Introduction
High-mobility group box 1 (HMGB1) is released by
activated cells, including cardiomyocytes and damaged or necrotic
cells, and serves as an inflammatory cytokine. In addition, HMGB1
serves an important role in multiple organ pathologies, including
cerebral, liver, lung and renal injury, and rheumatoid arthritis
(1). It is likely that the specific
properties of HMGB1 exhibit an effect on cardiomyocytes. Recently,
it has been suggested that HMGB1 is involved in cardiovascular
diseases (2), such as cardiac
fibrosis, myocardial ischemia-reperfusion injury, heart
transplantation, aortic valve calcification and sepsis-associated
myocardial dysfunction (3–7). Although these previous reports have
revealed that HMGB1 serves an essential role in the cardiovascular
system, in which our previous study (8) also reported that HMGB1 induced
cardiomyocyte hypertrophy, its physiological function in
cardiomyocytes requires further investigation.
Cardiac hypertrophy is induced by several factors,
including inflammatory factors and stresses, such as hypoxia
(9). This process is initially
considered beneficial, but it may progress to heart failure
(10,11) under prolonged stress (12). It has been suggested that cardiac
hypertrophy may be attenuated by controlling inflammation (13). HMGB1 acts as an inflammatory
cytokine and serves an important role in cardiovascular pathology
(14). Therefore, increased levels
of circulating HMGB1 may be associated with human heart disease
(15). For example, exogenous HMGB1
treatment in acute myocardial infarction induces cardiomyocyte
survival by attenuating apoptosis and AMP-activated protein
kinase-dependent autophagy (16).
Our previous study demonstrated that HMGB1 may induce cardiomyocyte
hypertrophy (8). In addition, HMGB1
may be partially derived from cardiac myocytes under pressure
overload and may serve a crucial role in cardiac dysfunction
(17). Notably, maintenance of
stable nuclear HMGB1 levels prevents heart hypertrophy and failure
by inhibiting DNA damage (18).
However, to the best of our knowledge, the direct effects of
exogenous HMGB1 treatment on cardiomyocytes remain elusive.
14-3-3 proteins are distributed ubiquitously in all
eukaryotic organisms and serve a major role in stress response in
several cells, including cardiomyocytes. The 14-3-3 protein family
includes several highly conserved acid proteins, named according to
their different isoforms (19). The
14-3-3 protein family includes several highly conserved acid
proteins, named according to their different isoforms (β, ε, η, γ,
τ, σ and ζ) detected in the cell cytoplasm and nucleus (20,21).
14-3-3η has been reported to serve an essential role in myocardial
metabolism (22,23). Additionally, depletion of 14-3-3η
increases cardiac hypertrophy, inflammation, fibrosis and apoptosis
(24). It has previously been
suggested that 14-3-3 proteins regulate the nuclear translocation
of the nuclear factor of activated T cells (NFAT) (25,26),
which in turn may induce pathological cardiac hypertrophy (27–29).
In addition, a recent study has revealed that simvastatin
upregulates 14-3-3 expression, which ultimately exerts beneficial
effects through cardioprotection against pressure overload
(30). In addition, 14-3-3 proteins
interact with PI3K and NFAT3-mediated transcription in
cardiomyocytes (19), which in turn
phosphorylates its downstream target, Akt, resulting in
physiological cardiac growth (31,32).
The aforementioned results indicate that 14-3-3 proteins may be
involved in the process of cardiac hypertrophy. However, whether
HMGB1 interacts with 14-3-3 proteins to affect cardiac hypertrophy
remains unclear, and, to the best of our knowledge, there are no
studies on the association between HMGB1 and 14-3-3 proteins in
cardiac hypertrophy.
Therefore, the present study hypothesized that HMGB1
may induce cardiomyocyte hypertrophy by regulating the 14-3-3 and
PI3K/Akt signaling pathways. To test this hypothesis, neonatal
mouse cardiomyocytes (NMCs) were isolated and treated with HMGB1.
Subsequently, NMCs were treated with wortmannin, a specific PI3K
inhibitor. Furthermore, to assess the nuclear translocation of NFAT
in NMCs, the effects were observed by confocal microscopy.
Materials and methods
Preparation of NMCs and treatment
NMCs were isolated from 100 C57BL/6 mice (male;
weight, 2±0.5 g; age, 1–3 days old), which were housed in a
pathogen free facility with 50% humidity at 22°C, with a 12-h
light/dark cycle and free access to food and water. Mice were
obtained from the Animal Center of the Fourth Military Medical
University (Xi'an China)]. The mice were sacrificed by decapitation
and heart cell isolation was performed using the Pierce Primary
Cardiomyocyte Isolation kit (Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. A monolayer of isolated
NMCs was plated at a density of 3×106 cells/plate. NMCs
were cultured in DMEM (Gibco; Thermo Fisher Scientific, Inc.)
containing 1% penicillin-streptomycin (Thermo Fisher Scientific,
Inc.) and 10% FBS (Thomas Scientific, Inc.) at 37°C in a humidified
atmosphere with 5% CO2. The presence of fibroblasts was
minimized by the addition of cardiomyocyte growth supplement
(dilution, 1:1,000; Thermo Fisher Scientific, Inc.). Subsequently,
NMCs were divided into the following four groups: The control
(Ctrl) group; the wortmannin (Wort; kindly provided by Dr Jun
L)-treated group; the recombinant HMGB1-treated (rHMGB1) group; and
the rHMGB1 plus Wort (rHMGB1+Wort) group. In addition, rHMGB1 and
wortmannin treatment of NMCs was performed according to our
previous study (8). NMCs were
treated with 200 ng/ml rHMGB1 (Sigma-Aldrich; Merck KGaA) or PBS
for 24 h in the rHMGB1 or Ctrl groups, respectively, and with 100
nmol/l wortmannin for 60 min prior to exposure to PBS or rHMGB1 in
the Wort or rHMGB1+Wort groups, respectively.
NMC protein synthesis measurement
Following treatment, NMCs were trypsinized, counted
using a cell counting chamber (Beckman Coulter, Inc.) and lysed for
further protein detection. Subsequently, protein concentration was
determined using a Bradford protein assay (Bio-Rad Laboratories,
Inc.). Finally, the protein synthesis per cell was calculated by
dividing the total amount of protein by the number of NMCs.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from NMCs using the
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). RNA samples were quantified spectrophotometrically at 260
nm. Subsequently, RT-qPCR was performed as previously described
(8). Briefly, RNA was exposed to
RNAse-free DNase I and 5 µg total RNA was reverse transcribed into
cDNA using oligo(dT) and M-MuLV reverse transcriptase (Promega
Corporation). The reverse transcription products served as
templates for PCR using gene-specific primers (Table I) (33). qPCR was subsequently performed using
a TaqMan™ Fast Advanced Master Mix (Applied Biosystems; Thermo
Fisher Scientific, Inc.) using the 7500 Sequence Detector Real-Time
PCR system (Applied Biosystems; Thermo Fisher Scientific, Inc.).
The thermocycling conditions were as follows: Initial step at 95°C
for 10 min, followed by 40 cycles at 95°C for 15 sec and 60°C for 1
min. Fluorescence signals of each gene were recorded during the
elongation phase of each PCR cycle. The melting curve analysis was
used to confirm the amplification specificity, and the RNA
abundance was expressed as ΔΔCq. GAPDH expression served as the
internal control, and each gene was quantified in duplicate.
