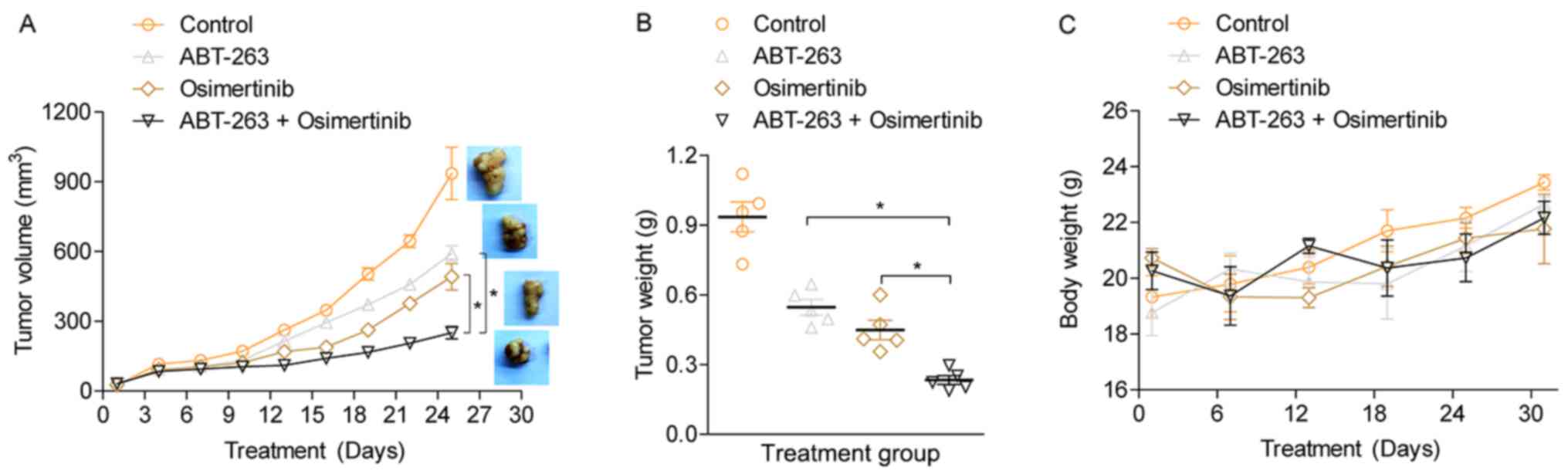
ABT-263 enhances antitumor effects of
osimertinib in vivo
To investigate the antitumor effects of osimertinib
and ABT-263 in vivo, a nude mouse subcutaneous tumor model was
established using HCC827/OR cells. Treatment of the tumor model
mice with either osimertinib or ABT-263 alone led to a slight
inhibition of tumor growth compared with the vehicle treated
control (Fig. 5A). However, the
combination of osimertinib with ABT-263 significantly inhibited
tumor growth compared with the single agent treatment groups. At
the end of the experiment, the tumors were harvested and weighed.
The combined treatment of osimertinib with ABT-263 significantly
suppressed tumor growth compared with the single-drug treatment
(Fig. 5B), further indicating the
potential synergistic antitumor effect of osimertinib and ABT-263
combined. In addition, it was observed that the body weight of
osimertinib-treated mice was slightly less compared with ABT-263
treated or vehicle treated mice. However, the difference of body
weight among four groups was not significant, suggesting that
treatment with the individual agents and the combined use of both
agents had slight toxic side effects on the mice (Fig. 5C). It was hypothesized that the
osimertinib, both as single and combined agents, may possess side
effect on gastrointestinal tract and affect mice's food intake. The
expression levels of Bcl-2, Bcl-xL and BimEL were detected in the
xenograft tumor tissues using immunohistochemistry (IHC) (Fig. S1). Consistent with the in vitro
experiments, the results showed that combined treatment using
osimertinib and ABT-263 notably increased the expression of BimEL.
However, the expression levels of Bcl-2 and Bcl-xL were but not
notably altered. This result indicated that ABT-263 enhanced
osimertinib-induced cell apoptosis by elevating BimEL
expression.
Discussion
EGFR TKIs have provided a significant benefit for
patients with NSCLC harboring EGFR mutations (21); however, the clinical efficacy of
TKIs is hindered by acquired resistance (22). Osimertinib, as a third generation
TKI, was discovered to overcome resistance to first and second
generation TKIs, such as gefitinib, erlotinib and afatinib, by
targeting the T790M mutation of EGFR (23–24).
