Introduction
Atherosclerosis is a chronic progressive
inflammatory disease and is the leading cause of mortality
worldwide due to the progression to myocardial infarction and
stroke, which are major clinical consequences (1,2). It is
widely accepted that the transformation of vascular smooth muscle
cells (VSMCs) from a quiescent contractile phenotype to a
proliferative migratory phenotype facilitates stable plaque
formation, contributing to the pathogenesis of atherosclerosis
(3). However, the molecular
mechanisms underlying the proliferation and migration of VSMCs in
the pathogenesis of atherosclerosis remain to be fully
understood.
MicroRNAs (miRNAs/miRs) are evolutionarily
conserved, non-coding small RNAs that can post-transcriptionally
modulate gene expression by binding to the 3′-untranslated region
(UTR) of target mRNA (4). miRNAs
have been shown to play pivotal roles in tumorigenesis, biological
metabolism and immune inflammation by modulating cell
proliferation, apoptosis, migration, invasion and differentiation
(5–8). Increasing evidence has revealed that
miRNAs may be closely associated with the pathogenesis of
atherosclerosis through the modulation of VSMC proliferation and
migration (9–12). Therefore, targeting miRNAs may
represent a potentially effective method for the treatment of
atherosclerosis.
Sustained Ca2+ influx mediated by
store-operated channels (SOCs) is triggered in response to
Ca2+ depletion by the endoplasmic reticulum (ER), and
has been discovered to serve an important role in numerous cellular
functions, such as cell proliferation and apoptosis (13). Stromal interaction molecule 1
(STIM1) has been reported to modulate SOCs by functioning as an ER
Ca2+ sensor and has also been reported to modulate the
proliferation of VSMCs (14,15).
Using bioinformatics technology, TargetScan (http://www.targetscan.org/vert_71) and miRDB
(http://www.mirdb.org/index.html), STIM1
was predicted as a target gene of miR-541-3p in the present study.
Thus, it was hypothesized that miR-541-3p may be involved in the
pathogenesis of atherosclerosis by targeting STIM1.
The present study aimed to determine the role of the
miR-541-3p/STIM1 axis in the biological behaviors of VSMCs.
Oxidized low-density lipoprotein (ox-LDL)-treated VSMCs were used
as an in vitro model of atherosclerosis, and VSMC viability
and migration were detected to investigate the pathogenesis of
atherosclerosis in vitro.
Materials and methods
Cell culture and treatment
Human VSMCs (cat. no. CRL-1999™) were purchased from
the American Type Culture Collection and cultured in F-12K medium
(Gibco; Thermo Fisher Scientific, Inc.), supplemented with 10% FBS
(Gibco; Thermo Fisher Scientific, Inc.). The cells were maintained
at 37°C in a humidified incubator with 5% CO2.
To construct the in vitro atherosclerosis
model, VSMCs were treated with 100 mg/l ox-LDL (cat. no. H7950;
Beijing Solarbio Science & Technology Co., Ltd.) for 24 or 48 h
at 37°C. The untreated VSMCs served as a control.
Cell transfection
miR-541-3p mimic and inhibitor (used to overexpress
and knockdown miR-541-3p expression in VSMCs, respectively), small
interfering RNA (siRNA/si) targeting STIM1 (si-STIM1) and the
negative controls (NCs), mimic-NC, inhibitor-NC and si-NC
(non-targeting sequences) were all synthesized by Shanghai
GenePharma Co., Ltd. The mimic (150 nM), inhibitor (100 nM),
si-STIM1 (50 nM) and the same amount of control vectors were
transfected into 1×106 cells using
Lipofectamine® 2000 transfection reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) at room temperature according to
the manufacturer's protocol. The transfection efficiency was
detected by RT-qPCR and western blotting 48 h post-transfection.