Finally, the RT-qPCR data were analyzed using the 2−ΔΔCq
method (34).
 | Table I.Primers used for reverse
transcription-quantitative PCR. |
Table I.
Primers used for reverse
transcription-quantitative PCR.
| Gene | Forward primer
(5′→3′) | Reverse primer
(5′→3′) |
|---|
| ANP |
AGGCAGTCGATTCTGCTT |
CGTGATAGATGAAGGCAGGAAG |
| BNP |
TAGCCAGTCTCCAGAGCAATTC |
TTGGTCCTTCAAGAGCTGTCTC |
| GAPDH |
CCTTCCGTGTTCCTACCCC |
GCCCAAGATGCCCTTCAGT |
Western blot analysis
NMCs were rinsed with PBS and lysed in buffer [Cell
Lysis buffer (10X); cat. no. 9803; Cell Signaling Technology, Inc.]
on ice. Subsequently, cell lysates were centrifuged (2,000 × g; 5–7
min; 4°C), and the supernatant was collected. Following a Bradford
assay to determine protein concentration, equal amounts of proteins
(20 µg) were diluted in sample buffer (Thermo Fisher Scientific,
Inc.), boiled and separated using 15% SDS-PAGE (Thermo Fisher
Scientific, Inc.). The separated proteins were transferred to a
nitrocellulose membrane (LI-COR Biosciences). Following blocking in
5 ml 1X Odyssey® blocking buffer (LI-COR Biosciences)
for 1 h at room temperature, the nitrocellulose membranes were
incubated with primary antibodies at 4°C for 12 h. Subsequently,
membranes were washed with 1X PBS-T (PBS with 0.1% Tween-20) and
incubated with secondary antibodies for 1 h at room temperature.
Finally, immunoreactive protein bands were analyzed by densitometry
using the Odyssey® infrared imaging system (version 2.1;
LI-COR Biosciences), and the specific protein bands were visualized
using an Odyssey® scanner, then analyzed using Image
Studio Lite version 5.0 (LI-COR Biosciences). The primary
antibodies used in the present study were all diluted 1:1,000 and
were: Anti-atrial natriuretic peptide (ANP; cat. no. ab126149;
Abcam), anti-pan14-3-3 (cat. no. ab32377; Abcam), anti-14-3-3η
(cat. no. ab206292; Abcam), anti-phospho-Akt (p-Akt) (cat. no.
ab38449; Abcam), anti-histone (cat. no. ab1791; Abcam),
anti-β-tubulin (cat. no. ab6160; Abcam), anti-NFAT3 (cat. no.
orb315632; Biorbyt Ltd.) and anti-β-actin (cat. no. SC-8432; Santa
Cruz Biotechnology, Inc.). The secondary antibodies were goat
anti-mouse IRDye® (1:10,000; cat. no. 926-68070; LI-COR
Biosciences), goat anti-rabbit IRDye® (1:10,000; cat.
no. 926-32211; LI-COR Biosciences) and goat anti-rat
IRDye® (1:10,000; cat. no. 926-32219; LI-COR
Biosciences). In addition, β-tubulin and histone protein levels
served as a loading control for the total protein lysate, the
cytosol and nuclear extracts, respectively.
Confocal microscopy observation
NFAT3 localization in NMCs was analyzed using
confocal microscopy. Briefly, NMCs were isolated and cultured on
laminin pre-coated Lab-Tek chamber slides (Thermo Fisher
Scientific, Inc.). Following treatment, cells (1×105
cells/well) were fixed with 4% paraformaldehyde at room temperature
for 30 min, permeabilized using 0.2% Triton X-100 and washed with
PBS. Subsequently, cells were incubated in 1 ml blocking reagent
(SuperBlock Blocking Buffer; Thermo Scientific™; Thermo Fisher
Scientific, Inc.) with primary rabbit anti-NFAT3 (1:200; cat. no.
sc-8405; Santa Cruz Biotechnology, Inc.) and primary mouse
anti-α-actinin antibodies (1:200; cat. no. A2543; Sigma-Aldrich;
Merck KGaA) in a cold room at 4°C for 24 h. Following extensive
washing, NMCs were incubated with Alexa Fluor 594-labeled goat
anti-rabbit (1:1,000; cat. no. Z25307; Invitrogen; Thermo Fisher
Scientific, Inc.) and Alexa Fluor 488-labeled goat anti-mouse
antibodies (1:1,000; cat. no. A20181; Invitrogen; Thermo Fisher
Scientific, Inc.) at 4°C for 30 min. Finally, cells were mounted
with mounting media containing
2-(4-amidinophenyl)-1H-indole-6-carboxamidine (DAPI, 1:1,000)
(Vector Laboratories, Inc.; Maravai LifeSciences). A Carl Zeiss 710
confocal microscope (Carl Zeiss AG; magnification, ×40) was used to
image the NFAT3, α-actinin and DAPI staining.
Cell extract preparation
The cytosol and nuclear extracts from NMCs were
prepared using an NE-PER extraction kit (Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol. The specificity of
the cytosol or nuclear extracts was verified by western blot
analysis by detecting β-tubulin or histone protein expression,
respectively.
Terminal deoxynucleotidyl transferase
dUTP nick-end-labeling (TUNEL) assay
The NMC apoptosis rate was detected using a TUNEL
assay kit (Roche Diagnostics), according to the manufacturer's
protocol. Briefly, following treatment NMCs (1×105
cells/well) were fixed with 4% paraformaldehyde at room temperature
for 30 min and permeabilized with 0.2% Triton X-100 at room
temperature for 30 min. The cells were subsequently incubated with
TUNEL reagent for 1 h at room temperature. Thereafter, the cells
were mounted with a mounting medium containing DAPI (1:1,000;
Vector Laboratories, Inc.) at room temperature for 10 min. The
TUNEL-positive and the total number of nuclei were counted in five
independent experiments (3 randomly selected fields of view) under
a Nikon Eclipse 80 fluorescent microscope (magnification, ×10;
Nikon Corporation).
Statistical analysis
All data were analyzed using GraphPad Prism 5
(GraphPad Software, Inc.) and presented as the mean ± SEM for
continuous variables. One-way ANOVA was used to assess differences
among groups followed by Tukey's post hoc test. P<0.05 was
considered to indicate a statistically significant difference. The
Ctrl group value was set as 1 to express the fold change.
Results
rHMGB1 induces NMC hypertrophy
Following exposure to rHMGB1 for 24 h, NMCs were
harvested and their cellular protein content was analyzed. The
results revealed that rHMGB1 treatment significantly increased the
protein content per cell compared with that in the Ctrl group
(Fig. 1A). Subsequently, the
protein levels of ANP, a pathological cardiac hypertrophy marker,
were detected using western blot analysis. Protein levels of ANP in
the rHMGB1 group were significantly increased compared with those
in the Ctrl group (Fig. 1B). In
addition, the mRNA levels of ANP and brain natriuretic peptide
(BNP), another pathological cardiac hypertrophy marker, were
upregulated in the rHMGB1 group compared with those in the Ctrl
group (Fig. 1C). Representative
confocal images of NMCs are presented in Fig. 1D. Therefore, the present results
suggest that rHMGB1 induces NMC hypertrophy.
