Introduction
Autophagy maintains cellular homeostasis via the
recycling of long-lived proteins and removal of damaged organelles
(1–3), such that it protects cardiomyocytes
from damage and death, as well as improves cardiac function in
several pathological conditions, including traumatic shock,
ischemia-reperfusion and coronary artery disease, amongst others
(4–9). However, previous studies have reported
that activated myocardial autophagy results in cardiomyocyte death
in rats following severe burns, instead of protecting cardiac
function (6,10). Although autophagy is active,
redundant autophagosome accumulation is found in various types of
pathological conditions, such as oxidative stress and acute
hypoxia, leading to subsequent autophagic cell death (11–14).
Eliminating autophagosome accumulation and enhancing the activity
of autophagy-related pathways significantly improves cardiomyocyte
viability (15). Thus, improvement
of autophagy flux is important for protecting cells from damage
when under stress.
Lysosomes, which contain hydrolases that degrade
endocellular macromolecules, are essential in the autophagy pathway
(16). Impaired lysosomal function
leads to autophagosome accumulation and subsequent cardiomyocyte
death, which has been observed in various pathological conditions
including hypoxia, ischemia-reperfusion and fibrosis (14,17,18).
Therefore, studying the mechanism of lysosomal acidification may
identify potential avenues for treatment of diseases that result
from excess cardiomyocyte death. In lysosomes, the activation of
hydrolases requires a low luminal pH (19). Transporters, such as vacuolar-type
H+-ATPase (v-ATPase) and Cl−, K+
and Na+ channels, participate in maintaining the luminal
pH (19,20).
H(+)/Cl(−) exchange transporter 7 (CLC-7) is a
2Cl−/1H+ antiporter that is primarily found
in late endosomes and lysosomes (21,22).
CLC-7 regulates Cl− conductance, which enables
osteoclast ruffled border ATPases to facilitate efficient proton
pumping (23). The ruffled border
delivers HCl and proteases to resorption lacuna between osteoclasts
and the bone (23,24). The low luminal pH caused by HCl
serves a fundamental role in dissolving and absorbing bone material
(23). Loss of CLC-7 transporters
leads to lysosomal storage diseases and osteopetrosis (18,23–25).
However, the mechanism of CLC-7 in lysosomes is contested. CLC-7
knockdown was shown to inhibit lysosomal acidification in HeLa
cells and primary mouse microglia, as well as isolated lysosomes
(19,21,26).
However, lysosomal pH changes in CLC-7 knockout mice were not
significant (18,23–25,27).
Furthermore, to the best of our knowledge, there are no studies on
the role of CLC-7 in autophagy. Rapamycin, a well-known activator
of autophagy, results in activation of autophagy by specifically
inhibiting mTOR function (1,28). In
the present study, a model of autophagy using rapamycin-treated
neonatal mouse cardiomyocytes was established, and the potential
roles of CLC-7 in lysosomal acidification and autophagy were
investigated.
Materials and methods
Chemicals and reagents
Antibodies against CLC-7 (cat. no. GTX55139;
GeneTex, Inc.), sequestosome 1 (p62; cat. no. 23214; Cell Signaling
Technology, Inc.), α-tubulin (cat. no. 11224–1-AP; ProteinTech
Group, Inc.), autophagy related 5 (ATG5; cat. no. 10181-2-AP;
ProteinTech Group, Inc.), microtubule associated protein 1 light
chain 3 (LC3; cat. no. PA1-46286; Thermo Fisher Scientific, Inc.),
lysosomal-associated membrane protein 1 (LAMP1; cat. no. ab25245;
Abcam), mTOR (cat. no. 2983; Cell Signaling Technology, Inc.),
phospho (p)-mTOR (Ser2448; cat. no. 5536; Cell Signaling
Technology, Inc.), Beclin-1 (cat. no. ab207612; Abcam) and
cathepsin D (cat. no. ab75852; Abcam) were utilized. In addition,
horseradish peroxidase (HRP)-conjugated anti-rabbit secondary
antibody (cat. no. SA00001-2; ProteinTech Group, Inc.), rapamycin
(cat. no. S1039; Selleck Chemicals) and chloroquine (CQ; cat. no.
S8808; Selleck Chemicals) were used in this study. Cell culture
reagents were purchased from Thermo Fisher Scientific, Inc.
RNAlater™ Solution (cat. no. AM7020; Thermo Fisher Scientific,
Inc.), TRIzol® reagent (cat. no. 15596018; Invitrogen;
Thermo Fisher Scientific, Inc.), QuantiNova™ reverse transcription
kit (cat. no. 205411; Qiagen, Inc.) and QuantiNova™
SYBR® Green PCR kit (cat. no. 208054; Qiagen, Inc.) were
used for reverse transcription-quantitative (RT-q)PCR. All other
reagents were provided by Sigma-Aldrich (Merck KGaA), unless
otherwise stated.
Primary cell culture
Ventricular muscles of neonatal C57BL/6J mice (age,
1 day old; male and female; weight, 1.5–1.8 g) from the
Experimental Animal Center of the Army Medical University, were
digested with trypsin and cultured following the protocols
published previously (29). Mice
were given food and water freely and were kept in an environmental
of temperature (20–22°C), humidity (50–60%) and a 12/12 h
light/dark cycle. According to the grouping, cellular survival
status and experimental repetitions, 126 neonatal mice were
sacrificed in this research. There were no other causes of
mortality of mice other than execution for the experiment
(extracting myocardium). Before execution, their skin condition
(redness of the skin) and autonomous movements were monitored, and
a warm environment was required. Euthanasia of mice was performed
by decapitation following anesthesia. Mice were anesthetized by
intraperitoneal injection of pentobarbital sodium (50 mg/kg). When
no spontaneous or stimulus-induced movement and squeaks were
detected, cerebral palsy was considered to be effective, and the
mice were decapitated. After soaking with 75% ethanol, the chest
was opened and the heart was extracted instantly. It took ~3 min
from drug injection to observing completely successful anesthesia,
and then to extracting the heart.
Cardiomyocytes were cultured in DMEM-F12 (Hyclone;
Cytiva), supplemented with 5-bromode-oxyuridine (Brdu; 31 mg/l;
Sigma-Aldrich; Merck KGaA), 10% (v/v) heat-inactivated FBS (Gibco;
Thermo Fisher Scientific, Inc.), penicillin G (100 U/ml) and
streptomycin (100 mg/ml; Sigma-Aldrich; Merck KGaA), and maintained
at 37°C in a 5% CO2 incubator. 5Brdu supplement can
effectively inhibit the growth of myocardial fibroblasts, but has
no obvious effect on cardiomyocytes vitality, and has been widely
used in the study of primary myocardial cell culture (29–31).
Treatment with reagents [rapamycin, CQ and small interfering
(si)RNA] for experimental analysis was performed after 3 days of
culture. The reagents treatment processes were mentioned in
following experimental methods.
Establishment of the autophagy
model
On the 3rd day of cell culture, cardiomyocyte
beating was viewed under an optical microscope (magnification,
×400). Fast and rhythmic cellular beats were indicative of a
healthy state (32). The medium was
replaced with rapamycin-containing (200 nM) or DMSO-containing
(same amount as the former) medium and cultured for different time
periods (37°C for 1, 3 and 6 h). Expression levels of
autophagy-related proteins were detected using western blotting to
determine successful establishment.
Autophagy blockage
CQ was used to block autophagy. The medium was
replaced with CQ-containing (100 nM) medium and cardiomyocytes were
pretreated for 2 h at 37°C prior to activation of autophagy with
rapamycin treatment. The blocking effect of CQ on cardiomyocytes
autophagy was detected using western blotting and lysosomal
acidification assay.
CLC-7 knockdown
The CLC-7-specific siRNAs were designed, synthesized
and verified by Shanghai GenePharma Co., Ltd. Non-targeting
controls (negative control siRNA) were used. The sequences used
were: CLC-7 siRNA1 (S1) sense, 5′-GCUCCUCUCCCUCAAGUAUTT-3′ and
antisense, 5′-AUACUUGAGGGAGAGGAGCTT-3′; CLC-7 siRNA2 (S2) sense,
5′-GCAUCUACCAUGGAAAUAUTT-3′ and antisense,
5′-AUAUUUCCAUGGUAGAUGCTT-3′; and negative control siRNA sense,
5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense,
5′-ACGUGACACGUUCGGAGAATT-3′. Cells were transfected with siRNAs
(200 nM) or negative control siRNA (200 nM), using
Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) for 72 h at 37°C prior to reagent treatment.
Western blotting
Samples were homogenized in RIPA buffer (cat. no.
KGP702-100; Changchun Keygen Biological Products Co., Ltd.)
containing protease inhibitor at 4°C, and then centrifuged at
16,000 × g for 15 min at 4°C to extract soluble protein. Protein
extracts were quantified using the Bradford method and loaded on 8
or 12% SDS-gels (20 µg per lane), resolved using SDS-PAGE, and then
transferred to nitrocellulose membranes. Following blocking (5%
skim milk) for 2 h at room temperature, membranes were incubated
overnight at 4°C with primary antibodies against CLC-7 (1:500),
p-mTOR (1:1,000), mTOR (1:800), LC3 (1:2,000), p62 (1:1,000), ATG5
(1:800), Beclin1 (1:800), cathepsin D (1:1,000) or α-tubulin
(1:2,000). Subsequently, the membranes were incubated with
HRP-conjugated anti-rabbit secondary antibody (1:5,000) at room
temperature for 2 h. Signals were visualized using a Quantity One
system image analyzer (Bio-Rad Laboratories, Inc.) and densitometry
analysis was performed using imaging software (Quantity One 4.6.2;
Bio-Rad Laboratories, Inc.).