The sequences were as follows: Mimic, 5′-TGGTGGGCACAGAATCTGGACT-3′;
mimic-NC, 5′-UUCUCCGAACGUGUCACGUTT-3′; inhibitor,
5′-AGTCCAGATTCTGTGCCCACCA-3′; inhibitor-NC,
5′-ACAGGAUUGAGGGGGGGCCCU-3′; si-STIM1,
5′-GAGGTGCAATATTACAACATCAAGA-3′; si-NC,
5′-GAGAACGTTATAACACTACATGAGA-3′.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from VSMCs using the RNApure
Tissue & Cell kit (CoWin Biosciences), according to the
manufacturer's protocol. Total RNA (1 µg) was reverse transcribed
into cDNA for miRNA and mRNA detection using stem-loop primers and
random primers using EasyScript® First-Strand cDNA
Synthesis SuperMix (Beijing Transgen Biotech Co., Ltd.) according
to the manufacturer's protocol, respectively. qPCR was subsequently
performed using a TaqMan Universal Master mix II kit (Thermo Fisher
Scientific, Inc.) on a Bio-Rad detection system (Bio-Rad
Laboratories, Inc.). GAPDH and U6 served as the internal references
for the normalization of mRNA and miRNA expression, respectively.
The relative expression was calculated using the 2−ΔΔCq
method (16). PCR thermocycling
conditions were as follows: Initial denaturation at 95°C for 30
sec, followed by 39 cycles at 95°C for 15 sec and 60°C for 30 sec,
and a final step at 72°C for 5 min. The following primer pairs were
used: Stem-loop primer (used for RT of miR-541-3p),
5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAGTCCA-3′;
miR-541-3p forward, 5′-TGGTGGGCACAGAATC-3′ and reverse,
5′-GTGCAGGGTCCGAGGT-3′; U6 forward, 5′-CTCGCTTCGGCAGCACA-3′ and
reverse, 5′-AACGCTTCACGAATTTGCGT-3′; STIM1 forward,
5′-GTTCTGAAGGCTACGGGACC-3′ and reverse, 5′-GTCAGAAGGGGGTGTGTCAG-3′;
GAPDH forward, 5′-CCACTAGGCGCTCACTGTTCTC-3′ and reverse,
5′-ACTCCGACCTTCACCTTCCC-3′.
Western blotting
Total protein was extracted from VSMCs using RIPA
lysis buffer (Sangon Biotech Co., Ltd.) supplemented with protease
inhibitor (Beijing Solarbio Science & Technology Co., Ltd.).
Total protein was quantified using a BCA protein kit (Bio-Rad
Laboratories, Inc.) and 30 µg protein from each sample was loaded
and separated via SDS-PAGE on 10% gels. The separated proteins were
subsequently transferred onto polyvinylidene difluoride membranes
(EMD Millipore) and blocked with 5% non-fat milk for 1 h at room
temperature. The membranes were then incubated with the following
primary antibodies overnight at 4°C: Anti-STIM1 (cat. no. 4916;
Cell Signaling Technology, Inc.; 1:3,000 dilution), anti-VEGF (cat.
no. 2463; Cell Signaling Technology, Inc. 1:3,000 dilution),
anti-matrix metalloproteinase (MMP)9 (cat. no. 13667; Cell
Signaling Technology, Inc.; 1:3,000 dilution), anti-phosphorylated
(p)-PI3K p85 (cat. no. 17366; Cell Signaling Technology, Inc.;
1:2,000 dilution), anti-PI3K p85 (cat. no. 4292; Cell Signaling
Technology, Inc.; 1:3,000 dilution), anti-p-AKT1 (cat. no. 9018;
Cell Signaling Technology, Inc.; 1:2,000 dilution), anti-AKT1 (cat.
no. 2938; Cell Signaling Technology, Inc.; 1:3,000 dilution) and
anti-GAPDH (cat. no. 2118; Cell Signaling Technology, Inc.; 1:2,000
dilution). Following the primary antibody incubation, the membranes
were incubated with the goat anti-rabbit IgG HRP-linked secondary
antibodies (cat. no. sc-2004; Santa Cruz Biotechnology, Inc.;
1:5,000 dilution) for 1 h at room temperature. Protein expression
levels were determined using a western blotting imaging and
quantitative system (Gel Doc XR+; Bio-Rad Laboratories, Inc.)
following incubation with the ECL western blotting substrate (EMD
Millipore). Densitometric analysis was performed using ImageJ
software (version 1.6.0; National Institutes of Health) after
background subtraction, with GAPDH expression levels acting as the
internal reference control.
