Introduction
Rheumatoid arthritis (RA) is one of the most common
articular diseases, affecting 0.5 to 1% of the world population and
resulting in disability due to joint destruction (1). Recently, biologic agents targeting
proinflammatory cytokines have greatly improved the treatment of
patients with RA. However, approximately 30% of RA patients are
resistant to these therapies, suggesting that other factors are
involved in RA physiopathology (2).
Fibroblast-like synoviocytes (FLS) play a unique role in both
inflammation and joint destruction. FLS are resistant to apoptosis
and consequently overgrow, promoting the synthesis of molecules
that mediate joint destruction and inflammation (3,4). In
vitro studies have demonstrated far greater proliferation and
cytokine production in synovial cells derived from RA than those
derived from osteoarthritis (OA) patients (5,6).
However, the mechanism that regulates synovial cell outgrowth
remains incompletely understood.
Our previous studies implicated transcription
factors, such as NF-κB and Jun proto-oncogene, AP1 transcription
factor subunit (JUN), in the regulation of FLS proliferation,
through the recruitment of the coactivator cAMP-responsive
element-binding protein (CREB)-binding protein (CBP) (7,8). CBP
is involved in multiple cellular processes and functions. CBP acts
as a transcriptional coactivator and a histone acetyltransferase
(HAT) by interacting with several transcription factors, including
CREB (9,10), and a variety of nuclear hormone
receptors (11). After binding
various transcription factors, CBP associates with RNA helicase A
(RHA)/DExH-box helicase 9, resulting in the recruitment of RNA
polymerase II (Pol II) complexes (12).
A subset of helix-loop-helix (HLH) proteins called
inhibitor of DNA binding/differentiation (ID) proteins function as
global regulators of cell fate determination. They play pivotal
roles in the coordinated regulation of gene expression during cell
growth, cell cycle control, differentiation, tumorigenesis
(13,14), and function by directly associating
with and modulating the activity of several families of
transcriptional regulators (15–17).
ID proteins contain an HLH region for dimerization but lack a basic
DNA binding domain. Therefore, these proteins act as transcription
dominant-negative repressors by dimerizing with and sequestering
ubiquitously expressed class A E-box HLH proteins (18), and in some cases, class B
(tissue-specific) HLH proteins (17).
Grap2 cyclin-D interacting protein (GCIP)/cyclin D1
binding protein 1 (CCNDBP1) was originally identified by yeast
two-hybrid screening. It is expressed in all human tissues and
particularly highly expressed in the heart, muscles, peripheral
blood leukocytes, kidneys, and brain-all associated with limited
cell differentiation and/or proliferation (19). Like ID proteins, GCIP possesses an
HLH domain but no basic domain. The amino acid sequence of the GCIP
HLH domain shares little identity with that of the ID proteins;
however, it has 78%homology with MAID, the maternal ID-like
protein. GCIP and MAID also functionally inhibit E12/myogenic
differentiation 1 activities (20).
Transient expression of GCIP reduces the phosphorylation of RB
transcriptional corepressor 1 (RB1) by cyclin-dependent protein
kinases, and represses E2F transcription factor 1-mediated
transcription (21). Recently, GCIP
was shown to suppress hepatocyte growth, as well as cancer growth
(22–24). However, the nuclear functions of
GCIP are not fully understood.
In the present study, we aimed to clarify the
molecular mechanism controlling FLS growth, and identify GCIP as a
CBP interacting protein. Our results demonstrate that GCIP
represses CREB-dependent transcription by inhibiting interactions
between CBP and Pol II, suggesting a novel inhibitory mechanism
used by ID-family HLH proteins.
Materials and methods
Plasmids and antibodies
The coding sequence for full-length GCIP was
PCR-amplified from pACT-GCIP, derived a previous yeast two-hybrid
screening. A series of deletion mutants were generated by PCR.
Full-length and deletion mutant versions of GCIP were
inserted into pGEX-5X-1 (GE Healthcare) for GST pulldown assays.
For transient transfection, these fragments were inserted into
pcDNA3-HA. The sequences of all generated plasmids were confirmed
by sequencing analysis. The RHA and CBP plasmids, PKA wild-type
(wt), PKA mutant, lacking kinase activity, pGAL4-CREB and pGAL4
expression vectors, Som-Luc, pG5b-Luc, and NF-κB-Luc reporter
plasmids, and the control plasmid RSV-β-gal have all been
previously described (12,25,26).
The following antibodies were used: anti-FLAG (M2), anti-HA (12CA5
and 3F10), anti-β-actin, and anti-cyclin D1 from Sigma-Aldrich
(Merck KGaA), anti-Pol II (Progen Biotechnik GmbH), anti-CBP
(Upstate Biotechnology, Inc.) and anti-His and anti-GST (GE
Healthcare). Anti-GCIP rabbit polyclonal antiserum was generated
against GST-GCIP (Tanpaku Seisei Kougyou). Anti-CBP rabbit
polyclonal antiserum and anti-RHA antibodies have been previously
described (12).
Cell culture, transient transfection
and stable cell line generation
Patients with RA were receiving stable doses of
methotrexate (6–10 mg/week) before joint replacement surgery.
Written informed consent was obtained from all patients prior to
collection of joint tissue samples. RA and OA samples were
collected from Bayside Misato Medical Center (Kochi, Japan).
Samples were collected from 6 patients with RA (age range, 64–78
years; mean age, 68.5 year; sex, female) between February 2012 and
May 2013. Samples were collected from 6 patients with OA (age
range, 67–80 years; mean age, 71.7 years; sex, female) between
February 2012 and April 2012. Human FLS were obtained from patients
with RA and OA by standard methods as previously described
(27). Briefly, the synovial tissue
was minced and digested with collagenase (Sigma-Aldrich; Merck
KGaA). The adherent cells were cultured in dishes in Dulbecco's
modified Eagle's medium (DMEM). 293 cells, 293T cells and FLS were
cultured in Dulbecco's modified Eagle's medium (DMEM) as previously
described (26,28). 239 cells were transfected with
pcDNA3-HA GCIP plasmid or pcDNA3-HA plasmids (control). 293 cells
stably expressing HA-GCIP or HA alone were selected and maintained
in DMEM containing 400 µg/ml G418. Transient transfection assays
were performed with 293 cells. Cells were lysed with cell lysis
buffer (Toyo Ink Group) 24 h after transfection, and luciferase
activities were measured. Reporter activity was induced by
co-transfection with the PKA expression vector. −293 cells were
transfected with 100 ng of CRE-Luc or pG5B-Luc reporter plasmid, 50
ng of wild-type or catalytically inactive PKA expression vector
(PKAwt or PKAmut, respectively), 50 ng of RSV-β-gal control
plasmid. For assay with the pG5B-Luc reporter plasmid, cells were
co-transfected with 100 ng of GAL4-CREB expression vector. 293
cells transfected with NF-κB-Luc were treated with 100 ng/ml TPA or
10 ng/ml TNF-α for 24 h. Cells were lysed with a passive lysis
buffer (Promega Corporation) 48 h after transfection and luciferase
activities were measured and normalized to the activity of
RSV-β-gal. All experiments were performed in triplicate. To ensure
equal amounts of DNA, empty plasmids were added to each
transfection (20).
GST pulldown assays
GST fusion proteins were expressed and purified
using glutathione (GSH) Sepharose beads (GE Healthcare).