Immunofluorescence analysis
The samples were primary cardiomyocytes cultured on
thin glass sheets. Immunofluorescence staining was performed as
described previously (6,29). Each sample was rinsed four times (5
min each time) at room temperature with 0.01 M PBS before
incubation with antibodies. Samples were fixed with 4%
paraformaldehyde for 15 min at room temperature. Blocking was
performed by immersing samples in 3% BSA (Sigma-Aldrich; Merck
KGaA) for 1 h at room temperature. The primary antibodies used were
rabbit anti-CLC-7 (1:100) and rat anti-LAMP1 (1:500). The samples
were incubated with primary antibodies at 4°C overnight, washed in
PBS and incubated with secondary antibodies for 1 h at room
temperature. The secondary antibodies used were goat polyclonal
secondary antibody to rat IgG H&L (Alexa Fluor 488) (1:500;
cat. no. ab150157; Abcam) or goat polyclonal secondary antibody to
rabbit IgG H&L (Alexa Fluor 633) (1:100; cat. no. A-21070;
Thermo Fisher Scientific, Inc.). Nuclei were stained with DAPI
(1:200) for 2 min at room temperature. The quantification of
co-localization was performed as described previously (33). Briefly, co-localization levels were
quantified by measuring the intensity of regions where red and
green fluorescence overlapped, using confocal software (LAS AF Lite
2.4.1; Leica Microsystems CMS GmbH) and ImageJ version 1.8.0
(National Institutes of Health). Co-localization was identified
when the maxima of two overlapping peaks were shifted <20 nm. To
assess lysosome-autophagosome fusion, primary antibodies against
LC3B (1:200) and LAMP1 (1:500) were utilized (34). LAMP1 localizes in the outer membrane
of lysosomes, whereas LC3B localizes in both the outer or inner
membranes of autophagosomes (35).
Therefore, double-positive LC3 and LAMP1 (yellow) puncta are used
to evaluate lysosome-autophagosome fusion (34). All samples were viewed under a Leica
TCS SP5 laser confocal scanning microscope (magnification, ×600;
Leica Microsystems GmbH), at the same voltage (220 V) to ensure
comparability of fluorescence intensity. Every image was captured
at a single confocal plane. In each group of samples, three fields
of view were chosen randomly, all cells in each field were observed
and quantified.
RT-qPCR
After removing medium, cell samples were immersed in
RNAlater™ Solution and RNA was extracted using TRIzol®
reagent. cDNA was synthesized using a QuantiNova™ reverse
transcription kit according to the manufacturer's protocol. The
reaction system was as follows: 50°C for 15 min, 85°C for 5 sec.
Using QuantiNova™ SYBR® Green PCR kit, qPCR was
performed on an Applied Biosystems® 7500 Real-Time PCR
system (Applied Biosystems; Thermo Fisher Scientific, Inc.). The
reaction conditions were as follows: 95°C for 30 sec; 40 cycles of
95°C for 10 sec and 60°C for 30 sec; 95°C for 15 sec, 60°C for 60
sec, 95°C for 15 sec. The sequences of the primers used were: LC3,
forward, 5′-CTGCCTGTCCTGGATAAGACCA-3′ and reverse,
5′-CTGGTTGACCAGCAGGAAGAAG-3′; and p62 forward,
5′-GCTCTTCGGAAGTCAGCAAACC-3′ and reverse,
5′-GCAGTTTCCCGACTCCATCTGT-3′; and β-actin forward,
5′-CATGTACGTTGCTATCCAGGC-3′ and reverse,
5′-CTCCTTAATGTCACGCACGAT-3′. The 2−ΔΔCq method was used
for data analysis (36).
Cell Counting Kit-8 (CCK-8) viability
assay
The CCK-8 assay was performed following the
manufacturer's instructions. Following treatment, medium in each
well (96-well plate) was replaced with 100 µl medium containing 10
µl CCK-8 reagent (Gen-view Scientific, Inc.) and incubated at 37°C
for 4 h. Optical density was measured using an automatic microplate
reader (Thermo Fisher Scientific, Inc.) at 450 nm.
Assessment of lactate dehydrogenase
(LDH) leakage
The leakage of LDH was determined using a
CytoTox-ONETM Homogeneous Membrane Integrity assay (Promega
Corporation). Briefly, 100 µl reaction buffer was transferred to
each well (96-well plate) and the samples were incubated at room
temperature for 15 min, then 25 µl stop buffer was added. The
plates were shaken for 10 sec and the fluorescence intensity was
measured at 560/580 nm. The reported data are given in percentages
of the results of the respective control groups.
Lysosomal acidification assay
Lysosomal acidification was measured using
LysoTracker and LysoSensor Probes (Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. The probe stock solution
was diluted to a working concentration (LysoTracker, 50 nM;
LysoSensor, 1 µM) in the medium. Cardiomyocytes were cultured in
medium containing the probe for 1 h at 37°C. The loading solution
was replaced with fresh medium and cardiomyocytes were observed
under a Leica TCS SP5 laser confocal scanning microscope
(magnification, ×600).
Transmission electron microscopy
(TEM)
TEM was used to detect autolysosomes or
autophagosomes in cardiomyocytes. Briefly, freshly prepared samples
were fixed overnight at 4°C with 2.5% glutaraldehyde in 0.1 M
phosphate buffer (pH 7.4) and 1% osmium tetroxide (pH 7.4). Samples
were dehydrated with a series of acetone (90 and 100%) solutions
and embedded in Epon (4 h) at room temperature. Before TEM, thin
sections (60 nm) were cut and double-stained at room temperature
with uranyl acetate (20 min) and lead citrate (5 min). Samples were
viewed and imaged (indicated magnification, 80 kx) using a Philips
TECNAI10 electron microscope (Philips Medical Systems, Inc.).
Statistical analysis
SPSS version 13.0 (SPSS, Inc.) was used for
statistical analysis. Data are presented as the mean ± standard
deviation of ≥3 independent repeats. An unpaired Student's t-test
or one-way ANOVA with Bonferroni's post-test was used to compare
differences between groups, based on the number of groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Rapamycin treatment activates
autophagy in the myocytes
Rapamycin is widely used in autophagy research
(37,38). To establish a stable cell autophagy
model, cardiomyocytes were treated with rapamycin, and the
expression levels of autophagy-related proteins were detected. DMSO
is an effective solvent for rapamycin (39); thus, DMSO groups, serving as control
(vehicle) groups, were studied in parallel to rapamycin-treated
groups. Following treatment with rapamycin for different periods of
time (1, 3 and 6 h), p-mTOR/mTOR ratio was decreased (P<0.05 and
P<0.01; Fig. 1A and B),
suggesting functional inhibition of mTOR. Moreover, the increase in
the expression levels of autophagy-related proteins, including
Beclin1, ATG5 and LC3 further confirmed activation of autophagy
(P<0.05 and P<0.01; Fig. 1A and
B).
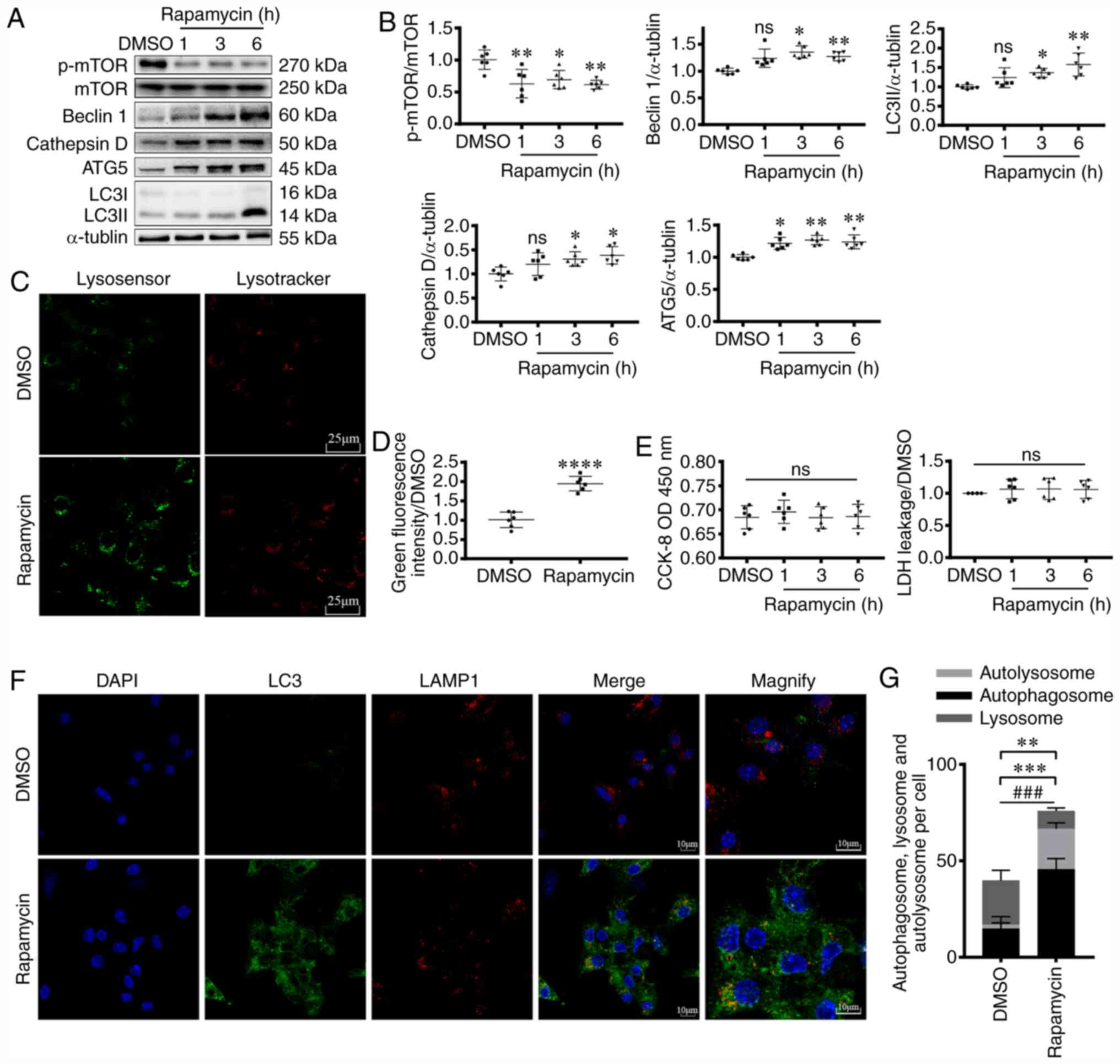 | Figure 1.Rapamycin treatment activates
myocardial autophagy pathway. (A and B) Western blotting, (C, D, F
and G) fluorescence staining and (E) CCK-8 and LDH analyses showing
the induction of myocardial autophagy following rapamycin
treatment. (A) Western blotting was used to assess p-mTOR (270
kDa), mTOR (250 kDa), Beclin1 (60 kDa), Cathepsin D (50 kDa), ATG5
(45 kDa) and LC3 (I: 16 kDa, II: 14 kDa) expression levels in
cardiomyocytes following rapamycin treatment for 1, 3 or 6 h.