Dual luciferase gene reporter
assay
The putative binding sites between miR-541-3p and
STIM1 were predicted using TargetScan (http://www.targetscan.org/vert_71) and miRDB
(http://www.mirdb.org/index.html). The
wild-type (WT) and mutant (MUT) type of the 3′-UTR of STIM1, which
were cloned into the pGL3 vector (Promega Corporation), were
constructed by Shanghai GenePharma Co., Ltd.; the resulting
plasmids were denoted as STIM1-WT or STIM1-MUT reporter plasmids,
respectively. VSMCs (1×105) were co-transfected with WT
(0.2 µg) or MUT (0.2 µg) and miR-541-3p mimic (150 nM) or mimic-NC
(150 nM), as well as the Renilla luciferase vector (0.2 µg;
control) using Lipofectamine 2000 transfection reagent. A total of
48 h post-transfection, the relative luciferase activity normalized
to Renilla luciferase activity was measured using a Dual
Luciferase Reporter assay system (Promega Corporation) according to
the manufacturer's protocol.
Cell Counting Kit-8 (CCK-8) assay
VSMCs were seeded into 96-well plates at a density
of 3×103 cells/well and cultured at 37°C overnight,
followed by transfection with different vectors as aforementioned.
After incubation at 37°C for 24 or 48 h, cell culture medium was
replaced with 10 µl CCK-8 reagent (Beyotime Institute of
Biotechnology) and 90 µl fresh medium containing 10% FBS, and
incubated at 37°C for a further 4 h. The optical density (OD) value
was measured at a wavelength of 450 nm using a microplate reader
(model 680; Bio-Rad Laboratories, Inc.). Cell viability (%) = OD
(treatment group)/OD (control group) ×100.
Transwell migration assay
Transwell chambers (BD Biosciences) were used to
determine the cell migration ability. Briefly, ~3×105
VSMCs were resuspended in serum-free medium and seeded into the
upper chamber of the Transwell plates. The lower chambers were
filled with 600 µl medium supplemented with 15% FBS. After