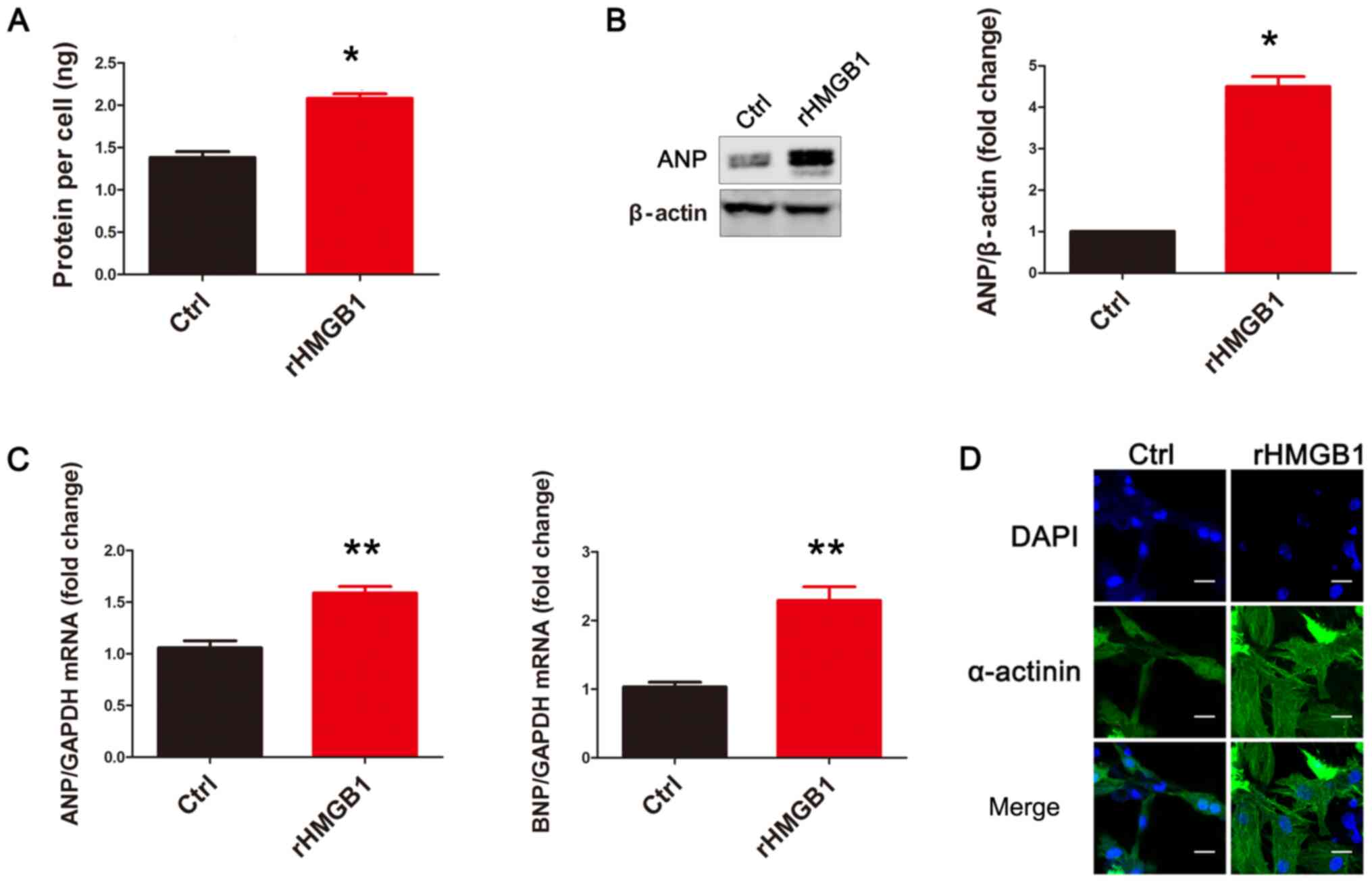 | Figure 1.rHMGB1 induces NMC hypertrophy.
rHMGB1 significantly increased (A) the protein content per cell,
(B) ANP protein levels and (C) the mRNA levels of ANP and BNP in
NMCs (n=3). Data are presented as the mean ± standard error of the
mean for continuous variables. One-way ANOVA was used to assess
differences among different groups. (D) Representative confocal
images from the Ctrl and rHMGB1 groups (scale bar, 20 µm). Cells
were stained with DAPI (blue signal) and α-actinin (green signal)
to identify nuclei and cardiomyocytes, respectively. rHMGB1,
recombinant high-mobility group box 1; NMC, neonatal mouse
cardiomyocytes; ANP, atrial natriuretic peptide; BNP, brain
natriuretic peptide; Ctrl, control; DAPI,
2-(4-amidinophenyl)-1H-indole-6-carboxamidine. *P<0.05,
**P<0.01 vs. Ctrl group. |
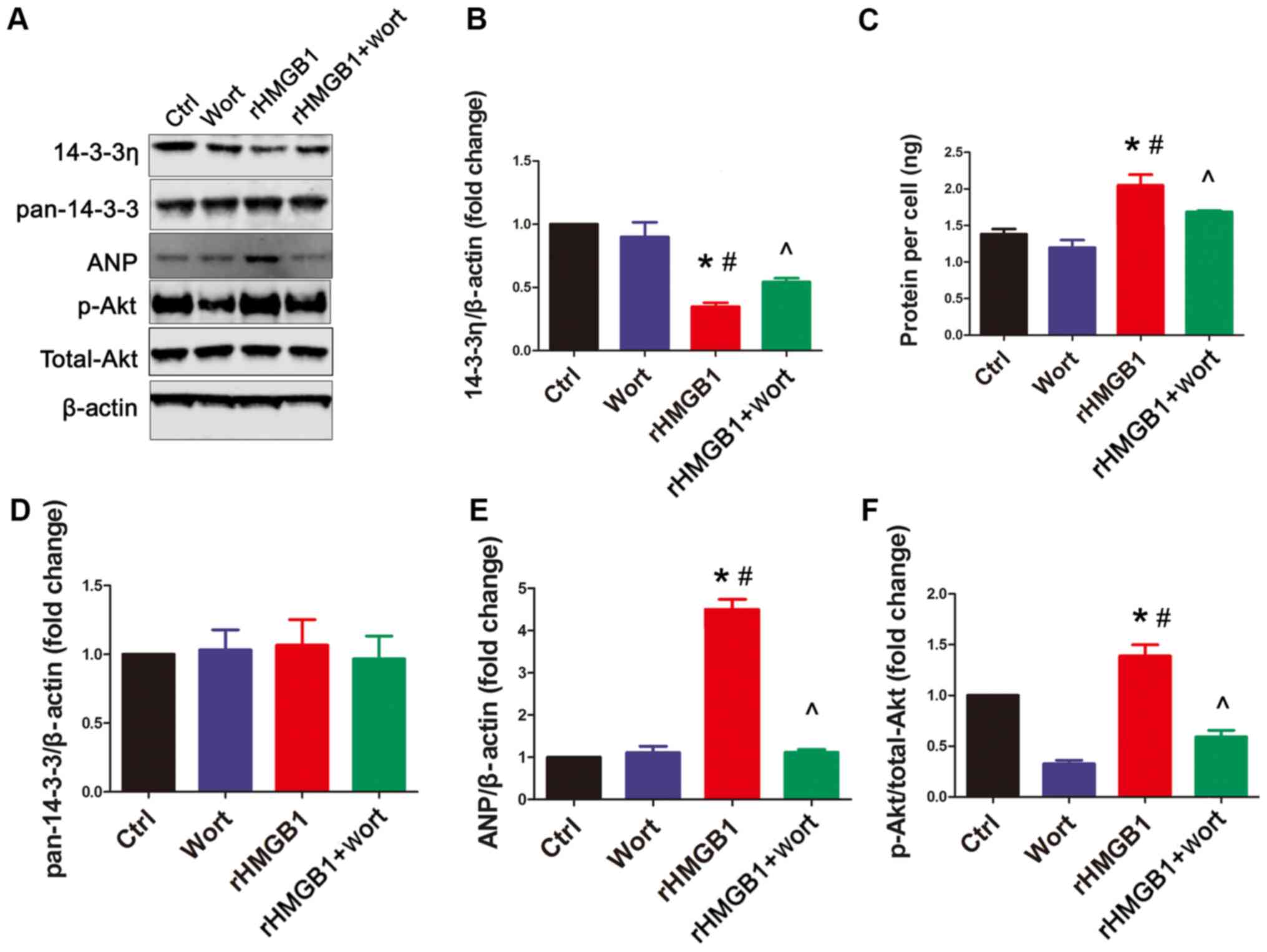
rHMGB1 decreases 14-3-3η protein
levels, which can be partially reversed by wortmannin
In the present study, western blot analysis was
performed using anti-pan14-3-3 (an antibody against endogenous
total 14-3-3 proteins) and anti-14-3-3η antibodies, to determine
whether rHMGB1 could regulate their expression in NMCs. Following
rHMGB1 treatment, 14-3-3η protein levels were decreased (Fig. 2A and B) and p-Akt levels were