α-tubulin (55 kDa) levels were measured as the loading control. (B)
Graphs representing the results. ANOVA. *P<0.05, **P<0.01 vs.
DMSO group. n=6. (C) Lysosomal acidification in the absence (DMSO)
or presence of 200 nM rapamycin (6 h) were measured using
LysoTracker and LysoSensor. Indicators show pH-dependent increase
of green fluorescence intensity upon lysosomal acidification. (D)
Quantification of results. Scale bar, 25 µm. Unpaired t-test.
****P<0.0001 vs. DMSO. n=6. (E) CCK-8 and LDH analysis of
cardiomyocyte viability following rapamycin treatment. ANOVA. n=6.
(F) Confocal images of LC3+ puncta (green dots) and
LAMP1+ puncta (red dots) in cardiomyocytes. (G) Graph
demonstrates the mean number of autolysosomes (yellow dots) and
autophagosomes (green dots). Scale bar, 10 µm. Unpaired t-test.
**P<0.01 vs. DMSO, comparing the number of autolysosome +
autophagosome; ***P<0.001 vs. DMSO, comparing the number of
autolysosome; ###P<0.001 vs. DMSO, comparing the
number of autophagosome. n=5. CCK-8, Cell Counting Kit-8; LDH,
lactate dehydrogenase; p-phospho-; ns, not significant; ATG5,
autophagy related 5; LC3, microtubule associated protein 1 light
chain 3; LAMP1, lysosomal-associated membrane protein 1; OD,
optical density. |
To observe autophagy more directly, following
rapamycin treatment for 6 h, immunofluorescence staining was
performed (Fig. 1F and G). The
number of autophagosomes (green) was significantly increased
(P<0.001), in line with the higher number of autolysosomes
(yellow, P<0.001). Additionally, total autophagosome and
autolysosome counts suggested enhanced autophagy (P<0.01).
Lysosomes serve pivotal roles in autophagy, and
their acidification level is also an important indicator of
autophagy (19). In the present
study, following rapamycin treatment, the increase in green
fluorescence intensity suggested that rapamycin treatment activated
lysosomal function (P<0.0001; Fig.
1C and D). Cathepsin D, a lysosomal protease, is stable at low
pH levels, and indirectly reflects lysosomal acidity (40). Following 3 and 6 h of rapamycin
treatment, an increase in the protein expression levels of
cathepsin D was observed (P<0.05; Fig. 1A and B), further suggesting that
rapamycin enhanced lysosomal function.
To investigate the effects of autophagy on cell
status, cardiomyocyte viability was assessed using CCK-8 and LDH
assays. The results demonstrated that rapamycin did not
significantly affect cell viability in early autophagy (within 6 h)
under normal conditions (Fig. 1E).
Taken together, the results suggested that rapamycin treatment
activated autophagy in cardiomyocytes.
Lysosomal acidification promotes
myocardial autophagy
Since enhanced autophagy and activated lysosomal
acidification were observed following rapamycin treatment, the role
of lysosomes in cardiomyocyte autophagy was further examined
(Fig. 2A-I). CQ, a lysosomal
acidification inhibitor (41) was
used to pretreat cardiomyocytes before rapamycin treatment and
autophagy was monitored. Cathepsin D protein expression was reduced
following CQ treatment (P<0.01; Fig.
2A and B), which was in line with the changes in lysosomal
green fluorescence intensity (P<0.01; Fig. 2D and E). These findings suggest that
lysosomal acidification was inhibited by CQ. Along with decreased
lysosomal acidification, the LC3II and p62 expression levels were
increased (P<0.01; Fig. 2A and
C). The increase in LC3II and p62 expression levels may have
been due to increased production or reduced degradation; thus, the
mRNA expression levels were assessed. No significant changes in
mRNA expression levels following CQ treatment were observed,
eliminating the possibility of gene expression variations (Fig. 2F). These results indicated that
lysosomal weakening of acidification induced autophagosome
accumulation and obstructed autophagy.
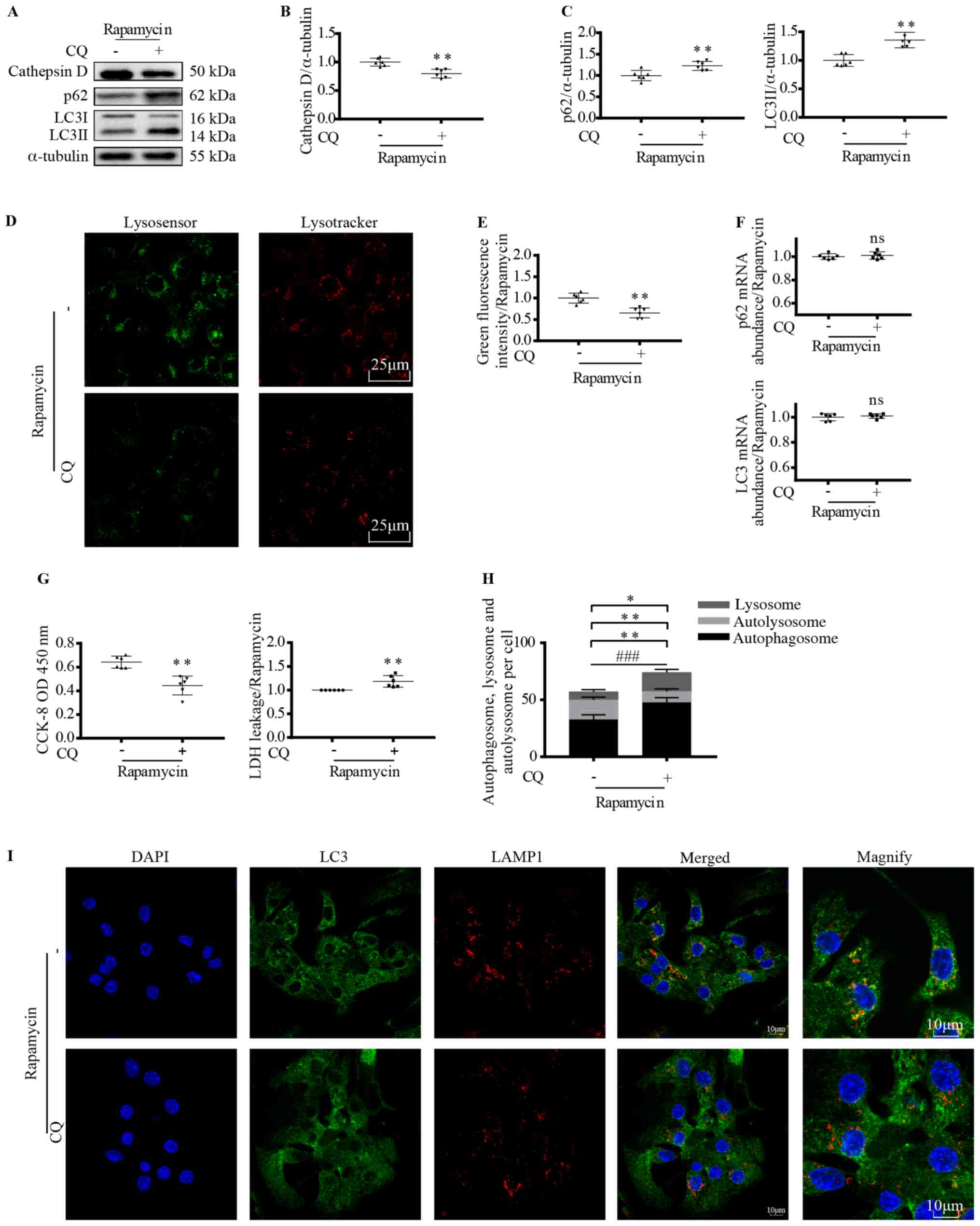 | Figure 2.Lysosomal acidification promotes
myocardial autophagy. Cardiomyocytes were treated with 200 nM
rapamycin for 6 h with or without pretreatment with CQ (100 nM for
2 h). (A) Western blotting and semi-quantitative analysis of (B)
cathepsin D, (C) LC3II and p62 protein expression levels. Unpaired
t-test. **P<0.01 vs. untreated control. n=6. (D) Lysosomal
acidification was measured using LysoTracker and LysoSensor. (E)
Graph shows quantification of green fluorescence intensity. Scale
bar, 25 µm. Unpaired t-test. **P<0.01 vs. untreated control.
n=6. (F) Analysis of LC3 and p62 gene expression levels using
reverse transcription-quantitative PCR. Unpaired t-test. n=6. (G)
Cardiomyocyte viability was evaluated using a CCK-8 and LDH assay.