35S-labeled GCIP or cell extracts were incubated with
GST fusion proteins bound to the resin in 1 ml of buffer A (20 mM
HEPES, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM DTT, 0.1% NP-40, 5%
glycerol, 1 mM Na3VO4, 5 mM NaF, 1 µg/ml
aprotinin and 1 µg/ml leupeptin) for 4 h at 4°C. After washing with
buffer A, bound proteins were resolved by SDS-PAGE and exposed to
an X-ray film.
Immunoprecipitation
293T cells were transfected with HA-GCIP and
FLAG-CBP expression vectors. After 48 h, the cells were lysed in 1
ml of lysis buffer (20 mM HEPES, pH 7.5, 100 mM KCl, 1 mM EDTA, 1
mM DTT, 0.1% NP-40, 5% glycerol and protease inhibitors). The
lysates were mixed with 1 µg of anti-HA antibody (3F10), anti-FLAG
antibody (M2), or anti-CBP antiserum conjugated to protein
G-Sepharose beads (GE Healthcare). After 4 h of incubation at 4°C,
the beads were washed three times with lysis buffer. Bound proteins
were resolved by SDS-PAGE and analyzed by western blotting.
Immunofluorescence
Staining was performed as previously described
(28). Briefly, cells were
permeabilized with 0.2% Triton X-100, then incubated with rat
anti-GCIP (1:100) and rabbit anti-CBP (1:100) primary antibodies
followed by Alexa Fluor 594 anti-mouse and Alexa Fluor 488
anti-rabbit secondary antibodies (1:1,000; Molecular Probes).
Samples were imaged on a Zeiss LSM 510 laser scanning confocal
microscope.
Western blotting
The cells were lysed in 1 ml lysis buffer (20 mM
HEPES, pH 7.5; 100 mM KCl; 1 mM EDTA; 1 mM DTT; 0.1% NP-40; 5%
glycerol and protease inhibitors). Proteins were quantified using
the Lowry Assay (Bio-Rad Laboratories, Inc.) and 30 µg proteins
were separated by 10% SDS-PAGE, and transferred onto a PVDF
membrane. The membrane was incubated in 5% skim milk/TBS-T for 1 h
at room temperature and then with rabbit polyclonal anti-GCIP
antibody, which was generated against GST-GCIP (1:200; Tanpaku
Seisei Kougyou) at 4°C overnight. The membrane was washed three
times and incubated with HRP-conjugated anti-rabbit secondary
antibody (1:1,000; cat. no. A9169; Sigma-Aldrich; Merck KGaA) for 1
h at room temperature. Proteins were detected by ECL plus (GE
Healthcare) and exposed to an X-ray film. Band intensity was
measured using ImageJ software (version 1.53f; National Institutes
of Health).
RNAi, proliferation assays and reverse
transcription-quantitative PCR (RT-qPCR)
GCIP siRNAs were purchased from Ambion and
transfected into cells with Lipofectamine 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.). Briefly, 24 h before transfection, cells
growing exponentially were trypsinized and transferred to a 96-well
plate. Proliferation was determined by assaying viable cell numbers
using the Cell Counting Kit-8 (Dojindo Molecular Technologies)
according to the manufacturer's protocol and the BrdU Cell
Proliferation Assay Kit (Merck Millipore). RT-qPCR was performed
using the LightCycler 480 Probes Master Mix (Roche Diagnostics).
Expression levels were normalized to the 18S rRNA gene levels. Two
sets of primers/probes were used for PCR: GCIP,
5′-GAAGCCACGACTCTGACCAT-3′ and 5′-GATGGCAGCATGGACTTGT-3′ (probe
#86), 18S rRNA, 5′-GCAATTATTCCCCATGAACG-3′ and
5′-GGGACTTAATCAACGCAAGC-3′ (probe #48).
Statistical analysis
All data are expressed as the means standard
deviation (SD) and were analyzed using Excel Statistics 2012
version 1.00 (SSRI Japan Co., Ltd., Tokyo). One-way analysis of
variance with a Tukey-Kramer post hoc analysis was used to compare
data among multiple groups. Differences between two groups were
examined using unpaired Student's t-test. P<0.05 was considered
to indicate a statistically significant difference.
Study approval
The human experimental protocols in this study
(approval nos. 2728 and 2729) were approved by the Ethics Review
Committee of Tokyo Medical University. Written informed consent was
obtained from all patients prior to the collection of joint tissue
samples. Of note, all the experiments were performed in accordance
with the relevant guidelines and regulations.
Results
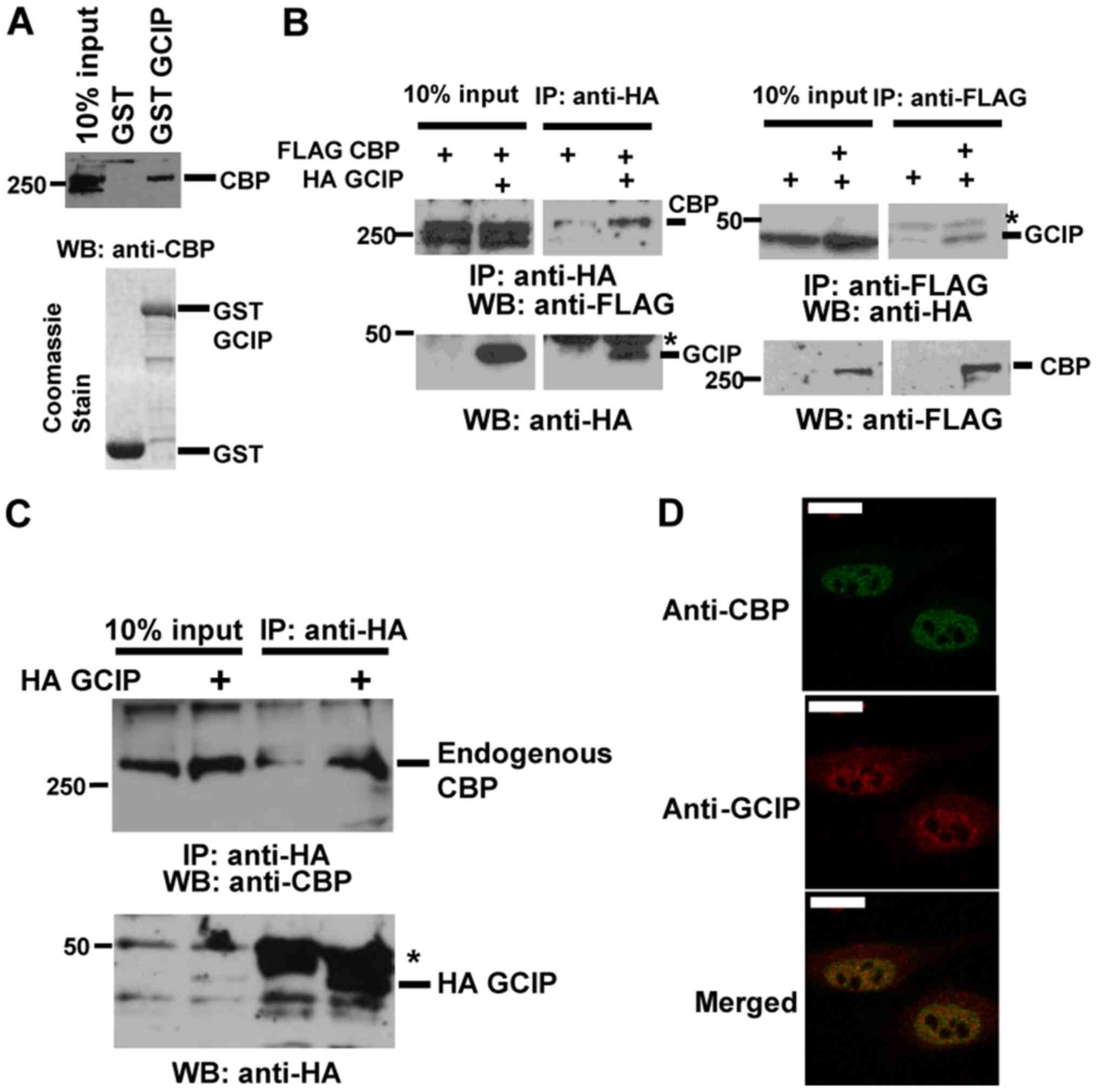
GCIP interacts with CBP
To identify proteins that interact with CBP, we
performed a yeast two-hybrid screen using the CBP C/H3 domain as
bait (29) and a library of
FLS-derived cDNAs as preys, and obtained clones expressing GCIP.