incubation at 37°C for 48 h, the cells remaining on the top of the
membranes were removed using cotton buds, while cells on the lower
membrane were fixed with −20° C-precooled absolute methanol for 15
min and stained with 0.2% crystal violet (Beijing Solarbio Science
& Technology Co., Ltd.) for 5 min all at room temperature. The
stained cells were subsequently counted under a light microscope
(magnification, ×200) to assess cell migration.
Statistical analysis
Data were presented as the mean ± SD. Statistical
analysis was performed using SPSS 21.0 software (IBM Corp.). Each
experiment was performed in triplicate. Statistical differences
between two groups and multiple groups were performed using an
unpaired Student's t-test or one-way ANOVA followed by a
Bonferroni's post hoc test, respectively. P<0.05 was considered
to indicate a statistically significant difference.
Results
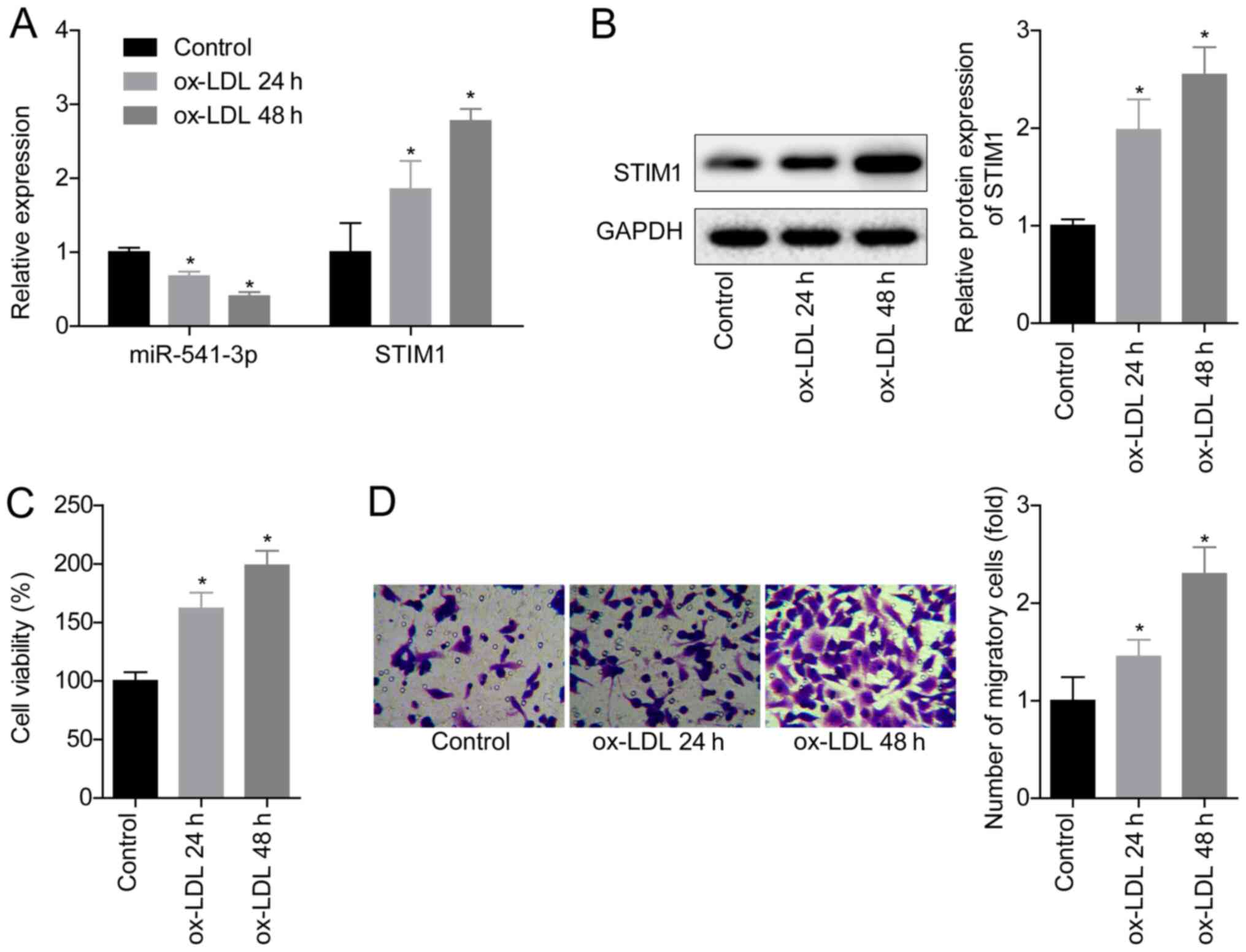
Ox-LDL treatment downregulates the
expression levels of miR-541-3p and upregulates STIM1 expression
levels
To determine the role of the miR-541-3p/STIM1 axis
in the pathogenesis of atherosclerosis, the expression levels of
miR-541-3p and STIM1 in VSMCs following the treatment with ox-LDL
were analyzed. The results demonstrated that ox-LDL treatment
significantly downregulated miR-541-3p expression levels and
significantly upregulated STIM1 expression levels compared with the
untreated VSMCs in the control group in a time-dependent manner
(Fig. 1A and B). VSMC viability was
significantly increased following treatment with 100 mg/l ox-LDL
for 24 and 48 h compared with the control group (Fig. 1C), as was the cell migratory ability
(Fig. 1D). These results indicated
that ox-LDL treatment may downregulate miR-541-3p expression and
upregulate STIM1 expression, which may be involved in process of
atherosclerosis.