Unpaired t-test. **P<0.01 vs. untreated control. n=6. (H) Mean
number of autolysosomes (yellow dots) and autophagosomes (green
dots) were quantified. (I) Confocal images show LC3 co-localization
with LAMP1. Scale bar, 10 µm. Unpaired t-test. *P<0.05,
autolysosome + autophagosome; **P<0.01, lysosome, autolysosome;
###P<0.001, autophagosome. n=5. CQ, chloroquine; LC3,
microtubule associated protein 1 light chain 3; CCK-8, Cell
Counting Kit-8; LDH, lactate dehydrogenase; LAMP1,
lysosomal-associated membrane protein 1; ns, not significant; OD,
optical density. |
Immunofluorescence results further confirmed these
findings (Fig. 2H and I). The
proportion of uncombined autophagosomes (green; P<0.001) and
lysosomes (red; P<0.01) was increased, while the proportion of
autolysosomes was decreased (yellow; P<0.01) following CQ
treatment, suggesting reduced autophagosome-lysosome fusion and
accumulation of autophagy substrates. The significant increase in
the total amount of autophagosomes and autolysosomes (P<0.05)
also indicated enhanced substrate accumulation. Additionally,
cardiomyocyte viability was decreased following CQ treatment, as
shown in the CCK-8 and LDH assays (P<0.01; Fig. 2G).
The aforementioned results demonstrated that
insufficient lysosomal function results in autophagosome
accumulation and cardiomyocyte damage, indicating that lysosomal
acidification is crucial for functional cardiomyocyte
autophagy.
CLC-7 is recruited to lysosomes during
cardiomyocyte autophagy
Subsequently, changes in CLC-7 expression were
determined using western blotting (Fig.
3A and B). There was a slight, but insignificant increase in
CLC-7 expression following rapamycin treatment. Results of the
immunofluorescence analysis also showed similar results (Fig. 3C and D).
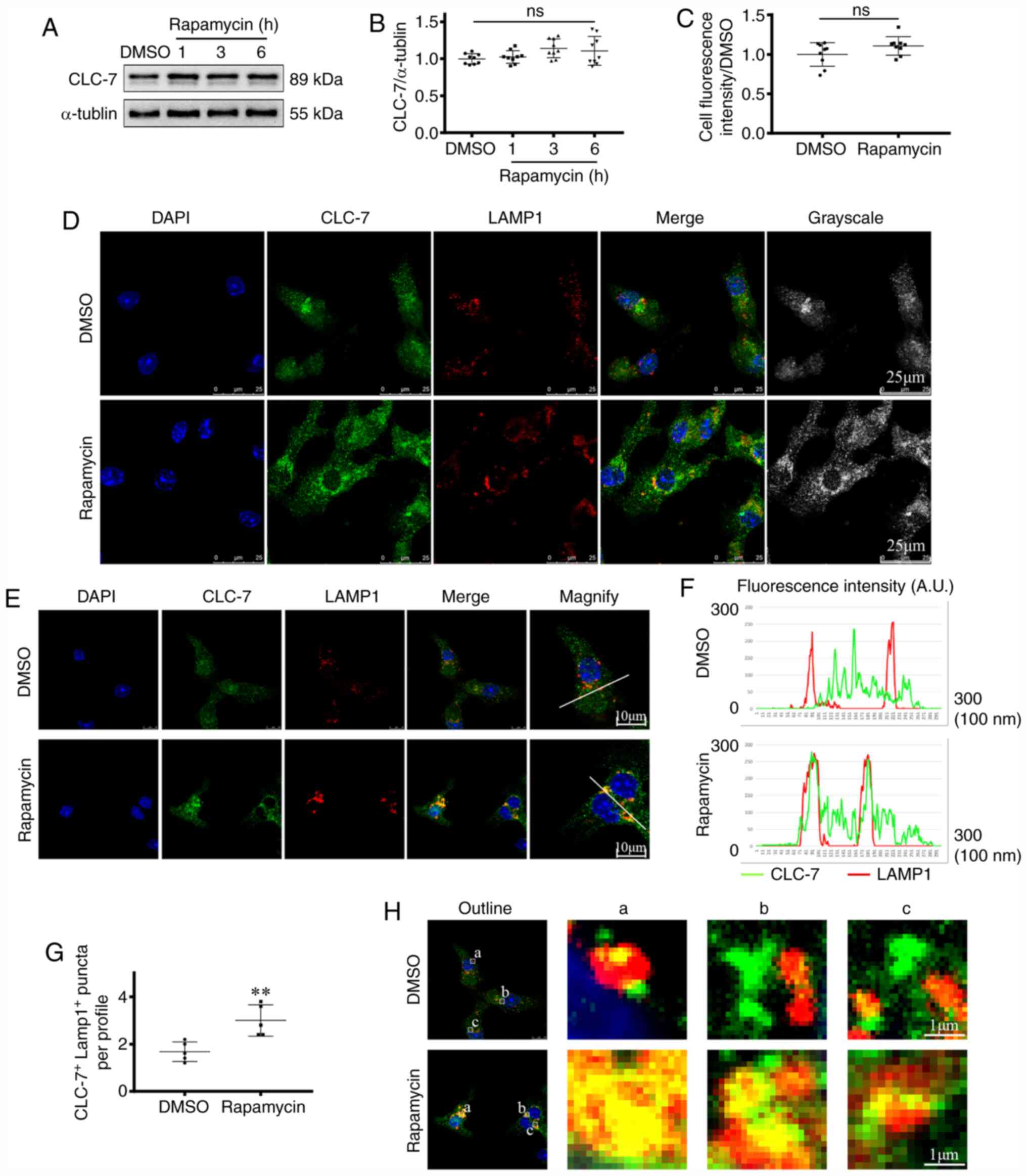 | Figure 3.CLC-7 is recruited to lysosomes
during cardiomyocyte autophagy (A and B) Western blotting and (C-H)
fluorescence staining showing myocardial CLC-7 protein expression
levels and localization following rapamycin treatment.
Immunofluorescent confocal micrographs of cardiomyocytes stained
for anti-CLC-7 (green), anti-LAMP1 (red) and DAPI for the nuclei
(blue). (A) Protein expression levels of CLC-7 were detected by
western blotting and (B) semi-quantified. ANOVA, n=9. (C)
Quantitative fluorescence analysis of total CLC-7, as detecting by
(D) immunofluorescent staining. Scale bar, 25 µm. Unpaired t-test.
n=9. (E) Amplified views show co-localization levels of CLC-7 and
LAMP1. Scale bar, 10 µm. (F) The graph shows the fluorescence
intensity peaks along profiles crossing CLC-7+
LAMP1+ puncta indicated in magnified micrograph in panel
(E). (G) Co-localization levels were quantified by measuring
fluorescence intensity overlaps, using confocal software. Unpaired
t-test. **P<0.01 vs. DMSO group. n=5. (H) Lysosomes in the
perinuclear region were viewed and shown in magnified images a, b
and c. Scale bar, 1 µm. LAMP1, lysosomal-associated membrane
protein 1; CLC-7, H(+)/Cl(−) exchange transporter 7; ns, not
significant. |
Extensive lysosomal recruitment of CLC-7 in
rapamycin-treated groups was observed (Fig. 3E-H). LAMP1 was used as the marker of
lysosomes (27). CLC-7
co-localization with LAMP-1 was increased in the rapamycin-treated
groups compared with the DMSO group. In the DMSO group, CLC-7 was
distributed diffusely throughout the cytosol. There was either
partial or no co-localization between the two proteins. By
contrast, in the rapamycin-treated groups, CLC-7 localized to the
perinuclear region and was concentrated in lysosomes (Fig. 3E). Co-localization levels were
quantified by measuring red and green fluorescence intensity
overlaps, as described previously (33). Complete co-localization was observed
when the maxima of two overlapping peaks were shifted <20 nm. An
increase in overlapping of fluorescence intensity peaks was
observed in rapamycin-treated groups compared with the DMSO groups
(P<0.01; Fig. 3F-G). These
findings suggested that there was increase in intracellular CLC-7
located in the lysosomes following rapamycin treatment. Thus, CLC-7
protein expression in the lysosomes in the perinuclear region was
subsequently observed (Fig. 3H). In
rapamycin-treated groups, increased CLC-7-lysosome co-localization
(yellow) was observed, in which the green puncta fused with the red
puncta completely. By contrast, in the DMSO groups, partial
co-localization was viewed, and various green puncta were
distributed besides the red puncta. Together, these findings
suggested that CLC-7 was recruited to lysosomes during
cardiomyocyte autophagy.