Consistently with this result, CBP interacted with GCIP in
vitro (Fig. 1A). To verify the
interaction in vivo, we transiently transfected 293T cells
with HA-GCIP and FLAG-CBP and performed immunoprecipitation
followed by immunoblotting. FLAG-CBP coimmunoprecipitated with
HA-GCIP, and HA-GCIP coimmunoprecipitated with FLAG-CBP (Fig. 1B). To further investigate the
physiological interaction between GCIP and CBP, we transiently
transfected 293T cells with HA-GCIP and performed
immunoprecipitation followed by immunoblotting. As showed in
Fig. 1C, endogenous CBP
coimmunoprecipitated with HA-GCIP. In addition, we performed
replicated the same experiment using FLS. Results show that the
endogenous CBP interacted with HA-GCIP (Fig. S1A). Next, we investigated the
subcellular localization of these proteins by immunofluorescence.
Endogenous GCIP displayed both nuclear and cytoplasmic localization
in 293 cells (Fig. 1D), while
endogenous CBP was observed in the nucleus. The nuclear dots of
GCIP partially overlapped with endogenous CBP. These results
indicate that GCIP physically interacts with CBP in the
nucleus.
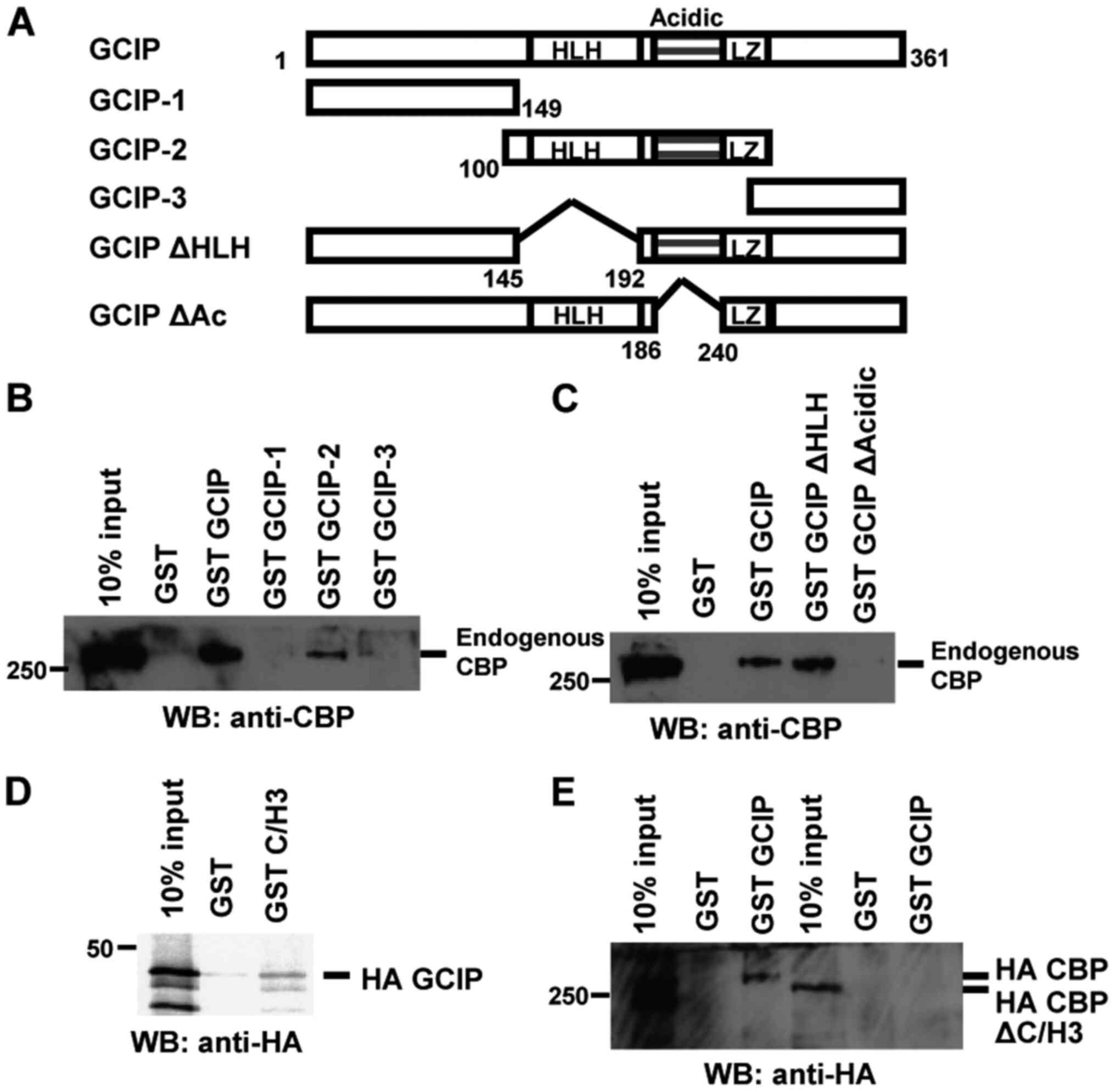
Mapping the interaction domains of
GCIP and CBP
GCIP contains an HLH domain in its central region.
An aspartic/glutamic acid-rich domain (Acidic) and a potential
leucine zipper (LZ) motif were detected in protein the C-terminal,
near the HLH domain (Fig. 2A). To
determine which portion of GCIP mediates its interaction with CBP,
we performed GST pulldown assays using several GCIP deletion
mutants and nuclear extracts from 293 cells. As showed in Fig. 2B, CBP bound to the central region of
GCIP containing the HLH, Acidic and LZ domains. We then determined
the minimal region required for CBP binding. Full-length GCIP and
GCIP ΔHLH interacted with CBP, while GCIP ΔAcidic did not (Fig. 2C). To map the regions of CBP that
associate with GCIP, we performed GST pulldown assays using the
C/H3 domain of CBP. The C/H3 domain was sufficient to bind GCIP
(Fig. 2D), and GCIP interacted with
full-length CBP but not CBP ΔC/H3 (Fig.
2E). Taken together, these results demonstrate that the CBP
C/H3 domain and the GCIP Acidic region are required for the
CBP-GCIP interaction.