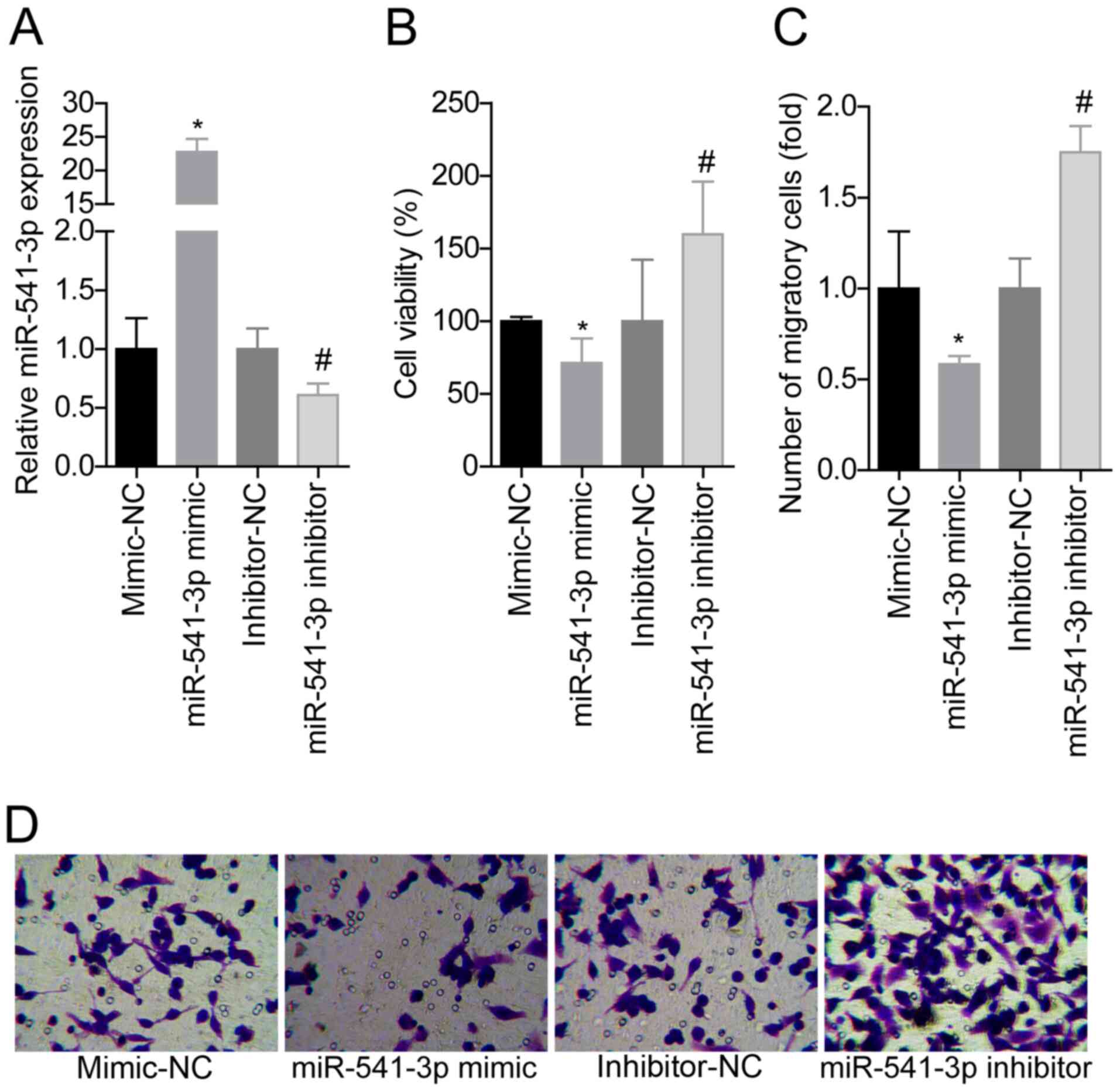
Overexpression of miR-541-3p inhibits
the viability and migration of VSMCs
The effects of miR-541-3p on cell viability and
migration in ox-LDL (100 mg/l; 24 h)-treated VSMCs were analyzed
using gain- and loss-of-function assays. Since ox-LDL treatment for
24 h caused significant differences in cell viability and
migration, this timepoint was assessed in subsequent experiments.
miR-541-3p expression levels were significantly downregulated
following the transfection of VSMCs with the miR-541-3p inhibitor,
and significantly upregulated following transfection with the
miR-541-3p mimic compared with the respective NCs (Fig. 2A). The cell viability (Fig. 2B) and migratory abilities (Fig. 2C and D) were significantly weakened
following the overexpression of miR-541-3p in VSMCs compared with
the mimic-NC-transfected VSMCs, whereas knockdown of miR-541-3p
expression exacerbated ox-LDL-induced stimulation of cell viability
and migration compared with the inhibitor-NC-transfected VSMCs
(Fig. 2B-D). These results
suggested that miR-541-3p may suppress the ox-LDL-induced induction
of cell viability and migration in VSMCs.