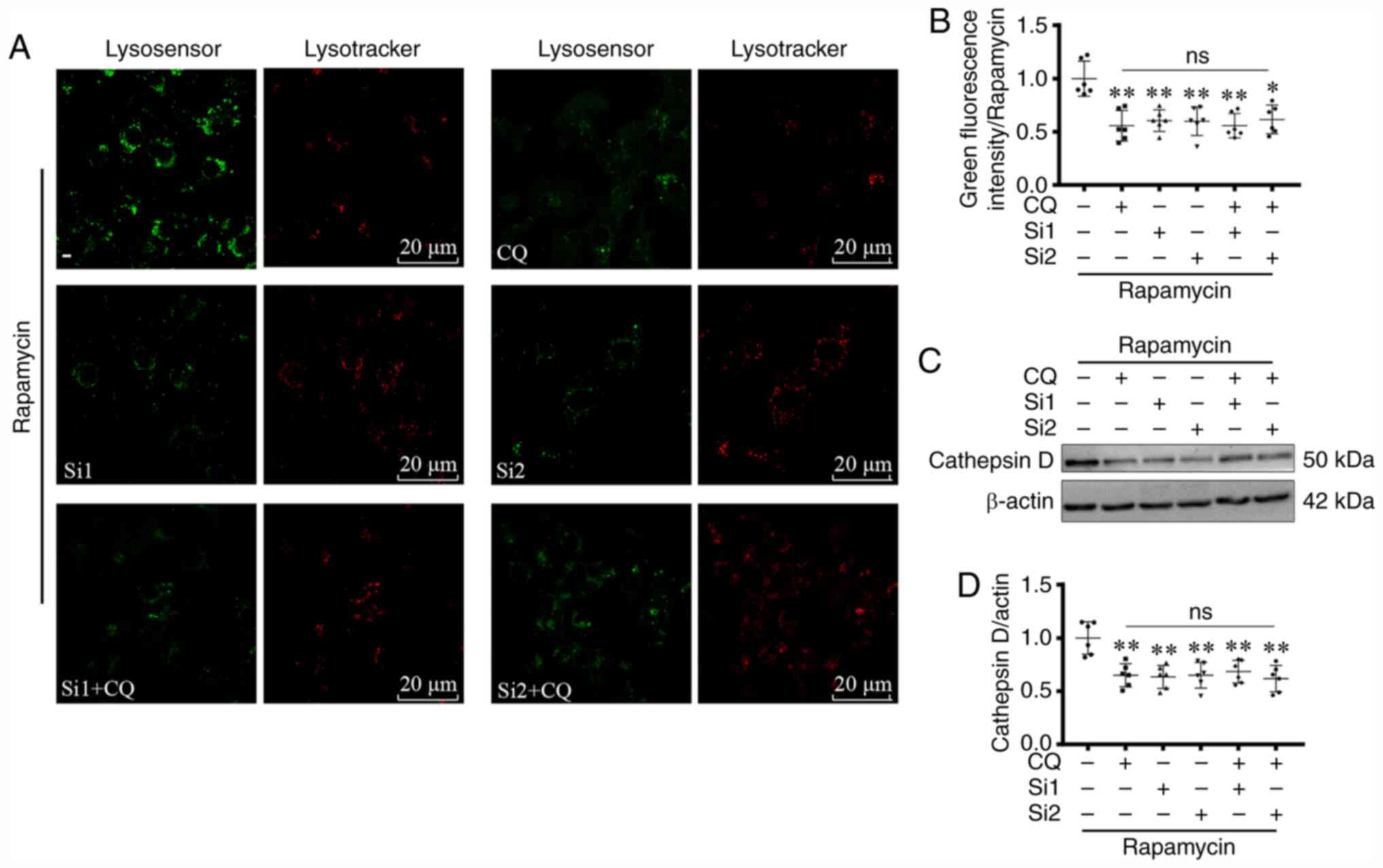
Autophagy substrates accumulate
following CLC-7 silencing
As enhanced CLC-7-LAMP1 co-localization was observed
following rapamycin treatment, it was hypothesized that CLC-7 may
participate in lysosomal acidification and autolysosome digestion.
Therefore, CLC-7 expression was knocked down using CLC-7 siRNAs
(Si1 and Si2). Following siRNA transfection, CLC-7 protein
expression levels were significantly decreased as detected via
western blotting (P<0.01; Figs. 4A,
B, D, F and S1) and
immunofluorescence observation (P<0.05; Fig. 4G and H). Along with decreased
cellular CLC-7 levels, deficient CLC-7-lysosome co-localization was
also observed (Fig. 4H).
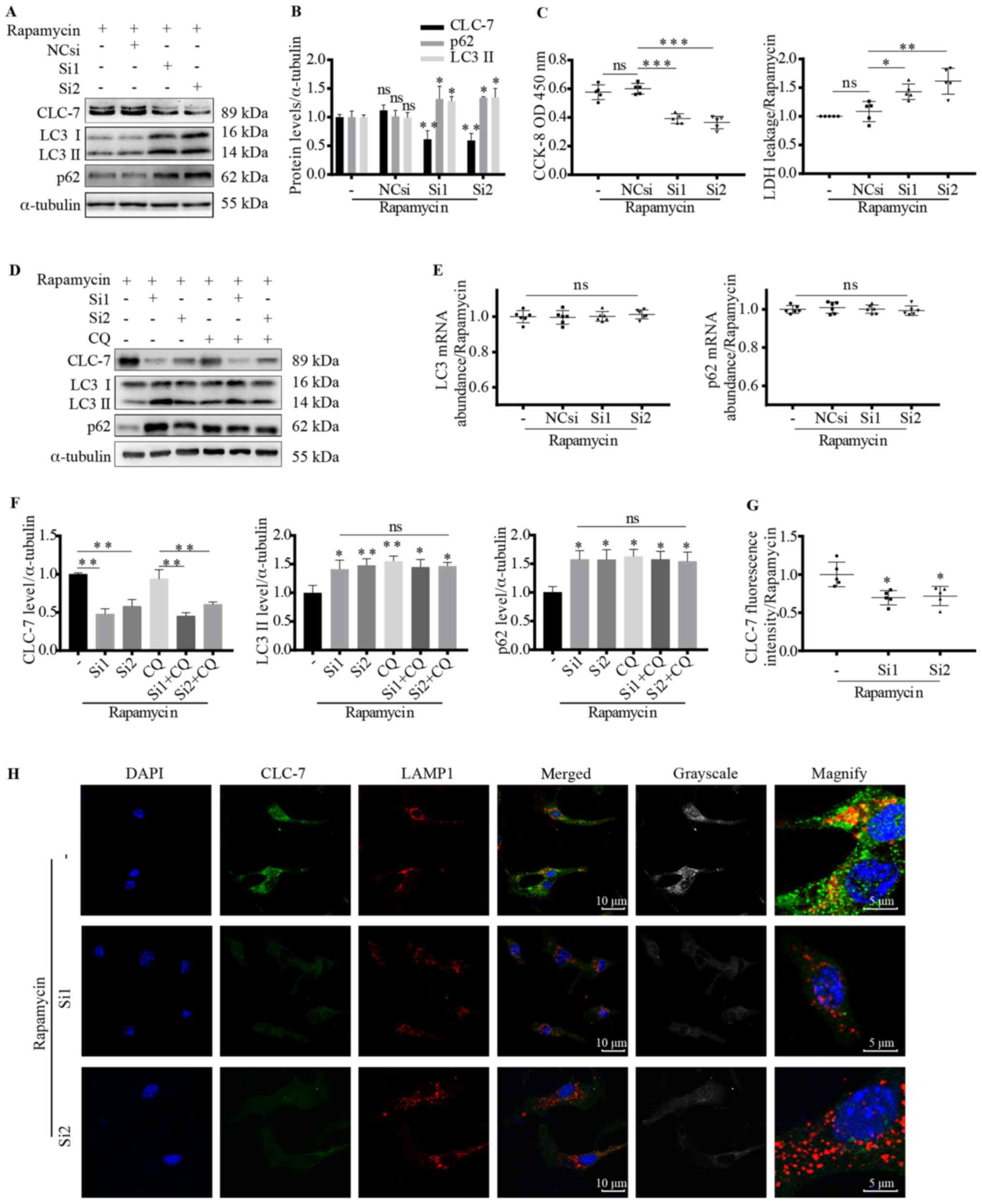 | Figure 4.Autophagy substrates accumulate
following CLC-7 silencing. (A, B, D and F) Expression levels of
autophagy-related proteins and CLC-7 were measured using western
blotting, (E) RT-qPCR and (G and H) immunofluorescence. (C) Cell
viability was assessed using CCK-8 and LDH assays following
transfection with CLC-7 siRNAs with or without CQ pretreatment. (A)
Western blot analysis of (B) expression levels of autophagy-related
proteins following transfection with CLC-7 siRNAs or NCsi.
*P<0.05, **P<0.01 vs. NCsi group. n=3. (C) Cardiomyocyte
viability analysis using CCK-8 and LDH assays. *P<0.05,
**P<0.01, ***P<0.001. n=5. (D) Western blot analysis of (F)
expression levels of autophagy-related proteins following
transfection with CLC-7 siRNAs with or without CQ (100 nM for 2 h)
pretreatment. *P<0.05, **P<0.01 vs. untreated control. n=3.
(E) LC3 and p62 mRNA expression levels were assessed using RT-qPCR.
ANOVA, n=6. (G) Quantification of (H) immunofluorescence analysis
of the localization and protein expression levels of CLC-7
following transfection with CLC-7 siRNAs. Scale bar, 5 or 10 µm.
ANOVA. *P<0.05 vs. untreated control. n=5. CLC-7, H(+)/Cl(−)
exchange transporter 7; siRNA, small interfering RNA; CQ,
chloroquine; CCK-8, Cell Counting Kit-8; LDH, lactate
dehydrogenase; NCsi, negative control siRNA; LC3, microtubule
associated protein 1 light chain 3; RT-qPCR, reverse
transcription-quantitative PCR; ns, not significant; OD, optical
density. |
Immunoblotting results demonstrated that CLC-7 was
involved in autophagy (Fig. 4A and
B). Moreover, following rapamycin treatment, LC3II and p62
expression levels were increased in the siRNA-transfected groups,
suggesting accumulation of autophagy substrates (P<0.05;
Fig. 4A and B). To investigate the
effect of CLC-7 on substrate generation or digestion, gene
expression levels of LC3 and p62 were assessed. However, increased
LC3 and p62 protein levels were not associated with increased
transcription (Fig. 4E). CQ, the
lysosomal function inhibitor, was used to further determine the
target of CLC-7. CLC-7 knockdown increased LC3 II and p62 protein
levels (P<0.05 and P<0.01; Fig.