GCIP inhibits CBP-mediated
transcriptional activation
CBP activates transcription via CREB (9,10) and
NF-κB (30). To examine whether
GCIP plays a role in CBP-mediated transactivation, we used a
somatostatin-luciferase (Som-Luc) reporter that contains an
endogenous somatostatin promoter with a cAMP response element (CRE)
(31). Protein kinase
cAMP-activated catalytic subunit alpha (PKA) induced Som-Luc
reporter activity and co-transfection with GCIP repressed Som-Luc
activity in a dose-dependent manner in both 293 cells and
RA-derived FLS (Figs. 3A and
S1B). To rule out the possible
effects of GCIP on CRE-binding proteins other than CREB (32), reporter assays were performed with
G5b-Luc as an artificial state in comparison with Som-Luc as an
endogenous state. Instead of Som-Luc, CREB fused to GAL4-DBD
(GAL4-CREB) and pG5b-Luc, which has five copies of the GAL4
DNA-binding site upstream of Luc (12), were co-transfected with GCIP into
293 cells. The results were similar to those with Som-Luc (Fig. 3B). Next, we examined the effect of
GCIP on NF-κB-mediated transactivation using NF-κB-Luc (30). The reporter was activated by phorbol
ester 12-O-tetradecanoylphorbol-13-acetate (TPA) or TNF-α.
Co-transfection with GCIP repressed NF-κB activity in a
dose-dependent manner (Fig. 3C). In
addition, GCIP also repressed NF-κB activity in FLS (Fig. S1C). Next we tested whether the
GCIP-CBP interaction was required for GCIP-mediated transcriptional
repression. Bothe GCIP and GCIP ΔHLH repressed CREB-dependent
transcription, while GCIP ΔAcidic, which could not bind to CBP, had
no effect (Fig. 3D). To confirm the
repression activity of GCIP on CREB-mediated transcription, we
further performed knockdown experiments using siRNAs. Knockdown of
GCIP induced the expression of endogenous cyclin D1, one of
CREB-target genes (Fig. 3E). These
results indicate that GCIP represses CREB- and NF-κB-mediated
transcription via interaction with CBP.
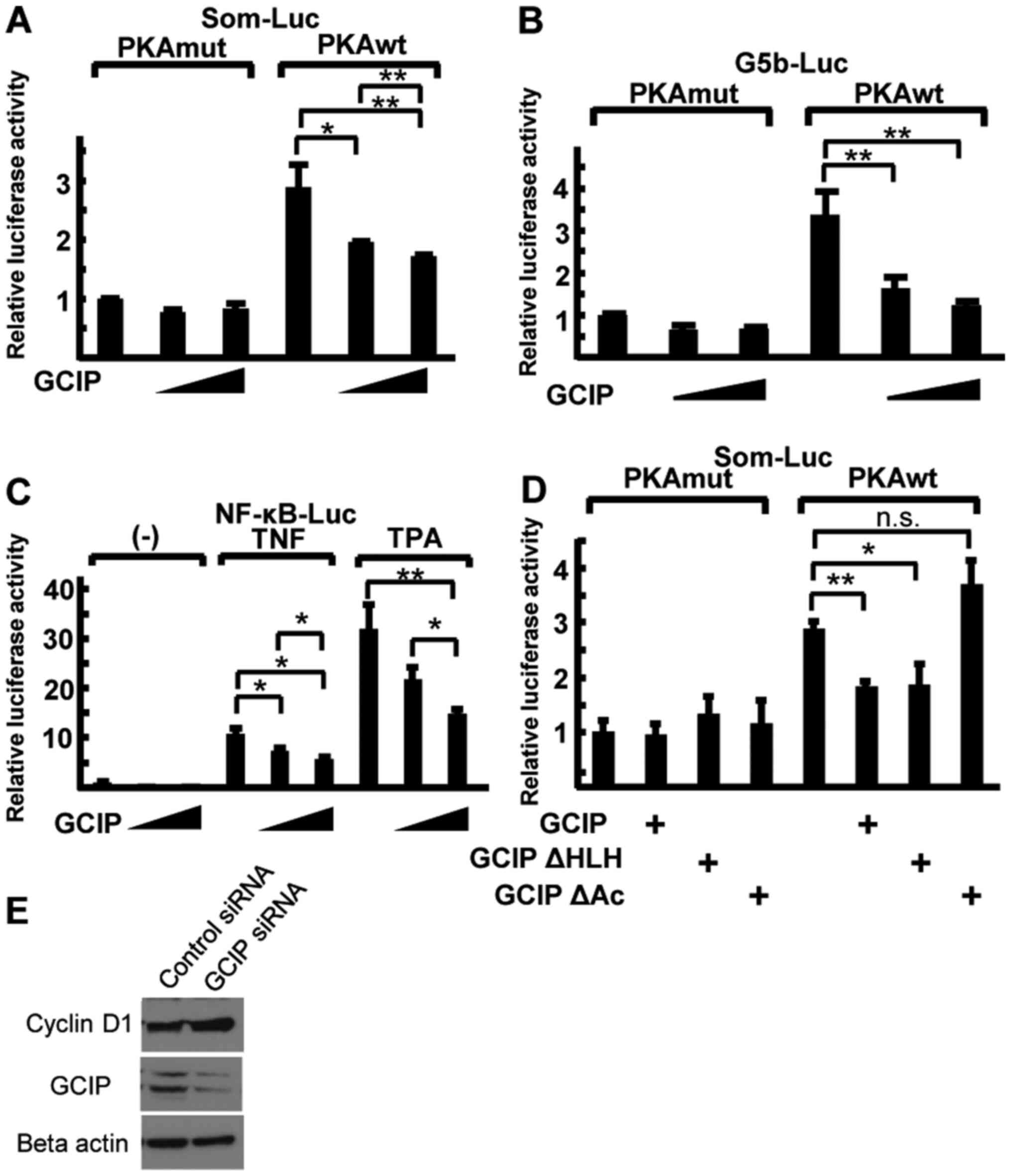 | Figure 3.GCIP represses CBP-mediated
transcription. (A and B) Reporter assays in 293 cells using Som-Luc
(A) and GAL4-CREB, with a G5b-Luc reporter containing five GAL4
recognition sites. (B) Reporter activity was induced by the
co-transfection of either WT or mutant PKA (PKAwt and PKAmut,
respectively). The luciferase activity of cells transfected with
empty vector and PKAmut was set to 1. (C) 293 cells were
co-transfected with NF-κB-Luc and GCIP. After transfection, cells
were treated with 100 ng/ml TPA or 10 ng/ml TNF-α. (D) 293 cells
were co-transfected with Som-Luc and plasmids expressing various
GCIP mutants. The luciferase activity of cells co-transfected with
empty vector and PKAmut was set to 1. (E) Whole-cell lysates of FLS
were analyzed by immunoblotting with the indicated antibodies. Data
were analyzed by performing a Tukey-Kramer post hoc analysis and
expressed as mean ± SD. *P<0.05, **P<0.01, n.s., not
significant. These experiments were repeated at least three times.