increased (Fig. 2A and F) in NMCs
compared with those in the Ctrl group (Fig. 2). However, rHMGB1 treatment had no
effect on the total endogenous 14-3-3 protein levels (Fig. 2A and D). Furthermore, wortmannin, a
specific PI3K inhibitor, significantly inhibited the
rHMGB1-mediated effects on 14-3-3η protein levels and Akt
phosphorylation (Fig. 2A, B and F).
However, wortmannin alone had no significant effect on 14-3-3η
levels compared with the Ctrl group (Fig. 2A and B). Additionally, no changes to
pan14-3-3 protein levels were observed among the different groups
(Fig. 2D). Finally, the effects of
rHMGB1 treatment on the protein content per cell and ANP protein
levels were inhibited by wortmannin (Fig. 2A, C and E). Overall, the present
results suggest that rHMGB1 regulates 14-3-3η protein levels and
hypertrophy in NMCs partially through the PI3K/Akt signaling
pathway.
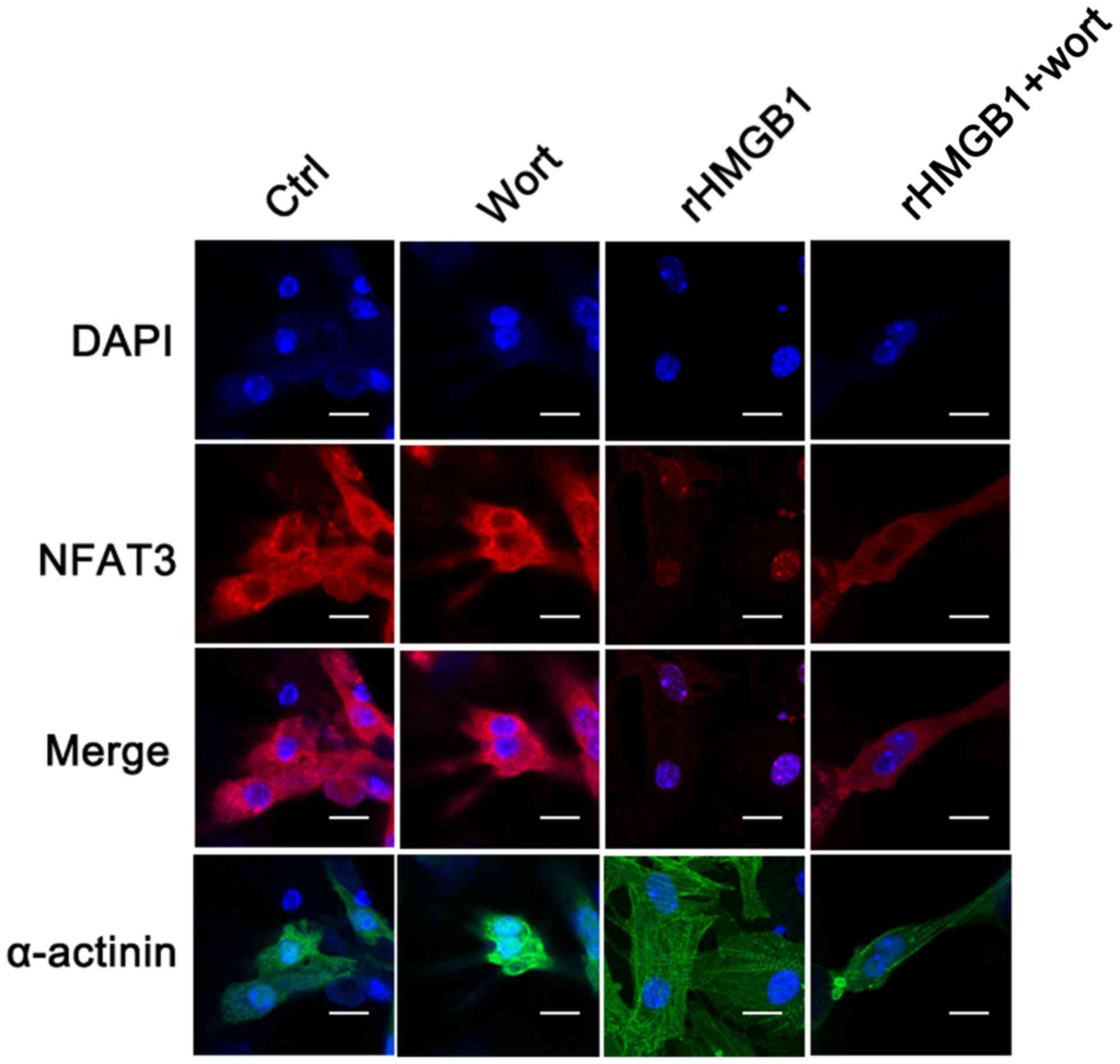
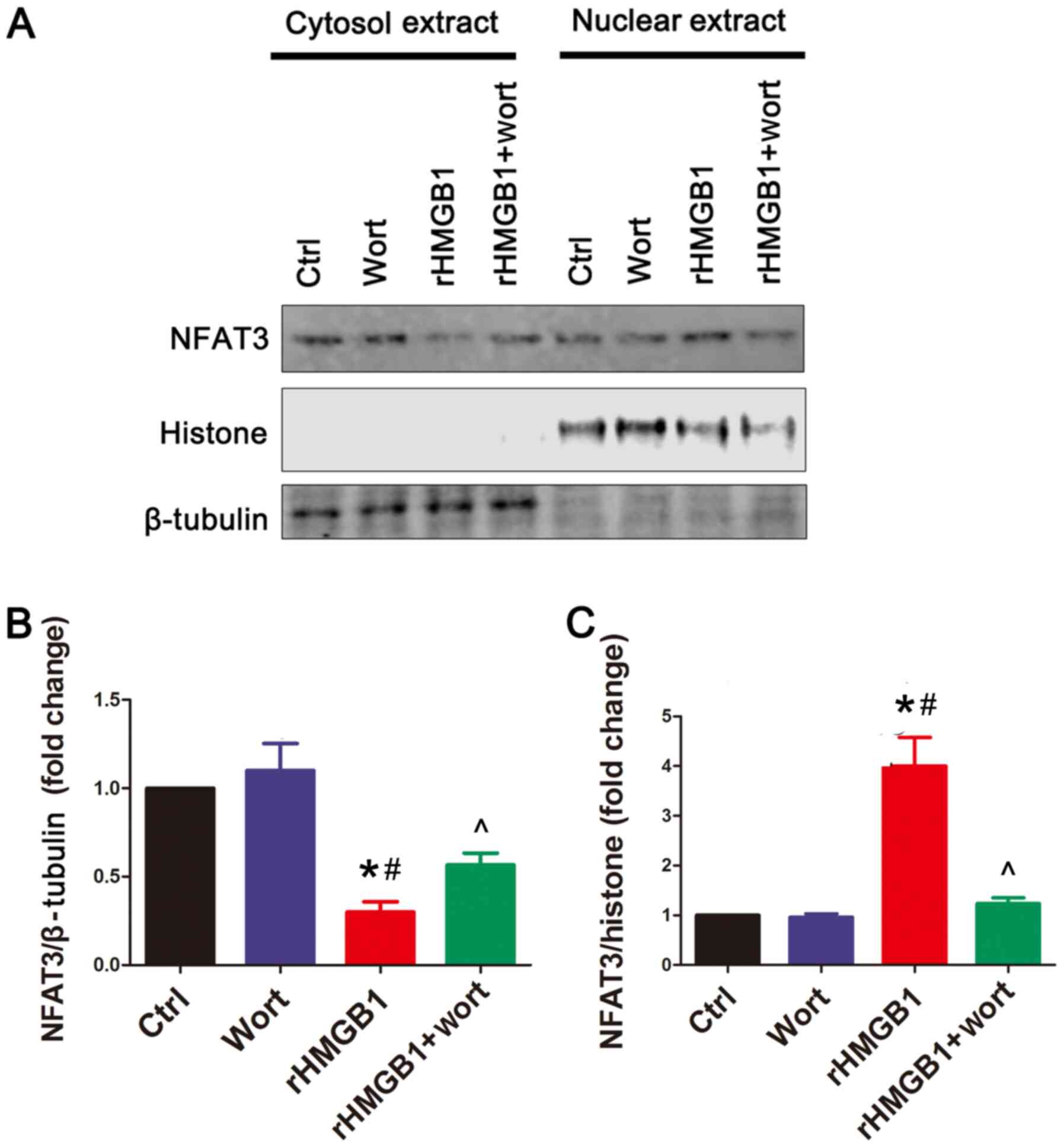
rHMGB1 induces NFAT3 nuclear
translocation, which can be inhibited by wortmannin
In the present study, confocal microscopy was used
to detect the subcellular location of NFAT3 and to determine
whether NFAT3 is translocated to the nucleus following rHMGB1