4F), but there was no further increase when cells were
additionally treated with CQ. Cell viability was measured using
CCK-8 and LDH assays. Following rapamycin treatment, CLC-7
knockdown decrease cardiomyocyte viability (P<0.05, P<0.01
and P<0.001; Fig. 4C),
suggesting CLC-7 may be important for cardiomyocyte survival and
was involved in autophagy.
CLC-7 knockdown decreased the co-localization of LC3
(green) and LAMP1 (red) (Fig. 5A and
B). In CLC-7-knockdown cells, the proportion of uncombined
autophagosomes increased (P<0.01), whereas the proportion of
autolysosomes (yellow) decreased (P<0.05 and P<0.01). These
trends were not further altered when cells were additionally
treated with CQ (Fig. 5B). These
findings suggested that CLC-7 knockdown blocked autophagy, and
lysosomes served as the primary targets. Furthermore, the
ultrastructure was observed via TEM (Fig. 5C). Following rapamycin treatment (6
h), in control groups, there was an increase in digested autophagy
substrates visible in autolysosomes (black arrow), and an increase
in undigested organelles within the lumina (black triangle)
following CLC-7 knockdown or CQ treatment. The results indicated
that CLC-7 knockdown impaired autophagy substrate clearance.
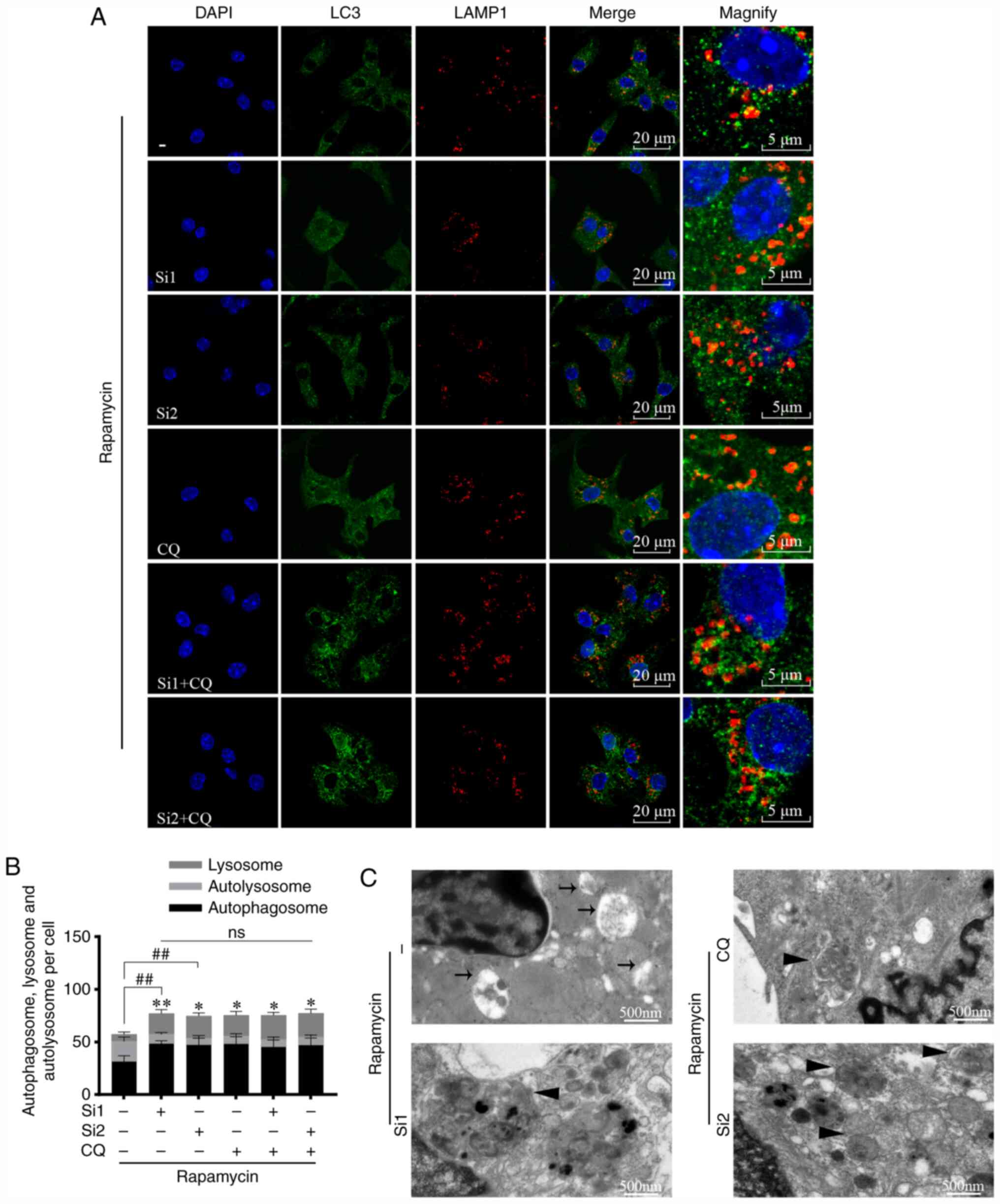 | Figure 5.CLC-7 silencing causes autophagosome
accumulation. Cardiomyocytes were transfected with 200 nM CLC-7
siRNAs or pretreated with 100 nM CQ for 2 h, then treated with 200
nM rapamycin for 6 h. (A) Cardiomyocytes were incubated with
antibodies against LC3 (green) and LAMP1 (red) and viewed via
confocal microscopy. Scale bar, 5 µm. (B) Quantitative results are
shown in the bar chart. Autolysosomes (yellow dots) and
autophagosomes (green dots) were counted per cell in three
microscopic fields per group. The mean number was calculated.
ANOVA. *P<0.05, **P<0.01, autolysosome;
##P<0.01, autophagosome. n=5. (C) Electron micrograph
showing the morphology of autophagosomes and autolysosomes in
cardiomyocytes; triangles represent autophagosomes, arrows
represent autolysosomes. Scale bar, 500 nm. Independent experiments
were repeated three times. CLC-7, H(+)/Cl(−) exchange transporter
7; siRNA, small interfering RNA; NCsi, negative control siRNA; LC3,
microtubule associated protein 1 light chain 3; LAMP1,
lysosomal-associated membrane protein 1; CQ, chloroquine; ns, not
significant. |
CLC-7 silencing results in decreased
lysosomal acidification
Since CLC-7 knockdown and CQ treatment appeared to
exhibit a partial same effect on autophagy, the role of CLC-7 in
lysosomes, specifically on lysosomal acidification, was assessed
using LysoTracker and LysoSensor Probes. The green fluorescence
levels were decreased in CLC-7 siRNA-transfected cells,
irrespective of CQ treatment (P<0.05 and P<0.01; Fig. 6A and B). The difference between
transfected cells treated with or without CQ was not significant.
These findings indicated that lysosomal acidification was weakened
in the CLC-7 siRNA transfected cells, and CQ did not have further
effect on acidification in the knockdown cells. Changes in
cathepsin D protein expression levels also provided similar results
(P<0.01; Fig. 6C and D),
suggesting weak lysosomal acidification. Collectively, the results
suggested that CLC-7 promoted lysosomal acidification in autophagy
in cardiomyocytes.
Discussion
The present study demonstrated that CLC-7 was
recruited to lysosomes in cardiomyocytes during autophagy. CLC-7
knockdown attenuated lysosomal acidification, thereby blocking
autolysosome self-degradation. These findings suggested that CLC-7
participated in the regulation of myocardial lysosomal
acidification-mediated autophagy. Furthermore, in previous studies
(23,24,27),
the pathological changes of CLC-7 were examined in chronic
diseases, whereas, to the best of our knowledge, there are no
studies on the role of CLC-7 in autophagy. The present study
provided evidence that antiporters may serve a more pronounced role
during early autophagy. Autophagy occurs under various acute stress
responses, such as hypoxia, ischemia and oxidative stress (1). This indicates that the role of CLC7
should be evaluated in both relatively protracted or genetic
diseases, but also in certain conditions associated with acute
stress.
Studies have confirmed that CLC-7 is located in the
endoplasmic reticulum in the perinuclear region or is diffused in
the cytoplasm in an inactivated state, and transferred to lysosomes
where it becomes functionally activated (25,26).
Inefficient CLC-7 delivery to lysosomes aggravates Alzheimer's
disease (26), while aberrant CLC-7
function leads to osteoarthritis (42). In the present study, lysosomal
recruitment of CLC-7 was increased following activation of
autophagy in mouse cardiomyocytes. In inactivated autophagic
vesicles, increased diffuse staining of CLC-7 in cardiomyocytes was
observed, dissimilar to the punctate LAMP1 positive staining seen
in the activated autophagic vesicles. These results suggested that
CLC-7 was involved in the regulation of autophagy.
In the present study, autophagy was induced using
rapamycin, a specific mTOR inhibitor, as has been performed
previously (37). mTOR is the
initial switch for activation of autophagy (43). In this study, p-mTOR/mTOR was
decreased, while as the expression levels of autophagy-related
proteins were increased following rapamycin treatment. These
findings demonstrated that autophagy was induced. Additionally,
CLC-7 localization changed following induction of autophagy.
However, autophagy is an intricately connected process that is
involved in numerous complex conditions (1), and whether CLC-7 alone underlies the
aberrant autophagy in these pathological conditions requires
further investigation.