GCIP, grap2 cyclin D interacting protein; CBP, cAMP-response
element-binding protein-binding protein; TPA,
12-O-tetradecanoylphorbol-13-acetate; siRNA, small interfering RNA;
FLS, fibroblast-like synoviocytes. |
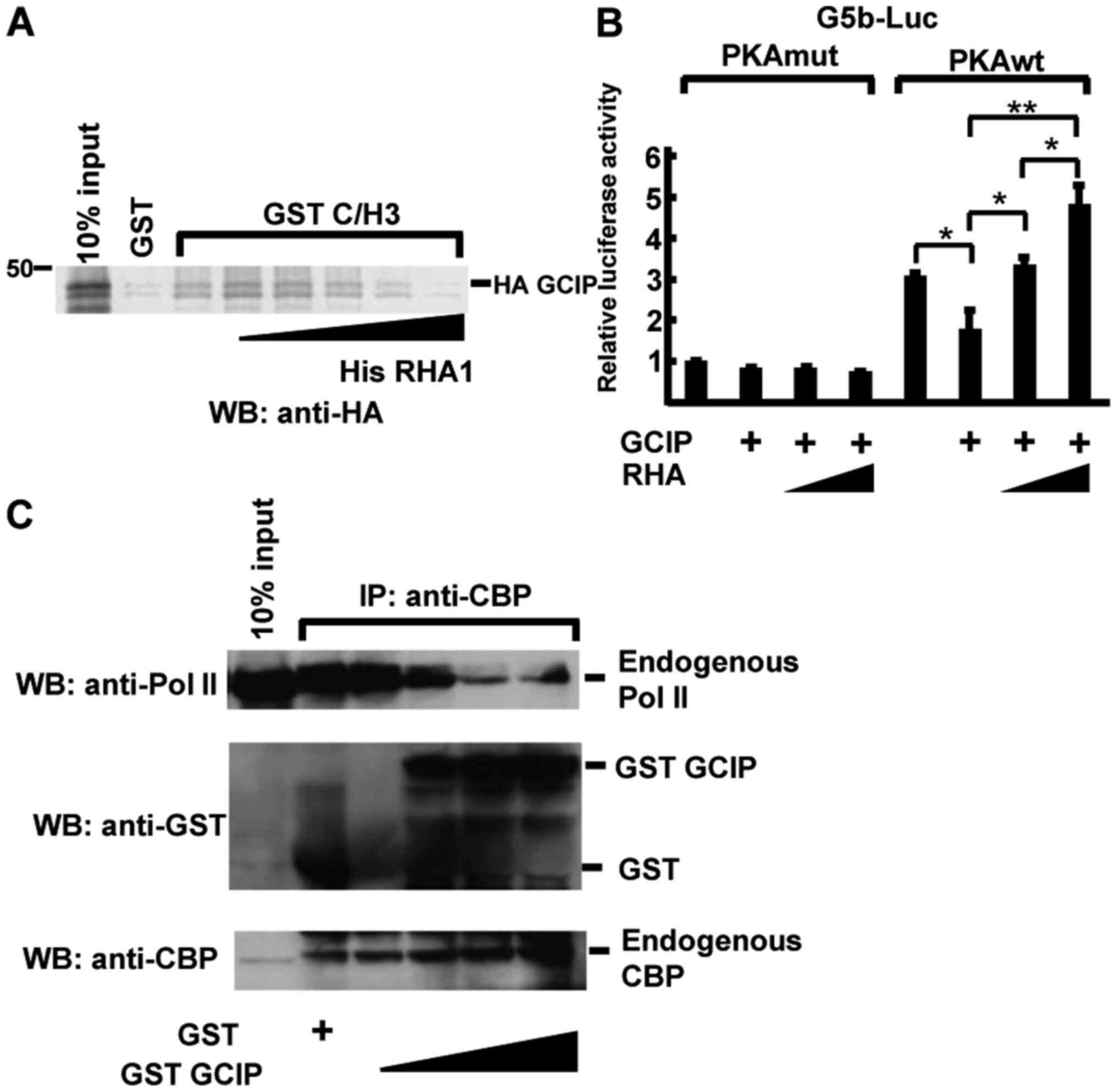
GCIP and the Pol II complex compete
for CBP binding
In CREB-dependent transcription, CBP associates with
phosphorylated CREB (Ser 133) (33)
and recruits Pol II complexes by binding RHA, which interacts with
the C/H3 domain (12). RHA
interacts with CBP via its N-terminal region (amino acids 1–262),
termed RHA1 (12). We hypothesized
that GCIP and RHA competitively bind CBP, resulting in the
repression of CREB-dependent transcription. To examine this, we
first performed an in vitro competitive binding assay to
address the possibility of direct competition between GCIP and RHA
for CBP binding. As shown in Fig.
4A, His-RHA1 directly competed with GCIP for association with
GST-C/H3 in a dose-dependent manner. Next, reporter assays were
performed with G5b-Luc in 293 cells co-transfected with RHA and
GCIP. In PKA-stimulated cells, GCIP repressed reporter activity
(Fig. 4B), which was recovered in a
dose-dependent manner in the context of RHA co-transfection
(Fig. 4B). To further examine
whether GCIP inhibits Pol II recruitment to CBP, we performed
immunoprecipitation assays (Fig.
4C). Pol II coimmunoprecipitated with endogenous CBP and the
addition of GST-GCIP decreased the formation of complexes between
CBP and Pol II (Fig. 4C). These
results demonstrate that GCIP inhibits Pol II complex recruitment
to CBP, resulting in the repression of CREB-dependent
transcription.
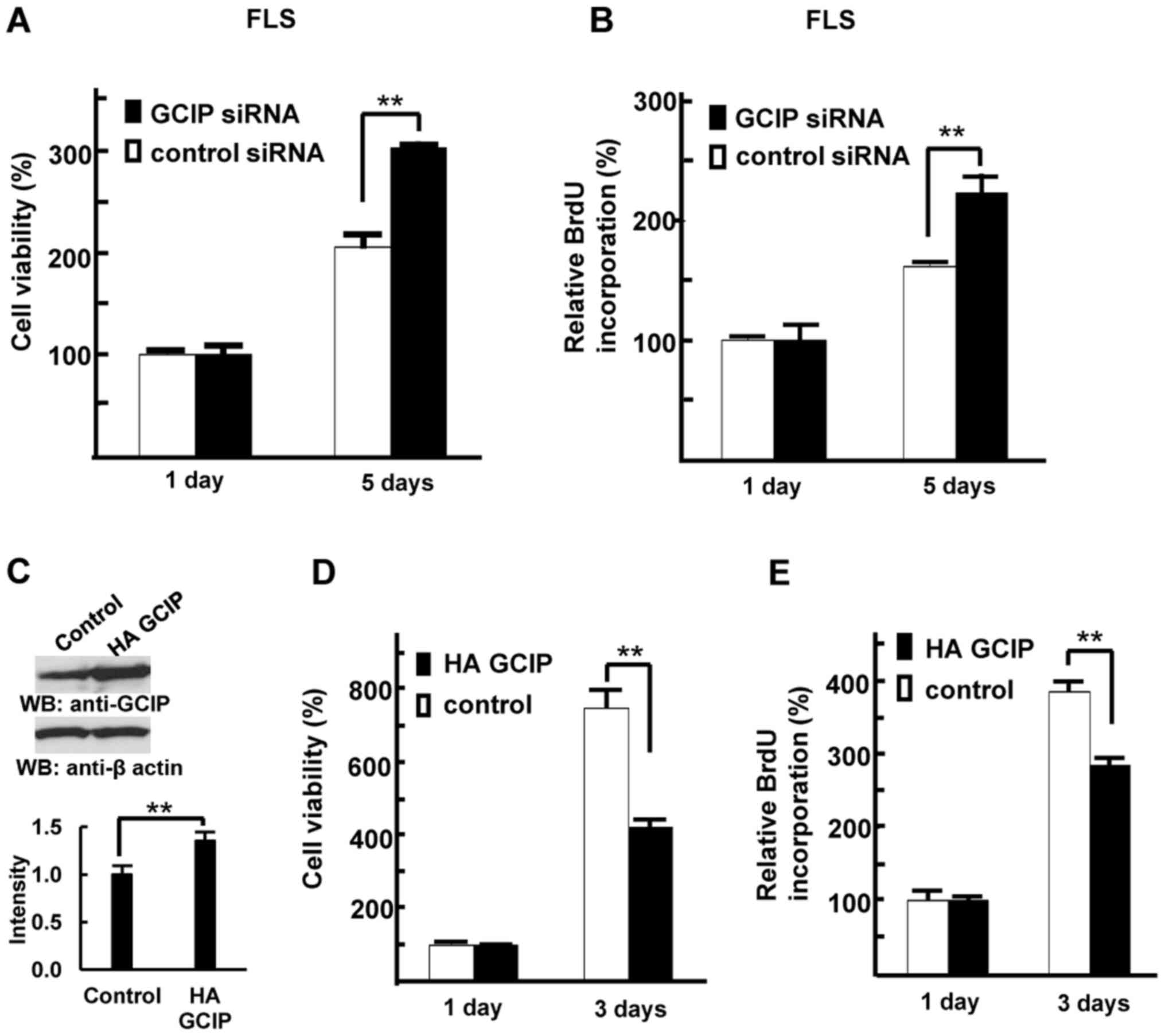
GCIP inhibits FLS growth
To investigate the role of GCIP in FLS growth, we
depleted GCIP from RA patients' FLS. Control or GCIP siRNAs
were transiently transfected into FLS, and their viability and
proliferation were measured. GCIP depletion resulted in increased
FLS viability compared with control cells (Fig. 5A). We next measured BrdU
incorporation in these cells. As shown in Fig. 5B, treatment with GCIP siRNA
enhanced FLS proliferation compared with control siRNA. Next, we
generated 293 cell lines stably expressing either HA-GCIP or a
control vector, as we were unable to generate FLS with stable GCIP
expression. First, we confirmed overexpression of GCIP (Fig. 5C). As shown in Fig. 5D and E, the growth rate of
GCIP-expressing 293 cells was lower than that of control cells.