treatment. In the rHMGB1 group, the levels of NFAT3 in the cytosol
were decreased, while those in the nuclei were increased compared
with the levels of NFAT3 in the Ctrl group (Fig. 3). Notably, wortmannin pretreatment
inhibited the nuclear translocation of NFAT3 following rHMGB1
treatment. The results were verified using western blot analysis,
demonstrating that rHMGB1 treatment upregulated and downregulated
NFAT3 protein levels in the NMC nuclear and cytoplasmic extracts,
respectively (Fig. 4). Furthermore,
wortmannin pretreatment reversed the effects of rHMGB1 treatment on
NFAT3 protein levels (Fig. 4). The
aforementioned results suggest that rHMGB1 may contribute to
cardiomyocyte hypertrophy by regulating NFAT3 translocation, which
may be affected by the PI3K/Akt signaling pathway.
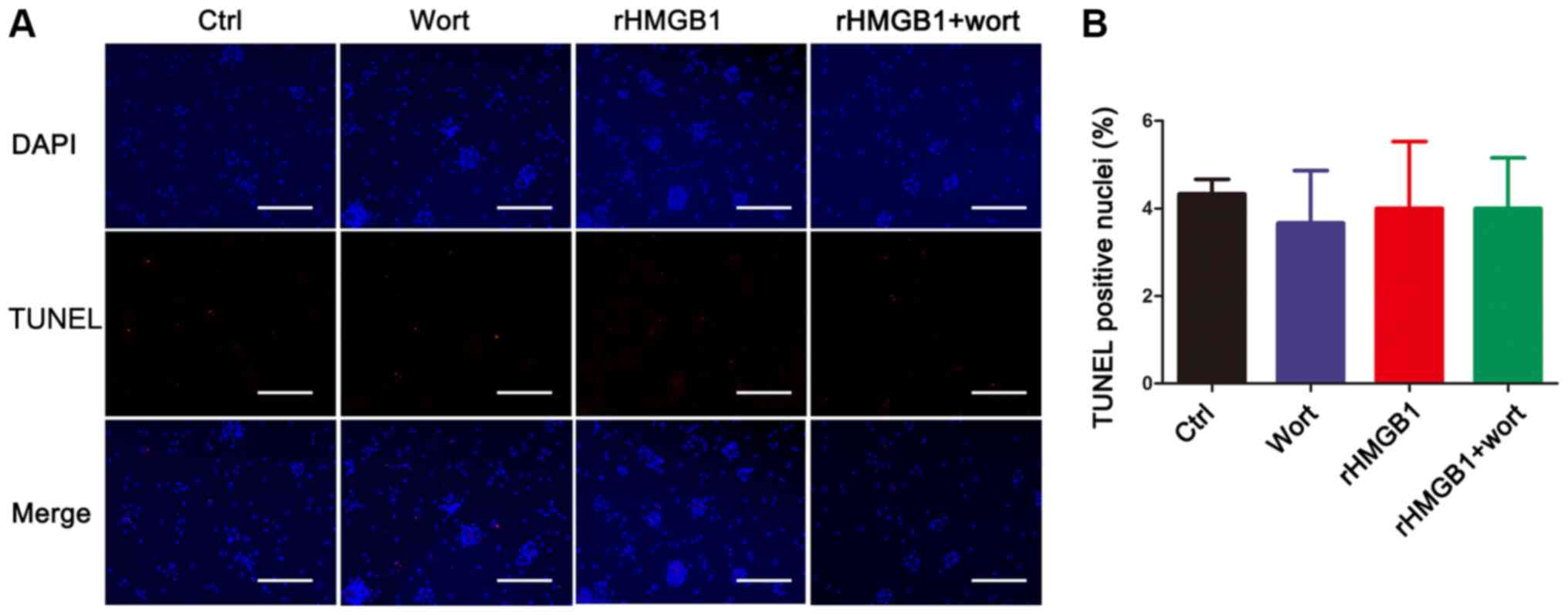
rHMGB1 has no effect on NMC
apoptosis
Cardiac hypertrophy may lead to cardiomyocyte
apoptosis during the heart failure process (35). In the present study, the apoptosis
rate was measured using the TUNEL assay to detect TUNEL-positive
nuclei and to determine whether rHMGB1 treatment induced NMC
apoptosis. However, no significant differences in the number of
TUNEL-positive nuclei were observed among the four groups (Fig. 5).