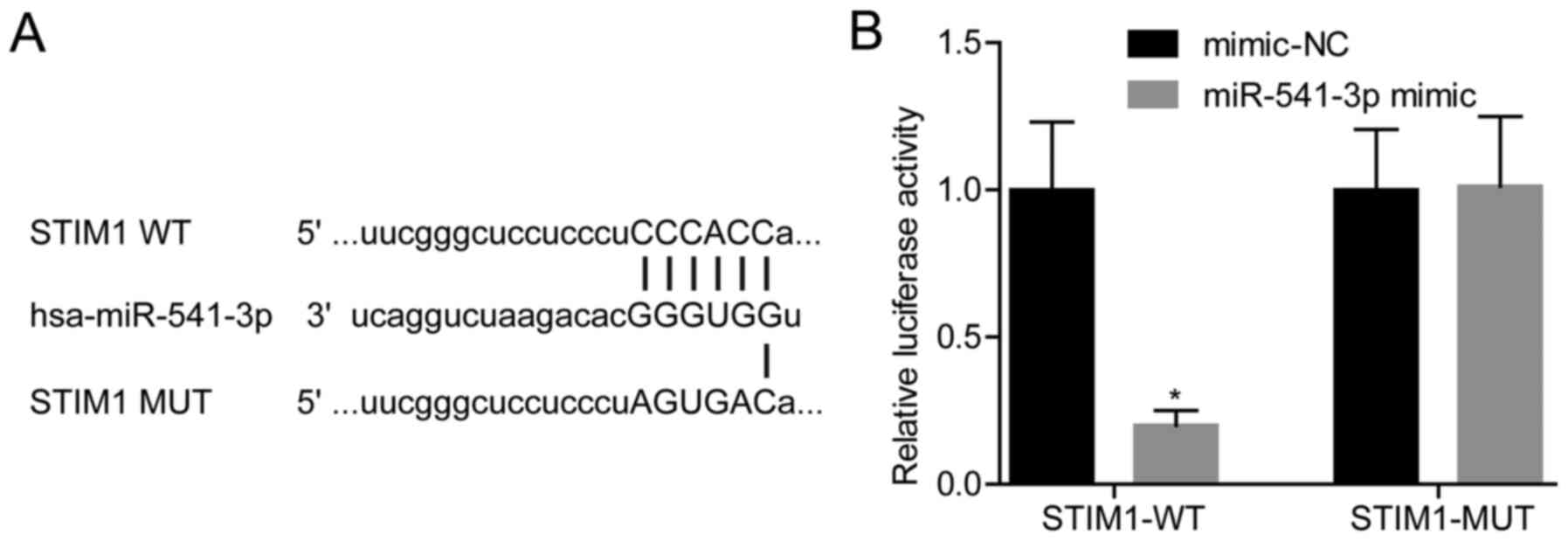
miR-541-3p targets STIM1 in VSMCs
As shown in Fig. 3A,
STIM1 was a putative target of miR-541-3p, which was confirmed by
the dual luciferase gene reporter assay using STIM1-WT or STIM1-MUT
reporter plasmids (Fig. 3B). The
results revealed that overexpression of miR-541-3p in VSMCs
transfected with STIM1-WT induced a significant reduction in
luciferase activity; however, the mutation in the binding site
between miR-541-3p and the 3′UTR of STIM1 abrogated this effect
(Fig. 3B). These results suggested
that miR-541-3p may target STIM1 in VSMCs.
Knockdown of miR-541-3p promotes
ox-LDL-induced VSMC viability and migration by targeting STIM1
CCK-8 and Transwell assays were subsequently
performed to investigate the role of the miR-541-3p/STIM1 axis in
ox-LDL-induced VSMC viability and migration. The transfection with
si-STIM1 significantly downregulated STIM1 expression at both the
mRNA and protein levels compared with the si-NC group (Fig. S1A and B). In addition, the
expression levels of STIM1 were significantly upregulated in VSMCs
in the miR-541-3p inhibitor + si-NC group as compared with in the
inhibitor-NC + si-NC group, while the expression levels were
significantly downregulated in cells transfected with si-STIM1 in
the inhibitor-NC + si-STIM1 group (Fig.
4A and B). Knockdown of STIM1 in the miR-541-3p inhibitor +
si-STIM1 group impaired the miR-541-3p inhibitor-induced
stimulatory effect on cell viability and migration in ox-LDL (100
mg/l; 24 h)-treated VSMCs as compared with the inhibitor-NC + si-NC
group (Fig. 4C and D), indicating
that knockdown of miR-541-3p may promote ox-LDL-induced VSMC
viability and migration by targeting STIM1.