These results indicate that GCIP negatively regulates cell
proliferation, suggesting that GCIP downregulation in the FLS of
patients with RA could result in FLS overgrowth.
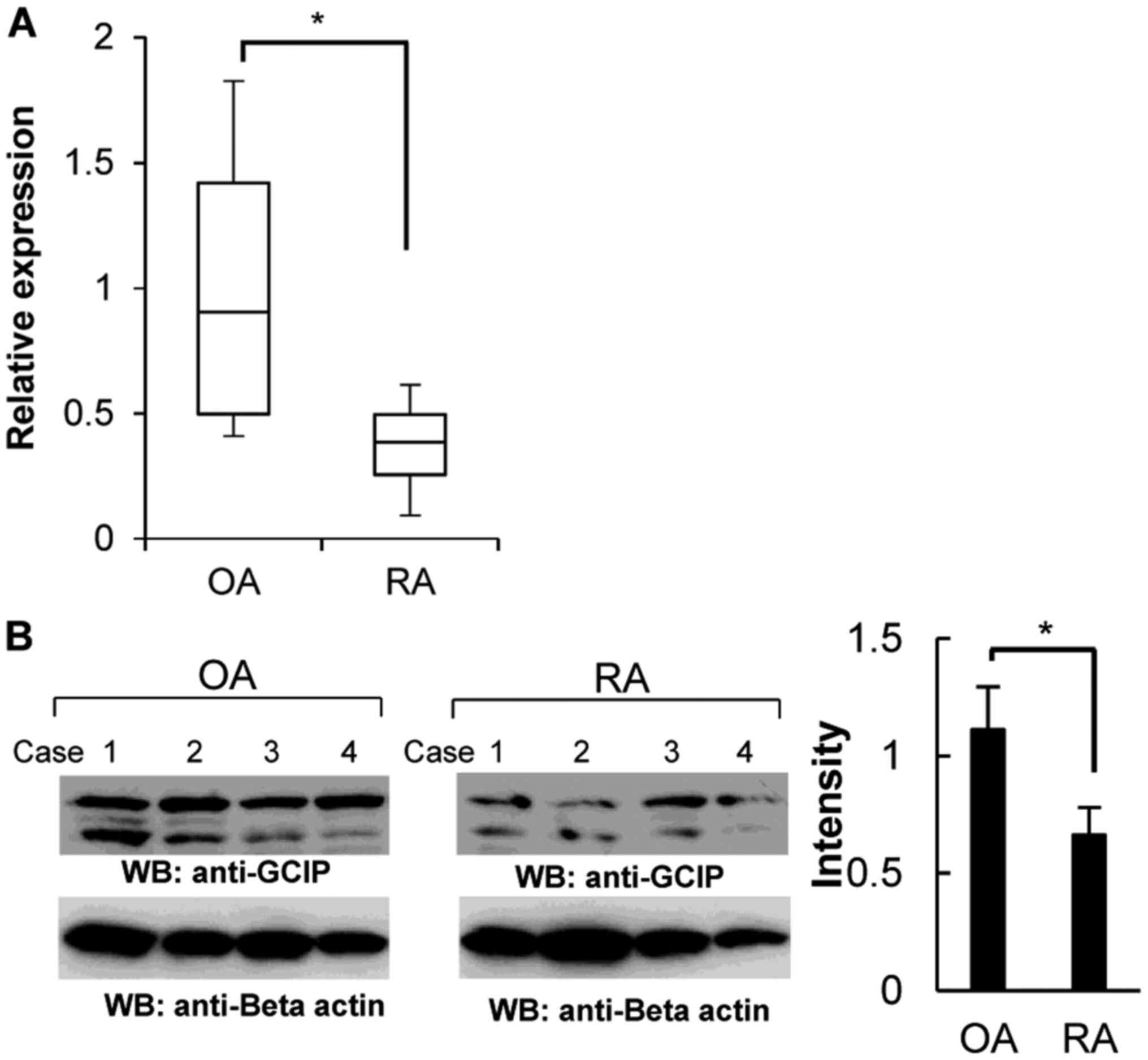
Downregulation of GCIP in FLS derived
from patients with RA
Based on the rapid proliferation of RA-derived FLS,
we hypothesized that the expression of GCIP might be low in
RA-derived FLS compared to osteoarthritis (OA)-derived FLS.
Therefore, we next investigated GCIP expression in FLS. As
shown in Fig. 6A, RA-derived FLS
displayed reduced GCIP transcript levels compared with those
observed in patients with OA. The GCIP protein level was also
significantly decreased in RA-derived FLS (Fig. 6B).
Discussion
Four members of the ID protein family are present in
mammalian cells (13). Outside
their conserved HLH domains, the ID proteins display extensive
sequence and function divergence (14,17).
ID proteins act as act as dominant-negative regulators by
dimerizing with different partners (14,17),
and mainly bind to basic HLH (bHLH) proteins, as well as a few
non-bHLH proteins such as RB1 (34,35)
and some transcription regulators, including the paired box
(13), ADD1/SREBP-1c (36), MIDA1 (37), and ETS-domain transcription factors
(38). However, to date, the
association between ID proteins and mediator proteins have not been
described. GCIP also possesses an HLH domain without a basic domain
and is related to the ID protein family (19). In this study, we found that GCIP
interacts with the coactivator CBP and represses CREB- and
NF-κB-dependent transcription through this interaction. To the best
of our knowledge, this study is the first to describe an
interaction between a transcriptional coactivator and an ID-like
protein.