Lysosomes are key components in autophagy flux
(44). In the present study,
acid-sensitive lysosomal tracker and cathepsin D were used to
detect lysosomal acidification. P62, an autophagy receptor, is a
marker reflecting autophagy levels, and its aggregation indicates
autophagosome accumulation (12,41,45).
LC3II, which is formed following cleavage of LC3I, is located on
autophagosome membranes during autophagy. Therefore, the levels of
LC3II are used as autophagy detecting tools (40,45).
Following inhibition of lysosomal acidification using CQ, lysosomal
acidification decreased and substrate LC3II and p62 protein
expression levels increased, suggesting autophagosome accumulation.
Thus, the present results demonstrated that lysosomal acidification
promoted autophagy substrate degradation; a finding that is in line
with previous studies (14,37,44).
LAMP1 is considered a lysosomal marker protein, and
as the autophagosomes fuse with lysosomes, LC3 and LAMP1
co-localized puncta are detectable in autolysosomes (40). In the present study, CQ treatment
was used as a positive control for blocking autophagy flux. CQ
disrupts autophagy by inhibiting lysosomal acidification (37). A previous study reported that
rapamycin activated autophagy and autophagosome-lysosome fusion in
CHO cells, whereas CQ increased lysosomal pH, and therefore blocked
the fusion (37). In the present
study, autophagosome-lysosome fusion was increased following
rapamycin treatment and was decreased after additional treatment
with CQ. CLC-7 silencing or CQ treatment reduced the number of
LC3+ LAMP1+ puncta, whereas the number of
LC3+ puncta increased, suggesting decreased
autophagosome-lysosome fusion and an accumulation of unfused
autophagosomes. Furthermore, the difference between the counts of
autophagosomes, and the abundance of LC3 and p62 mRNA expression
levels in the two treatment conditions was insignificant,
indicating a similarity in the effect of CLC-7 knockdown and CQ
treatment on autophagy. CQ did not further increase the of
autophagy substrates, which were accumulated after CLC-7 silencing.
Accumulation of the substrates was observed in the autophagosomes
following CLC-7 knockdown or CQ treatment using TEM. These results
further support our previous hypothesis that CLC-7 knockdown and CQ
treatment have similar effect on autophagy. Lysosomal acidification
was inhibited by CLC-7 knockdown, suggesting that lysosomal
acidification was the target, and CLC-7 silencing interfered with
autolysosome-mediated degradation by blocking lysosomal
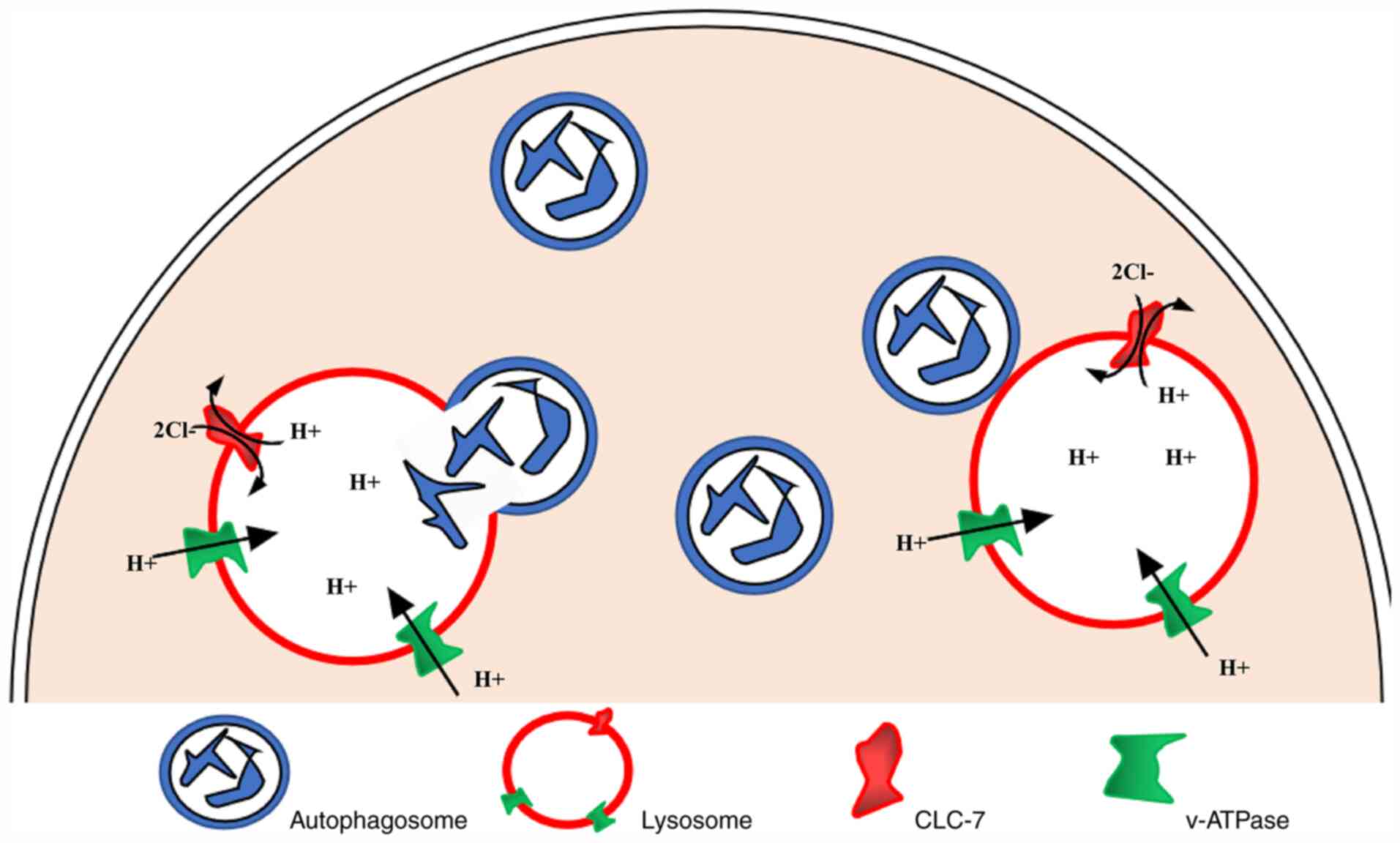
acidification. Previous studies have revealed that v-ATPase in
lysosome membrane transfers H+ into the lumen to acidify
lysosomes (16,19,46)
(as shown in the schematic diagram in Fig. 7). At the same time, the increase in
potential caused by the continuous accumulation of luminal
H+ hinders the further transfer of H+. CLC-7,
as a 2Cl−/1H+ antiporter, exports one
H+ and imports two Cl−, resulting in a net
negative charge to offset the increase in the luminal potential,
thereby promoting the continuous flow of H+ and lysosomal
acidification further (21,22).
However, the present study has the following
limitations. Although it has been clarified that CLC-7 is
indispensable for autophagy flux, further investigation should be
conducted. For instance, under stress conditions, such as oxidative
stress or acute hypoxia, it should be examined whether impaired
lysosomal acidification is caused by insufficient CLC-7 expression
or dysfunction, or a joint involvement. Secondly, the specific
molecular mechanism that causes insufficient CLC-7 expression or
dysfunction under such stress conditions needs to be clarified in
order to identify effective targets in improving lysosomal
acidification and autophagy. It should also be considered that
CLC-7, as a membrane protein, may directly participate in the
molecular interaction in autophagosome-lysosome fusion.
Additionally, CLC-7 ion transport function may require further
investigation using electrophysiological techniques.
In conclusion, to the best of our knowledge, the
present study was the first to examine the roles of CLC-7 in
autophagy. The present results suggested that CLC-7 was essential
for lysosomal acidification and lysosome-mediated autophagy in
cardiomyocytes. These findings increase the understanding of CLC-7
function in autophagy, and provide a novel insight into the role of
CLC-7, differing from that in CLC-7 deficiency-related diseases.
CLC-7 activation may be a novel and valuable therapeutic target for
improving autophagy. Additional studies may yield further insights
into organ therapy following acute stress and in autophagy-mediated
diseases.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported by the Key Program of
National Natural Science Foundation of China (grant no.
81430042).
Availability of data and materials
The datasets used and/or analyzed in the current
study are available from the corresponding authors on reasonable
request.
Authors' contributions
YH and DZ conceived the research and supervised the
study. JL, JW and YL designed the experiments with help from DZ and
XZ. JL and JW performed the experiments with help from XZ, QZ, RFY
and JJ. JL, JW and CD participated in the acquisition of raw data
and computed data and collated the data. JL, JW and XZ analyzed the
data. JL, JW and CD wrote, edited and revised the manuscript. All
authors participated in revising the manuscript. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Animal-based investigations were designed in
accordance with the Guide for the Care and Use of Laboratory
Animals published by the National Institutes of Health (NIH Pub.
No. 85-23, revised 1996). The present study was reviewed and
approved by the Animal Experiment Ethics Committee of the Third
Military Medical University in Chongqing, China. All experiments
involving animals were performed in accordance with the ethical
standards of the institution or practice at which the studies were
performed.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CLC-7
|
H(+)/Cl(−) exchange transporter 7
|
|
v-ATPase
|
vacuolar-type H+-ATPase
|
|
LC3
|
microtubule associated protein 1 light
chain 3
|
|
p62
|
sequestosome 1
|
|
LAMP1
|
lysosomal-associated membrane protein
1
|
|
CQ
|
chloroquine
|
References
|
1
|
Ravanan P, Srikumar IF and Talwar P:
Autophagy: The spotlight for cellular stress responses. Life Sci.