However, similar to other TKIs, NSCLC cells eventually develop
resistance to osimertinib (25). To
identify the underlying mechanism, an osimertinib-resistant cell
line HCC827/OR was established through the dosage-escalation of
osimertinib over 6 months. Compared with the osimertinib-sensitive
HCC827 parent cells, HCC827/OR cells were insensitive to the growth
inhibition and apoptosis-induced effects of osimertinib,
demonstrating IC50 values ~70-fold higher than HCC827
cells.
The inhibition of apoptosis has been identified as
one of the major mechanisms involved in the resistance of NSCLC
cells to osimertinib (13).
Apoptosis is a mechanism of programmed cell death, which is
triggered by at least two broad signaling pathways: The extrinsic
pathway and the intrinsic pathway. The extrinsic pathway is
activated by the interaction between specific ligands and death
receptors that belong to the tumor necrosis factor superfamily,
subsequently initiating the activation of the caspases cascade to
produce proteolysis of essential cell proteins. The intrinsic
pathway, also known as the mitochondrial apoptotic pathway, is
initiated by the release of cytochrome c and other proteins from
the mitochondria into the cytosol, inducing the formation of the
apoptosome complex and the protease hydrolysis cascade. Bcl-2
family proteins are well-known important regulators of the
mitochondrial apoptotic pathway, through keeping the balance
between pro- and antiapoptotic proteins. The upregulation of
antiapoptotic Bcl-2 family members, such as Bcl-2, Bcl-xL and
induced myeloid leukemia cell differentiation protein Mcl-1
(Mcl-1), have been identified in a number of solid tumor and
leukemia cell lines (26–29). In the present study, Bcl-2 and
Bcl-xL expression levels were upregulated in the
osimertinib-resistant cell line compared with the sensitive parent
cell line. In addition, the downregulation of Bcl-2 or Bcl-xL
expression levels by siRNA reversed the drug resistance to
osimertinib in HCC8278/OR cells. The downregulation of Bcl-2 was
also discovered to overcome the apoptotic inhibition-induced
resistance to first generation EGFR TKI gefitinib, as previously
reported (14,30), indicating the essential role of
Bcl-2 in the resistance of NSCLC cells to EGFR TKIs.
ABT-263 is an orally bioavailable Bcl-2 family
inhibitor with a high affinity for Bcl-2 and Bcl-xL (31). Based on the reported role of Bcl-2
and Bcl-xL in the resistance to osimertinib, it was hypothesized
that the combination of ABT-263 and osimertinib may overcome the
resistance of NSCLC cells to osimertinib. The results of the
present study demonstrated that ABT-263 re-sensitized HCC-827/OR
cells to osimertinib; the two agents exerted synergistic effects on
the inhibition of cell viability and colony formation ability.
Moreover, ABT-263 enhanced osimertinib-induced cell apoptosis by
elevating BimEL levels and releasing cytochrome c, thereby
resulting in the cleavage of caspase-3 and PARP-1 (32,33).
Bim is another proapoptotic Bcl-2 family member that regulates cell
apoptosis through binding to antiapoptotic members, such as Bcl-2
and Mcl-1, and inhibiting their activation, and/or directly
activating proapoptoic members, including Bax and Bcl-2 homologous
antagonist/killer (34). In the
present study, the downregulation of BimEL expression levels by
siRNA confirmed the contribution of this protein to the efficacy of
osimertinib- and ABT-263-induced apoptosis. Finally, ABT-263 was
observed to overcome the resistance of osimertinib in xenograft
tumor models and the expression levels of Bcl-2, Bcl-xL and BimEL
were also detected by IHC. Consistent with in vitro experiments,
results from IHC indicated that ABT-263 enhanced
osimertinib-induced cell apoptosis through elevating BimEL
levels.
As the first third-generation EGFR-TKI in clinical
application, osimertinib is primarily used for advanced NSCLC or
patients who relapse with poor Eastern Cooperative Oncology Group
scores, therefore it is hard to obtain pathological tissues
clinically from osimertinib-resistant patients. Hence, one of the
limitations of the present study was that the expression of Bcl-2
and Bcl-xL were not determined in the tissues of patients with
osimertinib-resistant NSCLC. In the future the tissues of patients
with osimertinib-resistant NSCLC will be examined and the results
from clinical samples will be reported.