Discussion
The present study demonstrated that rHMGB1 induced
NMC hypertrophy, decreased 14-3-3η protein levels and induced
translocation of NFAT3 from the cytoplasm to the nucleus. The
effects of rHMGB1 on NMC hypertrophy, 14-3-3η levels and NFAT3
translocation may be partially blocked by wortmannin, a specific
PI3K inhibitor. However, rHMGB1 alone did not affect NMC apoptosis.
Overall, the present results support the hypothesis that exogenous
HMGB1 induces cardiomyocyte hypertrophy via the
14-3-3/PI3K/Akt/NFAT signaling pathway.
HMGB1 is a ubiquitous nuclear protein that is
released from cell nuclei following tissue damage (36). Endogenous HMGB1 is located in the
nucleus (37) and is actively
secreted by innate immune cells (38) or other cells under stress conditions
(6). Therefore, HMGB1 is found in
cells and in the systemic circulation. It has been reported that
endogenous HMGB1 may serve an important role in myocardium
pathology (39). In addition,
patients with myocarditis display increased systemic HMGB1 levels,
suggesting its involvement in the pathogenesis of inflammatory
cardiomyopathy (2). However, the
effects of secreted HMGB1 on the cardiovascular system remain
unclear, and the effect of exogenous HMGB1 treatment on
cardiomyocytes requires further investigation. The results of the
present study revealed that rHMGB1 treatment increased ANP protein
levels, BNP and ANP mRNA synthesis, and protein content per cell,
highlighting its role in the induction of NMC hypertrophy. In the
present study, HMGB1 increased the intracellular levels of ANP and
BNP in cardiomyocytes. However, whether HMGB1 is able to enhance
the secretion of ANP or BNP was not the main purpose of the present
study; this should be analyzed in vivo in future studies.
The aforementioned results suggest that both endogenous and
exogenous HMGB1 serve a role in myocardial modifications, which is
consistent with a previous study demonstrating that exogenous HMGB1
treatment induced cardiomyocyte survival in a murine myocardial
infarction model (16).
In the present study, HMGB1 activated NFAT3 by
promoting its translocation to the nucleus, thereby upregulating
its nuclear expression. Additionally, it has been documented that
NFAT3 is regulated by 14-3-3 proteins (25,26),
exhibits inflammatory effects and induces pathological hypertrophy
in cardiac myocytes (27–29).
14-3-3 proteins interact with several proteins,
including PI3K, Akt and NFAT3 in diabetic cardiomyopathy (40). 14-3-3 proteins are dimeric
phosphoserine-binding molecules separated into several isoforms,
including the β, γ, ε, ζ, η, θ and σ isoforms (19). These proteins bind to their target
proteins and modify their function by altering their intracellular
localization and phosphorylation status (20). Several studies have concluded that
14-3-3 proteins, particularly the 14-3-3η isoform, are involved in
diabetic cardiomyopathy (40,41).
However, whether 14-3-3η is involved in rHMGB1-induced hypertrophy,
an independent predictor of cardiovascular morbidity and mortality,
requires further investigation. The results of the present study
revealed that the levels of total 14-3-3 proteins in NMCs did not
change in the presence of rHMGB1. However, the protein levels of
14-3-3η were significantly decreased in rHMGB1-treated NMCs,
resulting in significant induction of NMC hypertrophy. Therefore,
the present results suggest that 14-3-3η, and not total 14-3-3, may
serve a major role in rHMGB1-induced NMC hypertrophy.
NMCs were treated with a specific PI3K inhibitor
prior to exposure to rHMGB1. Notably, the levels of 14-3-3η were
partially preserved in rHMGB1-treated NMCs following wortmannin
pretreatment. Additionally, wortmannin partially inhibited NFAT3
nuclear translocation. The present results suggest that PI3K, Akt
and NFAT3 may interact with 14-3-3η and may influence
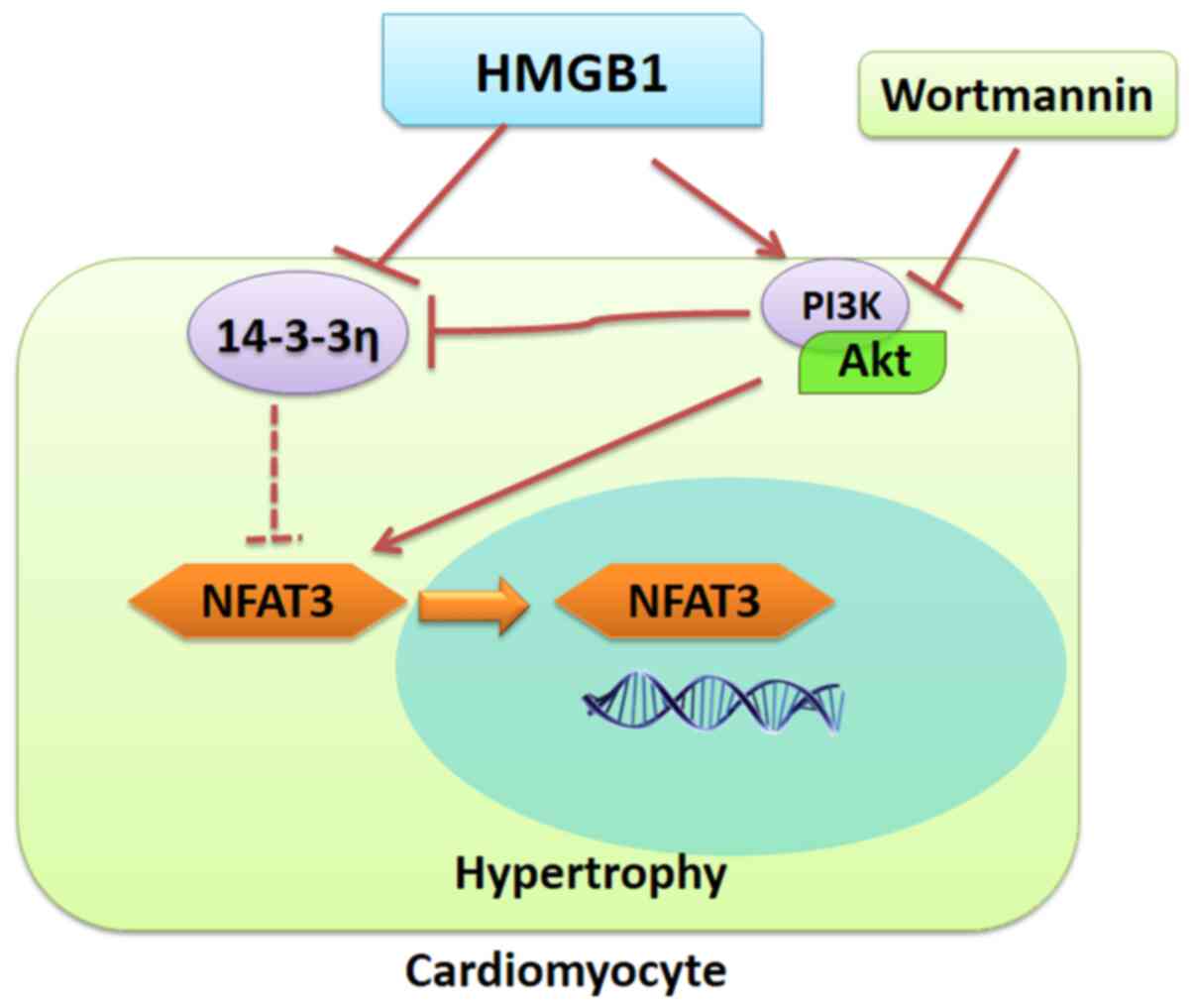
rHMGB1-induced hypertrophy. The proposed model of rHMGB1-mediated
regulation of NMC hypertrophy is illustrated in Fig. 6.