Previous studies demonstrate that CBP activates
CREB- and NF-κB-dependent transcription and functions in cell
growth of FLS (5,14). RHA is a cofactor that mediates the
interaction between transcriptional coactivator CBP and Pol II
complex (12). And RHA also
activates CREB- and NF-κB-dependent transcription. Therefore, we
think that RHA is involved in cell proliferation. In addition, GCIP
did not interact with RHA, and GCIP did not inhibit the interaction
between RHA and Pol II. Therefore, we hypothesize that the
inhibitory effect of GCIP on cell growth could be due to
competition for binding to CBP with RHA.
Recent studies have suggested that HAT inhibitors
represent important anti-inflammatory therapies (39,40).
FLS reportedly induce several inflammatory cytokines. A recent
study demonstrated that NF-κB regulates IL8 expression via CBP
(41). Besides, CBP inhibition
induces a TNF-α-dependent increase in NF-κB function and target
gene expression in LPS-stimulated FLS (42). NF-κB and JUN may also affect the
regulation of FLS proliferation by recruiting CBP (7,8,43). In
addition, CBP regulates cell growth via several transcription
factors, including CREB (9,10), JUN, Fos proto-oncogene, AP1
transcription factor subunit (44),
and a variety of nuclear hormone receptors (11). In this study, GCIP was identified
applying a yeast two-hybrid screening to a library of FLS-derived
cDNAs. GCIP partially colocalized with CBP and repressed CREB- and
NF-κB-dependent transcription by interacting with CBP. Therefore,
GCIP may be a key factor in synovial cell outgrowth and could be a
promising diagnostic and therapeutic target. Further analysis is
needed to resolve the role of GCIP in FLS growth, and to determine
whether GCIP modulation is a viable strategy to repress
RA-associated synovial cell overgrowth. Nevertheless, our findings
also indicate that the coactivator CBP is a functional target for
GCIP in the regulation of cell proliferation. This suggests that
GCIP targets not only bHLH proteins but also the coactivator CBP,
which unveils a novel inhibitory mechanism for an HLH protein.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr Minako Nakazawa
(Institute of Medical Science, St. Marianna University School of
Medicine) for insightful comments, and Ms. Sanae Shinkawa, Ms.
Yukari Nakagawa, Ms. Makiko Yui and Ms. Etsuko Nakamura (Institute
of Medical Science, St. Marianna University School of Medicine) for
technical assistance. The authors would like to thank Mr. Yu
Nakatani (Misato Marine Hospital) for collecting patients' samples
and aiding in performing the experiments.
Funding
The present study was supported in part by the Japan
Society for the Promotion of Science KAKENHI (grant nos. 23659176,
26670479, 16H05157 and 18K06972). This study was also supported in
part by funds provided through a program of the Strategic Research
Foundation at Private Universities (grant nos. S1411011 and
2014–2018) from the Ministry of Education, Culture, Sports, Science
and Technology of Japan (MEXT). Partial funding was also provided
via grants from the Naito Foundation, Natural Science Scholarship
Daiichi-Sankyo Foundation of Life Science, Mitsubishi, Tanabe
Pharma Corporation, Bureau of Social Welfare and Public Health,
Academic contribution of Pfizer, Eisai, Santen Pharmaceutical,
Abbvie, AYUMI Pharmaceutical Corporation, Takeda Science
Foundation, AstraZeneca (R&D Grant 2013), and the ONO Medical
Research Foundation and Industry-university cooperation
(BioMimetics Sympathies Inc.).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
HF, SA and TN conceived the project and designed the
experiments. HF, SA and TN performed the experiments and analyzed
the data. HF and TN wrote the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The human experimental protocols in the present
study (approval nos. 2728 and 2729) were approved by the Ethics
Review Committee of Tokyo Medical University. Written informed
consent was obtained from all patients prior to the collection of
joint tissue samples. In addition, we confirm that all the
experiments were performed in accordance with the relevant
guidelines and regulations.
Patient consent for publication
Consent for publication was obtained from
patients.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Firestein GS: Evolving concepts of
rheumatoid arthritis. Nature. 423:356–361. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
van Vollenhoven RF: Treatment of
rheumatoid arthritis: State of the art 2009. Nat Rev Rheumatol.
5:531–541. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Noss EH and Brenner MB: The role and
therapeutic implications of fibroblast-like synoviocytes in
inflammation and cartilage erosion in rheumatoid arthritis. Immunol
Rev. 223:252–270. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bartok B and Firestein GS: Fibroblast-like
synoviocytes: Key effector cells in rheumatoid arthritis. Immunol
Rev. 233:233–255. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bucala R, Ritchlin C, Winchester R and
Cerami A: Constitutive production of inflammatory and mitogenic
cytokines by rheumatoid synovial fibroblasts. J Exp Med.
173:569–574. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ribel-Madsen S, Bartels EM, Stockmarr A,
Borgwardt A, Cornett C, Danneskiold-Samsøe B and Bliddal H: A
synoviocyte model for osteoarthritis and rheumatoid arthritis:
Response to Ibuprofen, betamethasone, and ginger extract-a
cross-sectional in vitro study. Arthritis. 2012:5058422012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Asahara H, Fujisawa K, Kobata T, Hasunuma
T, Maeda T, Asanuma M, Ogawa N, Inoue H, Sumida T and Nishioka K:
Direct evidence of high DNA binding activity of transcription
factor AP-1 in rheumatoid arthritis synovium. Arthritis Rheum.
40:912–918. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fujisawa K, Aono H, Hasunuma T, Yamamoto
K, Mita S and Nishioka K: Activation of transcription factor
NF-kappa B in human synovial cells in response to tumor necrosis
factor alpha. Arthritis Rheum. 39:197–203. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Arias J, Alberts AS, Brindle P, Claret FX,
Smeal T, Karin M, Feramisco J and Montminy M: Activation of cAMP
and mitogen responsive genes relies on a common nuclear factor.
Nature. 370:226–229. 1994. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kwok RP, Lundblad JR, Chrivia JC, Richards
JP, Bächinger HP, Brennan RG, Roberts SG, Green MR and Goodman RH:
Nuclear protein CBP is a coactivator for the transcription factor
CREB. Nature. 370:223–226. 1994. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kamei Y, Xu L, Heinzel T, Torchia J,
Kurokawa R, Gloss B, Lin SC, Heyman RA, Rose DW, Glass CK and
Rosenfeld MG: A CBP integrator complex mediates transcriptional
activation and AP-1 inhibition by nuclear receptors. Cell.
85:403–414. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nakajima T, Uchida C, Anderson SF, Lee CG,
Hurwitz J, Parvin JD and Montminy M: RNA helicase A mediates
association of CBP with RNA polymerase II. Cell. 90:1107–1112.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Norton JD: ID helix-loop-helix proteins in
cell growth, differentiation and tumorigenesis. J Cell Sci.