In conclusion, the present study revealed that the
expression levels of Bcl-2 and Bcl-xL were upregulated in
osimertinib-resistant NSCLC cells compared with sensitive parent
cells. In addition, the combined treatment with osimertinib and
ABT-263 exerted synergistic inhibition on cell growth, and induced
an increased rate of cell apoptosis through the activation of the
mitochondrial apoptotic pathway. These findings may provide novel
evidence for the development of a combined therapeutic strategy for
the treatment of patients resistant to EGFR TKIs.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Scientific
Research Foundation from Huadong Hospital (grant no.
2019jc007).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YL and ZZ conceived and designed the study. YL, DB
and XZ completed molecular and cell biology experiments. YL and HZ
performed the in vivo study and analyzed the data. YL and ZZ
drafted the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The animal experimental protocol was approved by the
Animal Ethics Committee of Fudan University Shanghai Medical School
(Shanghai, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Torre LA, Siegel RL and Jemal A: Lung
cancer statistics. Adv Exp Med Biol. 893:1–19. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pao W and Chmielecki J: Rational,
biologically based treatment of EGFR-mutant non-small-cell lung
cancer. Nat Rev Cancer. 10:760–774. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nan X, Xie C, Yu X and Liu J: EGFR TKI as
first-line treatment for patients with advanced EGFR
mutation-positive non-small-cell lung cancer. Oncotarget.
8:75712–75726. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gazdar AF: Activating and resistance
mutations of EGFR in non-small-cell lung cancer: Role in clinical
response to EGFR tyrosine kinase inhibitors. Oncogene. 28 (Suppl
1):S24–S31. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee CC, Shiao HY, Wang WC and Hsieh HP:
Small-molecule EGFR tyrosine kinase inhibitors for the treatment of
cancer. Expert Opin Investig Drugs. 23:1333–1348. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yu HA, Arcila ME, Rekhtman N, Sima CS,
Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M and Riely GJ:
Analysis of tumor specimens at the time of acquired resistance to
EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers.
Clin Cancer Res. 19:2240–2247. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Su KY, Chen HY, Li KC, Kuo ML, Yang JC,
Chan WK, Ho BC, Chang GC, Shih JY, Yu SL, et al: Pretreatment
epidermal growth factor receptor (EGFR) T790M mutation predicts
shorter EGFR tyrosine kinase inhibitor response duration in
patients with non-small-cell lung cancer. J Clin Oncol. 30:433–440.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cross DA, Ashton SE, Ghiorghiu S, Eberlein
C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ,
et al: AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated
resistance to EGFR inhibitors in lung cancer. Cancer Discov.
4:1046–1061. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Soejima K, Yasuda H and Hirano T:
Osimertinib for EGFR T790M mutation-positive non-small cell lung
cancer. Expert Rev Clin Phar. 10:31–38. 2017. View Article : Google Scholar
|
|
11
|
Thress KS, Paweletz CP, Felip E, Cho BC,
Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, et
al: Acquired EGFR C797S mutation mediates resistance to AZD9291 in
non-small cell lung cancer harboring EGFR T790M. Nat Med.
21:560–562. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ricordel C, Friboulet L, Facchinetti F and
Soria JC: Molecular mechanisms of acquired resistance to
third-generation EGFR-TKIs in EGFR T790M-mutant lung cancer. Ann
Oncol. 29 (Suppl 1):i28–i37. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shi P, Oh Y-T, Deng L, Zhang G, Qian G,
Zhang S, Ren H, Wu G, Legendre B Jr, Anderson E, et al: Overcoming
acquired resistance to AZD9291, a third-generation EGFR inhibitor,
through modulation of MEK/ERK-dependent Bim and Mcl-1 degradation.
Clin Cancer Res. 23:6567–6579. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zou M, Xia S, Zhuang L, Han N, Chu Q, Chao
T, Peng P, Chen Y, Gui Q and Yu S: Knockdown of the Bcl-2 gene
increases sensitivity to EGFR tyrosine kinase inhibitors in the
H1975 lung cancer cell line harboring T790M mutation. Int J Oncol.