Furthermore, it has been reported that cardiomyocyte
hypertrophy may eventually lead to apoptosis (35). Therefore, a TUNEL assay was
performed to determine the rHMGB1-induced apoptosis rate in NMCs.
However, no statistically significant differences were observed in
the NMC apoptosis rate between the Ctrl and the rHMGB1-treated
groups. The present finding is consistent with a previous study
demonstrating that HMGB1 does not induce cardiomyocyte apoptosis
under normal conditions (42).
However, it has been reported that endogenous HMGB1 contributes to
ischemia-reperfusion-induced myocardial apoptosis (39). A potential explanation for these
contrasting findings may reside in the different environments or
distinct receptors on different cells. Therefore, further
investigation is required to investigate the role of exogenous
HMGB1 in different cell types.
In conclusion, the present study demonstrated that
extracellular HMGB1 treatment induced NMC hypertrophy potentially
through the 14-3-3η/PI3K/Akt/NFAT signaling pathway. Therefore,
14-3-3 may be an important factor that links HMGB1, PI3K/Akt and
NFAT3 in cardiomyocytes.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81300077).
Availability of data and materials
The datasets used and/or during the present study
are available from the corresponding author on reasonable
request.
Authors' contributions
FS conceived the study. FS, MS and JZ performed the
experiments. YL and JT analyzed the experimental data, assessed the
raw data and confirm the authenticity of all the raw data. FS
prepared the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present experimental procedures were performed
according to the ethical guidelines of the 1964 Declaration of
Helsinki. All animal experiments were approved by the Animal Care
and Welfare Ethics Committee of the Fourth Military Medical
University (Xi'an, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Biscetti F, Flex A, Alivernini S, Tolusso
B, Gremese E and Ferraccioli G: The role of high-mobility group
Box-1 and its crosstalk with microbiome in rheumatoid arthritis.
Mediators Inflamm. 2017:52303742017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bangert A, Andrassy M, Muller AM,
Bockstahler M, Fischer A, Volz CH, Leib C, Göser S, Korkmaz-Icöz S,
Zittrich S, et al: Critical role of RAGE and HMGB1 in inflammatory
heart disease. Proc Natl Acad Sci USA. 113:E155–E164. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dong LY, Chen F, Xu M, Yao LP, Zhang YJ
and Zhuang Y: Quercetin attenuates myocardial ischemia-reperfusion
injury via downregulation of the HMGB1-TLR4-NF-kB signaling
pathway. Am J Transl Res. 10:1273–1283. 2018.PubMed/NCBI
|
|
4
|
Zhang W, Tao A, Lan T, Cepinskas G, Kao R,
Martin CM and Rui T: Carbon monoxide releasing molecule-3 improves
myocardial function in mice with sepsis by inhibiting NLRP3
inflammasome activation in cardiac fibroblasts. Basic Res Cardiol.
112:162017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wu RN, Yu TY, Zhou JC, Li M, Gao HK, Zhao
C, Dong RQ, Peng D, Hu ZW, Zhang XW and Wu YQ: Targeting HMGB1
ameliorates cardiac fibrosis through restoring TLR2-mediated
autophagy suppression in myocardial fibroblasts. Int J Cardiol.
267:156–162. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shen W, Zhou J, Wang C, Xu G, Wu Y and Hu
Z: High mobility group box 1 induces calcification of aortic valve
interstitial cells via toll-like receptor 4. Mol Med Rep.
15:2530–2536. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lv Q, Li C, Mo Y and He L: The role of
HMGB1 in heart transplantation. Immunol Lett. 194:1–3. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Su FF, Shi MQ, Guo WG, Liu XT, Wang HT, Lu
ZF and Zheng QS: High-mobility group box 1 induces
calcineurin-mediated cell hypertrophy in neonatal rat ventricular
myocytes. Mediators Inflamm. 2012:8051492012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nehra S, Bhardwaj V, Kalra N, Ganju L,
Bansal A, Saxena S and Saraswat D: Nanocurcumin protects
cardiomyoblasts H9c2 from hypoxia-induced hypertrophy and apoptosis
by improving oxidative balance. J Physiol Biochem. 71:239–251.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fang X, Liu Y, Lu J, Hong H, Yuan J, Zhang
Y, Wang P, Liu P and Ye J: Protocatechuic aldehyde protects against
isoproterenol-induced cardiac hypertrophy via inhibition of the
JAK2/STAT3 signaling pathway. Naunyn Schmiedebergs Arch Pharmacol.
391:1373–1385. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang S, Han HM, Pan ZW, Hang PZ, Sun LH,
Jiang YN, Song HX, Du ZM and Liu Y: Choline inhibits angiotensin
II-induced cardiac hypertrophy by intracellular calcium signal and
p38 MAPK pathway. Naunyn Schmiedebergs Arch Pharmacol. 385:823–831.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zwadlo C, Schmidtmann E, Szaroszyk M,
Kattih B, Froese N, Hinz H, Schmitto JD, Widder J, Batkai S, Bähre
H, et al: Antiandrogenic therapy with finasteride attenuates
cardiac hypertrophy and left ventricular dysfunction. Circulation.
131:1071–1081. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Song L, Wang L, Li F, Yukht A, Qin M,
Ruther H, Yang M, Chaux A, Shah PK and Sharifi BG: Bone
marrow-derived tenascin-C attenuates cardiac hypertrophy by
controlling inflammation. J Am Coll Cardiol. 70:1601–1615. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gendy AM, Abdallah DM and El-Abhar HS: The
potential curative effect of rebamipide in hepatic
ischemia/reperfusion injury. Naunyn Schmiedebergs Arch Pharmacol.
390:691–700. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Raucci A, Di Maggio S, Scavello F,
D'Ambrosio A, Bianchi ME and Capogrossi MC: The Janus face of HMGB1
in heart disease: A necessary update. Cell Mol Life Sci.