113:3897–3905. 2000.PubMed/NCBI
|
|
14
|
Benezra R, Rafii S and Lyden D: The Id
proteins and angiogenesis. Oncogene. 20:8334–8341. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Benezra R, Davis RL, Lockshon D, Turner DL
and Weintraub H: The protein Id: A negative regulator of
helix-loop-helix DNA binding proteins. Cell. 61:49–59. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Christy BA, Sanders LK, Lau LF, Copeland
NG, Jenkins NA and Nathans D: An Id-related helix-loop-helix
protein encoded by a growth factor-inducible gene. Proc Natl Acad
Sci USA. 88:1815–1819. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Langlands K, Yin X, Anand G and Prochownik
EV: Differential interactions of Id proteins with
basic-helix-loop-helix transcription factors. J Biol Chem.
272:19785–19793. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sun XH, Copeland NG, Jenkins NA and
Baltimore D: Id proteins Id1 and Id2 selectively inhibit DNA
binding by one class of helix-loop-helix proteins. Mol Cell Biol.
11:5603–5611. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yao Y, Doki Y, Jiang W, Imoto M, Venkatraj
VS, Warburton D, Santella RM, Lu B, Yan L, Sun XH, et al: Cloning
and characterization of DIP1, a novel protein that is related to
the Id family of proteins. Exp Cell Res. 257:22–32. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Terai S, Aoki H, Ashida K and Thorgeirsson
SS: Human homologue of maid: A dominant inhibitory helix-loop-helix
protein associated with liver-specific gene expression. Hepatology.
32:357–366. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xia C, Bao Z, Tabassam F, Ma W, Qiu M, Hua
S and Liu M: GCIP, a novel human grap2 and cyclin D interacting
protein, regulates E2F-mediated transcriptional activity. J Biol
Chem. 275:20942–20948. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ma W, Stafford LJ, Li D, Luo J, Li X, Ning
G and Liu M: GCIP/CCNDBP1, a helix-loop-helix protein, suppresses
tumorigenesis. J Cell Biochem. 100:1376–1386. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ma W, Xia X, Stafford LJ, Yu C, Wang F,
LeSage G and Liu M: Expression of GCIP in transgenic mice decreases
susceptibility to chemical hepatocarcinogenesis. Oncogene.
25:4207–4216. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sonnenberg-Riethmacher E, Wustefeld T,
Miehe M, Trautwein C and Riethmacher D: Maid (GCIP) is involved in
cell cycle control of hepatocytes. Hepatology. 45:404–411. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Aratani S, Fujii R, Oishi T, Fujita H,
Amano T, Ohshima T, Hagiwara M, Fukamizu A and Nakajima T: Dual
roles of RNA helicase A in CREB-dependent transcription. Mol Cell
Biol. 21:4460–4469. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fujita H, Fujii R, Aratani S, Amano T,
Fukamizu A and Nakajima T: Antithetic effects of MBD2a on gene
regulation. Mol Cell Biol. 23:2645–2657. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fujita H, Aratani S, Yagishita N, Nishioka
K and Nakajima T: Identification of the inhibitory activity of
walnut extract on the E3 ligase Syvn1. Mol Med Rep. 18:5701–5708.
2018.PubMed/NCBI
|
|
28
|
Fujita H, Ohshima T, Oishi T, Aratani S,
Fujii R, Fukamizu A and Nakajima T: Relevance of nuclear
localization and functions of RNA helicase A. Int J Mol Med.
15:555–560. 2005.PubMed/NCBI
|
|
29
|
Yoshida E, Aratani S, Itou H, Miyagishi M,
Takiguchi M, Osumu T, Murakami K and Fukamizu A: Functional
association between CBP and HNF4 in trans-activation. Biochem
Biophys Res Commun. 241:664–669. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yamamoto K, Aono H, Nakajima T, Hasunuma T
and Nishioka K: Oligoclonal proliferation of human T-cell leukemia
virus type I infected lymphocytes in lesions of virus-induced
arthropathy. Biochem Biophys Res Commun. 208:1040–1045. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yao TP, Oh SP, Fuchs M, Zhou ND, Ch'ng LE,
Newsome D, Bronson RT, Li E, Livingston DM and Eckner R: Gene
dosage-dependent embryonic development and proliferation defects in
mice lacking the transcriptional integrator p300. Cell. 93:361–372.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mayr B and Montminy M: Transcriptional
regulation by the phosphorylation-dependent factor CREB. Nat Rev
Mol Cell Biol. 2:599–609. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Parker D, Ferreri K, Nakajima T, LaMorte
VJ, Evans R, Koerber SC, Hoeger C and Montminy MR: Phosphorylation
of CREB at Ser-133 induces complex formation with CREB-binding
protein via a direct mechanism. Mol Cell Biol. 16:694–703. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Iavarone A, Garg P, Lasorella A, Hsu J and
Israel MA: The helix-loop-helix protein Id-2 enhances cell
proliferation and binds to the retinoblastoma protein. Genes Dev.
8:1270–1284. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lasorella A, Iavarone A and Israel MA: Id2
specifically alters regulation of the cell cycle by tumor
suppressor proteins. Mol Cell Biol. 16:2570–2578. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Moldes M, Boizard M, Liepvre XL, Feve B,
Dugail I and Pairault J: Functional antagonism between inhibitor of
DNA binding (Id) and adipocyte determination and differentiation
factor 1/sterol regulatory element-binding protein-1c
(ADD1/SREBP-1c) trans-factors for the regulation of fatty acid
synthase promoter in adipocytes. Biochem J. 344:873–880. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Inoue T, Shoji W and Obinata M: MIDA1, an
Id-associating protein, has two distinct DNA binding activities
that are converted by the association with Id1: A novel function of
Id protein. Biochem Biophys Res Commun. 266:147–151. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yates PR, Atherton GT, Deed RW, Norton JD
and Sharrocks AD: Id helix-loop-helix proteins inhibit
nucleoprotein complex formation by the TCF ETS-domain transcription
factors. EMBO J. 18:968–976. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Balasubramanyam K, Varier RA, Altaf M,
Swaminathan V, Siddappa NB, Ranga U and Kundu TK: Curcumin, a novel
p300/CREB-binding protein-specific inhibitor of acetyltransferase,
represses the acetylation of histone/nonhistone proteins and
histone acetyltransferase-dependent chromatin transcription. J Biol
Chem. 279:51163–51171. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Choi KC, Jung MG, Lee YH, Yoon JC, Kwon
SH, Kang HB, Kim MJ, Cha JH, Kim YJ, Jun WJ, et al:
Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor,
inhibits EBV-induced B lymphocyte transformation via suppression of
RelA acetylation. Cancer Res. 69:583–592. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tong KM, Shieh DC, Chen CP, Tzeng CY, Wang
SP, Huang KC, Chiu YC, Fong YC and Tang CH: Leptin induces IL-8
expression via leptin receptor, IRS-1, PI3K, Akt cascade and
promotion of NF-kappaB/p300 binding in human synovial fibroblasts.
Cell Signal. 20:1478–1488. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Seong AR, Yoo JY, Choi K, Lee MH, Lee YH,
Lee J, Jun W, Kim S and Yoon HG: Delphinidin, a specific inhibitor
of histone acetyltransferase, suppresses inflammatory signaling via
prevention of NF-κB acetylation in fibroblast-like synoviocyte MH7A
cells. Biochem Biophys Res Commun. 410:581–586. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Khoa ND, Nakazawa M, Hasunuma T, Nakajima
T, Nakamura H, Kobata T and Nishioka K: Potential role of HOXD9 in
synoviocyte proliferation. Arthritis Rheum. 44:1013–1021. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bannister AJ and Kouzarides T: The CBP
co-activator is a histone acetyltransferase. Nature. 384:641–643.
1996. View Article : Google Scholar : PubMed/NCBI
|