42:2094–2102. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nakajima W, Sharma K, Hicks MA, Le N,
Brown R, Krystal GW and Harada H: Combination with vorinostat
overcomes ABT-263 (navitoclax) resistance of small cell lung
cancer. Cancer Biol Ther. 17:27–35. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Emi M, Kim R, Tanabe K, Uchida Y and Toge
T: Targeted therapy against Bcl-2-related proteins in breast cancer
cells. Breast Cancer Res. 7:R940–R952. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Beale PJ, Rogers P, Boxall F, Sharp SY and
Kelland LR: BCL-2 family protein expression and platinum drug
resistance in ovarian carcinoma. Br J Cancer. 82:436–440. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mu P, Nagahara S, Makita N, Tarumi Y,
Kadomatsu K and Takei Y: Systemic delivery of siRNA specific to
tumor mediated by atelocollagen: Combined therapy using siRNA
targeting Bcl-xL and cisplatin against prostate cancer. Int J
Cancer. 125:2978–2990. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Quadros MR, Connelly S, Kari C, Abrams MT,
Wickstrom E and Rodeck U: EGFR-dependent downregulation of Bim in
epithelial cells requires MAPK and PKC-delta activities. Cancer
Biol Ther. 5:498–504. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sukrithan V, Deng L, Barbaro A and Cheng
H: Emerging drugs for EGFR-mutated non-small cell lung cancer.
Expert Opin Emerg Drugs. 24:5–16. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu SG and Shih JY: Management of acquired
resistance to EGFR TKI-targeted therapy in advanced non-small cell
lung cancer. Mol Cancer. 17:382018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Santarpia M, Liguori A, Karachaliou N,
Gonzalez-Cao M, Daffinà MG, D'Aveni A, Marabello G, Altavilla G and
Rosell R: Osimertinib in the treatment of non-small-cell lung
cancer: Design, development and place in therapy. Lung Cancer
(Auckl). 8:109–125. 2017.PubMed/NCBI
|
|
24
|
Bollinger MK, Agnew AS and Mascara GP:
Osimertinib: A third-generation tyrosine kinase inhibitor for
treatment of epidermal growth factor receptor-mutated non-small
cell lung cancer with the acquired Thr790Met mutation. J Oncol
Pharm Pract. 24:379–388. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lazzari C, Gregorc V, Karachaliou N,
Rosell R and Santarpia M: Mechanisms of resistance to osimertinib.
J Thorac Dis. 12:2851–2858. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kitada S, Pedersen IM, Schimmer AD and
Reed JC: Dysregulation of apoptosis genes in hematopoietic
malignancies. Oncogene. 21:3459–3474. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yamaguchi R, Lartigue L and Perkins G:
Targeting Mcl-1 and other Bcl-2 family member proteins in cancer
therapy. Pharmacol Ther. 195:13–20. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Faber AC, Farago AF, Costa C, Dastur A,
Gomez-Caraballo M, Robbins R, Wagner BL, Rideout WM III, Jakubik
CT, Ham J, et al: Assessment of ABT-263 activity across a cancer
cell line collection leads to a potent combination therapy for
small-cell lung cancer. Proc Natl Acad Sci USA. 112:E1288–E1296.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sarosiek KA, Ni Chonghaile T and Letai A:
Mitochondria: Gatekeepers of response to chemotherapy. Trends Cell
Biol. 23:612–619. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lu M, Liu B, Xiong H, Wu F, Hu C and Liu
P: Trans-3,5,4-trimethoxystilbene reduced gefitinib resistance in
NSCLCs via suppressing MAPK/Akt/Bcl-2 pathway by upregulation of
miR-345 and miR-498. J Cell Mol Med. 23:2431–2441. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tse C, Shoemaker AR, Adickes J, Anderson
MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, et
al: ABT-263: A potent and orally bioavailable Bcl-2 family
inhibitor. Cancer Res. 68:3421–3428. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Saelens X, Festjens N, Vande Walle L, van
Gurp M, van Loo G and Vandenabeele P: Toxic proteins released from
mitochondria in cell death. Oncogene. 23:2861–2874. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sarosiek KA, Chi X, Bachman JA, Sims JJ,
Montero J, Patel L, Flanagan A, Andrews DW, Sorger P and Letai A:
BID preferentially activates BAK while BIM preferentially activates
BAX, affecting chemotherapy response. Mol Cell. 51:751–765. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shukla S, Saxena S, Singh BK and Kakkar P:
BH3-only protein BIM: An emerging target in chemotherapy. Eur J
Cell Biol. 96:728–738. 2017. View Article : Google Scholar : PubMed/NCBI
|