76:211–229. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Foglio E, Puddighinu G, Germani A, Russo
MA and Limana F: HMGB1 inhibits apoptosis following MI and induces
autophagy via mTORC1 inhibition. J Cell Physiol. 232:1135–1143.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang L, Liu M, Jiang H, Yu Y, Yu P, Tong
R, Wu J, Zhang S, Yao K, Zou Y and Ge J: Extracellular
high-mobility group box 1 mediates pressure overload-induced
cardiac hypertrophy and heart failure. J Cell Mol Med. 20:459–470.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Funayama A, Shishido T, Netsu S, Narumi T,
Kadowaki S, Takahashi H, Miyamoto T, Watanabe T, Woo CH, Abe J, et
al: Cardiac nuclear high mobility group box 1 prevents the
development of cardiac hypertrophy and heart failure. Cardiovasc
Res. 99:657–664. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liao W, Wang S, Han C and Zhang Y: 14-3-3
proteins regulate glycogen synthase 3beta phosphorylation and
inhibit cardiomyocyte hypertrophy. FEBS J. 272:1845–1854. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jia H, Liang Z, Zhang X, Wang J, Xu W and
Qian H: 14-3-3 proteins: An important regulator of autophagy in
diseases. Am J Transl Res. 9:4738–4746. 2017.PubMed/NCBI
|
|
21
|
Obsilova V, Kopecka M, Kosek D, Kacirova
M, Kylarova S, Rezabkova L and Obsil T: Mechanisms of the 14-3-3
protein function: Regulation of protein function through
conformational modulation. Physiol Res. 63 (Suppl 1):S155–S164.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sreedhar R, Arumugam S, Thandavarayan RA,
Karuppagounder V, Koga Y, Nakamura T, Harima M and Watanabe K: Role
of 14-3-3 η protein on cardiac fatty acid metabolism and macrophage
polarization after high fat diet induced type 2 diabetes mellitus.
Int J Biochem Cell Biol. 88:92–99. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sreedhar R, Arumugam S, Thandavarayan RA,
Giridharan VV, Karuppagounder V, Pitchaimani V, Afrin R, Miyashita
S, Nomoto M, Harima M, et al: Myocardial 14-3-3 η protein protects
against mitochondria mediated apoptosis. Cell Signal. 27:770–776.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sreedhar R, Arumugam S, Thandavarayan RA,
Giridharan VV, Karuppagounder V, Pitchaimani V, Afrin R, Harima M,
Nakamura M, Suzuki K, et al: Depletion of cardiac 14-3-3 η protein
adversely influences pathologic cardiac remodeling during
myocardial infarction after coronary artery ligation in mice. Int J
Cardiol. 202:146–153. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chhabra S, Fischer P, Takeuchi K, Dubey A,
Ziarek JJ, Boeszoermenyi A, Mathieu D, Bermel W, E Davey N, Wagner
G and Arthanari H: (15)N detection harnesses the slow relaxation
property of nitrogen: Delivering enhanced resolution for
intrinsically disordered proteins. Proc Natl Acad Sci USA.
115:E1710–E1719. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Faul C, Donnelly M, Merscher-Gomez S,
Chang YH, Franz S, Delfgaauw J, Chang JM, Choi HY, Campbell KN, Kim
K, et al: The actin cytoskeleton of kidney podocytes is a direct
target of the antiproteinuric effect of cyclosporine A. Nat Med.
14:931–938. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kumar S, Wang G, Liu W, Ding W, Dong M,
Zheng N, Ye H and Liu J: Hypoxia-induced mitogenic factor promotes
cardiac hypertrophy via calcium-dependent and hypoxia-inducible
Factor-1α mechanisms. Hypertension. 72:331–342. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Grund A, Szaroszyk M, Doppner JK,
Mohammadi MM, Kattih B, Korf-Klingebiel M, Gigina A, Scherr M,
Kensah G, Jara-Avaca M, et al: A gene therapeutic approach to
inhibit CIB1 ameliorates maladaptive remodeling in pressure
overload. Cardiovasc Res. 115:71–82. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gelinas R, Mailleux F, Dontaine J, Bultot
L, Demeulder B, Ginion A, Daskalopoulos EP, Esfahani H,
Dubois-Deruy E, Lauzier B, et al: AMPK activation counteracts
cardiac hypertrophy by reducing O-GlcNAcylation. Nat Commun.
9:3742018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Su F, Shi M, Zhang J, Zheng Q, Zhang D,
Zhang W, Wang H and Li X: Simvastatin protects heart from pressure
overload injury by inhibiting excessive autophagy. Int J Med Sci.
15:1508–1516. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
DeBosch B, Treskov I, Lupu TS, Weinheimer
C, Kovacs A, Courtois M and Muslin AJ: Akt1 is required for
physiological cardiac growth. Circulation. 113:2097–2104. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
O'Neill BT, Kim J, Wende AR, Theobald HA,
Tuinei J, Buchanan J, Guo A, Zaha VG, Davis DK, Schell JC, et al: A
conserved role for phosphatidylinositol 3-kinase but not Akt
signaling in mitochondrial adaptations that accompany physiological
cardiac hypertrophy. Cell Metab. 6:294–306. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gao RR, Wu XD, Jiang HM, Zhu YJ, Zhou YL,
Zhang HF, Yao WM, Li YQ and Li XL: Traditional Chinese medicine
Qiliqiangxin attenuates phenylephrine-induced cardiac hypertrophy
via upregulating PPARγ and PGC-1α. Ann Transl Med. 6:1532018.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lu F, Xing J, Zhang X, Dong S, Zhao Y,
Wang L, Li H, Yang F, Xu C and Zhang W: Exogenous hydrogen sulfide
prevents cardiomyocyte apoptosis from cardiac hypertrophy induced
by isoproterenol. Mol Cell Biochem. 381:41–50. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hwang JH, Chu H, Ahn Y, Kim J and Kim DY:
HMGB1 promotes hair growth via the modulation of prostaglandin
metabolism. Sci Rep. 9:66602019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Muller S, Ronfani L and Bianchi ME:
Regulated expression and subcellular localization of HMGB1, a
chromatin protein with a cytokine function. J Intern Med.
255:332–343. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tang D, Shi Y, Kang R, Li T, Xiao W, Wang
H and Xiao XZ: Hydrogen peroxide stimulates macrophages and
monocytes to actively release HMGB1. J Leukoc Biol. 81:741–747.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xu H, Yao Y, Su Z, Yang Y, Kao R, Martin
CM and Rui T: Endogenous HMGB1 contributes to
ischemia-reperfusion-induced myocardial apoptosis by potentiating
the effect of TNF-α/JNK. Am J Physiol Heart Circ Physiol.
300:H913–H921. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Watanabe K, Thandavarayan RA, Gurusamy N,
Zhang S, Muslin AJ, Suzuki K, Tachikawa H, Kodama M and Aizawa Y:
Role of 14-3-3 protein and oxidative stress in diabetic
cardiomyopathy. Acta Physiol Hung. 96:277–287. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Thandavarayan RA, Watanabe K, Ma M,
Veeraveedu PT, Gurusamy N, Palaniyandi SS, Zhang S, Muslin AJ,
Kodama M and Aizawa Y: 14-3-3 protein regulates Ask1 signaling and
protects against diabetic cardiomyopathy. Biochem Pharmacol.
75:1797–1806. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lin H, Shen L, Zhang X, Xie J, Hao H,
Zhang Y, Chen Z, Yamamoto H, Liao W, Bin J, et al: HMGB1-RAGE axis
makes no contribution to cardiac remodeling induced by
pressure-overload. PLoS One. 11:e1585142016.
|