188:53–67. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Glick D, Barth S and Macleod KF:
Autophagy: Cellular and molecular mechanisms. J Pathol. 221:3–12.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Deng M, Huang L and Zhong X: β-asarone
modulates Beclin1, LC3 and p62 expression to attenuate
Abeta40 and Abeta42 levels in APP/PS1 transgenic mice with
Alzheimer's disease. Mol Med Rep. 21:2095–2102. 2020.PubMed/NCBI
|
|
4
|
Huang YS: Myocardial damage and its
mechanism in burn patients. Zhonghua Zheng Xing Shao Shang Wai Ke
Za Zhi. 9:99–102. 1993.(In Chinese). PubMed/NCBI
|
|
5
|
Li Y, Ge S, Peng Y and Chen X:
Inflammation and cardiac dysfunction during sepsis, muscular
dystrophy, and myocarditis. Burns Trauma. 1:109–121. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xiao R, Teng M, Zhang Q, Shi XH and Huang
YS: Myocardial autophagy after severe burn in rats. PLoS One.
7:e394882012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Matsui Y, Takagi H, Qu X, Abdellatif M,
Sakoda H, Asano T, Levine B and Sadoshima J: Distinct roles of
autophagy in the heart during ischemia and reperfusion: Roles of
AMP-activated protein kinase and Beclin 1 in mediating autophagy.
Circ Res. 100:914–922. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Troncoso R, Vicencio JM, Parra V,
Nemchenko A, Kawashima Y, Del Campo A, Toro B, Battiprolu PK,
Aranguiz P, Chiong M, et al: Energy-preserving effects of IGF-1
antagonize starvation-induced cardiac autophagy. Cardiovasc Res.
93:320–329. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yan L, Vatner DE, Kim SJ, Ge H, Masurekar
M, Massover WH, Yang G, Matsui Y, Sadoshima J and Vatner SF:
Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci
USA. 102:13807–13812. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huang YS: Autophagy and hypoxic ischemic
myocardial damage after severe burn. Zhonghua Shao Shang Za Zhi.
34:3–7. 2018.(In Chinese). PubMed/NCBI
|
|
11
|
Marino G, Niso-Santano M, Baehrecke EH and
Kroemer G: Self-consumption: The interplay of autophagy and
apoptosis. Nat Rev Mol Cell Biol. 15:81–94. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Oh JM, Choi EK, Carp RI and Kim YS:
Oxidative stress impairs autophagic flux in prion protein-deficient
hippocampal cells. Autophagy. 8:1448–1461. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Funakoshi-Hirose I, Aki T, Unuma K,
Funakoshi T, Noritake K and Uemura K: Distinct effects of
methamphetamine on autophagy-lysosome and ubiquitin-proteasome
systems in HL-1 cultured mouse atrial cardiomyocytes. Toxicology.
312:74–82. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gonzalez P, Mader I, Tchoghandjian A,
Enzenmuller S, Cristofanon S, Basit F, Debatin KM and Fulda S:
Impairment of lysosomal integrity by B10, a glycosylated derivative
of betulinic acid, leads to lysosomal cell death and converts
autophagy into a detrimental process. Cell Death Differ.
19:1337–1346. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ma X, Liu H, Foyil SR, Godar RJ,
Weinheimer CJ, Hill JA and Diwan A: Impaired autophagosome
clearance contributes to cardiomyocyte death in
ischemia/reperfusion injury. Circulation. 125:3170–3181. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mindell JA: Lysosomal acidification
mechanisms. Annu Rev Physiol. 74:69–86. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Decker RS and Wildenthal K: Lysosomal
alterations in hypoxic and reoxygenated hearts. I. Ultrastructural
and cytochemical changes. Am J Pathol. 98:425–444. 1980.PubMed/NCBI
|
|
18
|
Weinert S, Jabs S, Supanchart C, Schweizer
M, Gimber N, Richter M, Rademann J, Stauber T, Kornak U and Jentsch
TJ: Lysosomal pathology and osteopetrosis upon loss of H+-driven
lysosomal Cl-accumulation. Science. 328:1401–1403. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ishida Y, Nayak S, Mindell JA and Grabe M:
A model of lysosomal pH regulation. J Gen Physiol. 141:705–720.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
DiCiccio JE and Steinberg BE: Lysosomal pH
and analysis of the counter ion pathways that support
acidification. J Gen Physiol. 137:385–390. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Graves AR, Curran PK, Smith CL and Mindell
JA: The Cl-/H+ antiporter ClC-7 is the primary chloride permeation
pathway in lysosomes. Nature. 453:788–792. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Leisle L, Ludwig CF, Wagner FA, Jentsch TJ
and Stauber T: ClC-7 is a slowly voltage-gated
2Cl(−)/1H(+)-exchanger and requires Ostm1 for transport activity.
EMBO J. 30:2140–2152. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kornak U, Kasper D, Bösl MR, Kaiser E,
Schweizer M, Schulz A, Friedrich W, Delling G and Jentsch TJ: Loss
of the ClC-7 chloride channel leads to osteopetrosis in mice and
man. Cell. 104:205–215. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kasper D, Planells-Cases R, Fuhrmann JC,
Scheel O, Zeitz O, Ruether K, Schmitt A, Poët M, Steinfeld R,
Schweizer M, et al: Loss of the chloride channel ClC-7 leads to
lysosomal storage disease and neurodegeneration. EMBO J.
24:1079–1091. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Steinberg BE, Huynh KK, Brodovitch A, Jabs
S, Stauber T, Jentsch TJ and Grinstein S: A cation counterflux
supports lysosomal acidification. J Cell Biol. 189:1171–1186. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Majumdar A, Capetillo-Zarate E, Cruz D,
Gouras GK and Maxfield FR: Degradation of Alzheimer's amyloid
fibrils by microglia requires delivery of ClC-7 to lysosomes. Mol
Biol Cell. 22:1664–1676. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lange PF, Wartosch L, Jentsch TJ and
Fuhrmann JC: ClC-7 requires Ostm1 as a beta-subunit to support bone
resorption and lysosomal function. Nature. 440:220–223. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gao G, Chen W, Yan M, Liu J, Luo H, Wang C
and Yang P: Rapamycin regulates the balance between cardiomyocyte
apoptosis and autophagy in chronic heart failure by inhibiting mTOR
signaling. Int J Mol Med. 45:195–209. 2020.PubMed/NCBI
|
|
29
|
Teng M, Jiang XP, Zhang Q, Zhang JP, Zhang
DX, Liang GP and Huang YS: Microtubular stability affects
pVHL-mediated regulation of HIF-1alpha via the p38/MAPK pathway in
hypoxic cardiomyocytes. PLoS One. 7:e350172012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lokuta A, Kirby MS, Gaa ST, Lederer WJ and
Rogers TB: On establishing primary cultures of neonatal rat
ventricular myocytes for analysis over long periods. J Cardiovasc
Electrophysiol. 5:50–62. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jiang XY, Zhang L, Yu C, Jiang H and Li J:
Research for a better method of neonatal rat cardiac myocytes,
primary culture and purification. Sichuan Da Xue Xue Bao Yi Xue
Ban. 46:301–304. 2015.(In Chinese). PubMed/NCBI
|
|
32
|
Ehler E, Moore-Morris T and Lange S:
Isolation and culture of neonatal mouse cardiomyocytes. J Vis Exp.
79:501542013.
|
|
33
|
Fuchsova B, Novak P, Kafkova J and Hozak
P: Nuclear DNA helicase II is recruited to IFN-alpha-activated
transcription sites at PML nuclear bodies. J Cell Biol.
158:463–473. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shi B, Huang QQ, Birkett R, Doyle R,
Dorfleutner A, Stehlik C, He C and Pope RM: SNAPIN is critical for
lysosomal acidification and autophagosome maturation in
macrophages. Autophagy. 13:285–301. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kawai A, Uchiyama H, Takano S, Nakamura N
and Ohkuma S: Autophagosome-lysosome fusion depends on the pH in
acidic compartments in CHO cells. Autophagy. 3:154–157. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Iwai-Kanai E, Yuan H, Huang C, Sayen MR,
Perry-Garza CN, Kim L and Gottlieb RA: A method to measure cardiac
autophagic flux in vivo. Autophagy. 4:322–329. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Namba DR, Ma G, Samad I, Ding D, Pandian
V, Powell JD, Horton MR and Hillel AT: Rapamycin inhibits human
laryngotracheal stenosis-derived fibroblast proliferation,
metabolism, and function in vitro. Otolaryngol Head Neck Surg.
152:881–888. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang J, Zhou W, Zhu S, Lin J, Wei P, Li
F, Jin P, Yao H, Zhang Y, Hu Y, et al: Persistency of enlarged
autolysosomes underscores nanoparticle-induced autophagy in
hepatocytes. Small. 13:16028762017. View Article : Google Scholar
|
|
41
|
Yang KC, Sathiyaseelan P, Ho C and Gorski
SM: Evolution of tools and methods for monitoring autophagic flux
in mammalian cells. Biochem Soc Trans. 46:97–110. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kurita T, Yamamura H, Suzuki Y, Giles WR
and Imaizumi Y: The ClC-7 chloride channel is downregulated by
hypoosmotic stress in human chondrocytes. Mol Pharmacol.
88:113–120. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rabanal-Ruiz Y, Otten EG and Korolchuk VI:
mTORC1 as the main gateway to autophagy. Essays Biochem.
61:565–584. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shen HM and Mizushima N: At the end of the
autophagic road: An emerging understanding of lysosomal functions
in autophagy. Trends Biochem Sci. 39:61–71. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mauvezin C and Neufeld TP: Bafilomycin A1
disrupts autophagic flux by inhibiting both V-ATPase-dependent
acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome
fusion. Autophagy. 11:1437–1438. 2015. View Article : Google Scholar : PubMed/NCBI
|