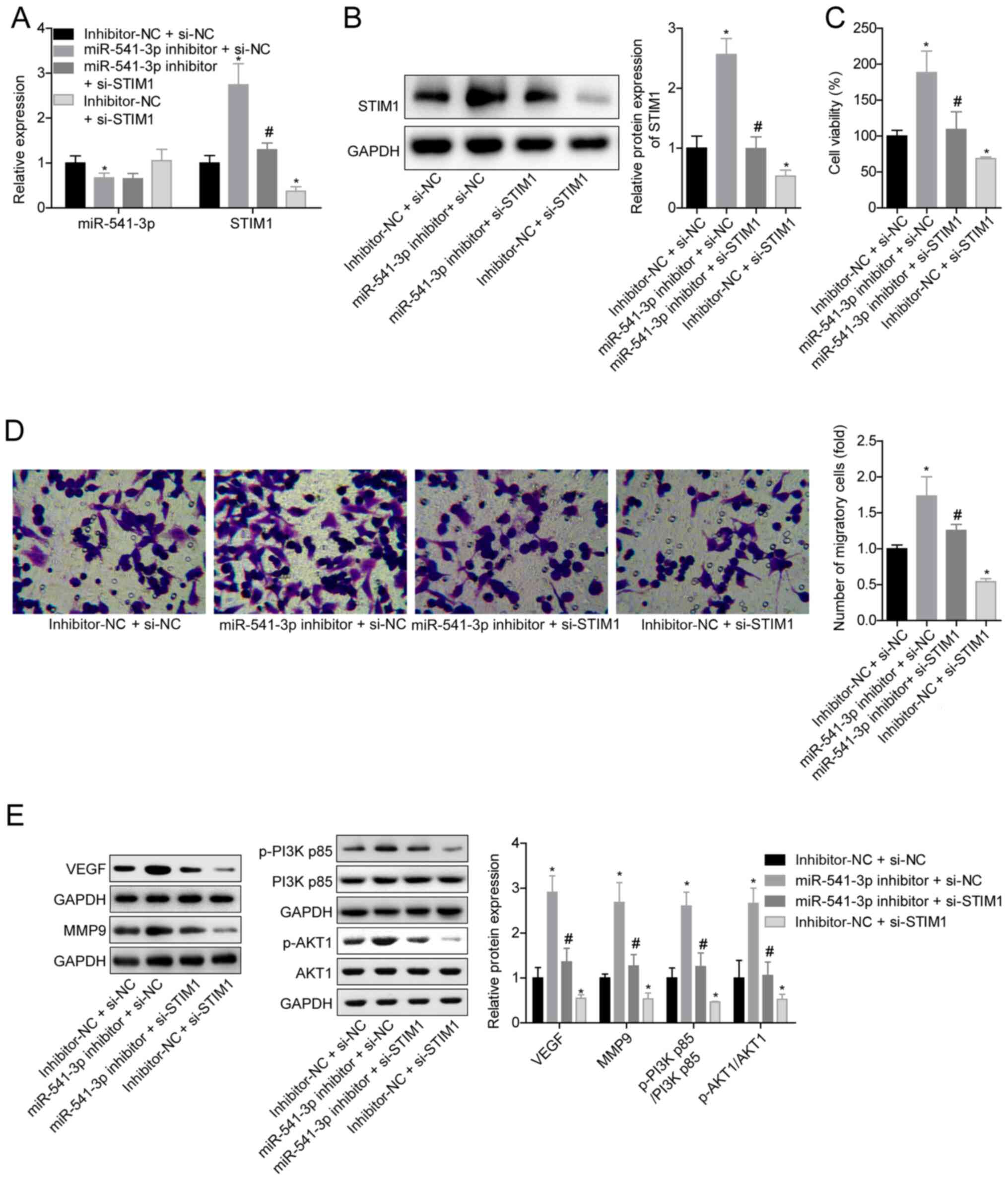 | Figure 4.Effects of the miR-541-3p/STIM1 axis
on cell viability and migration, and the activation of PI3K/AKT
signaling in VSMCs. VSMCs were pretreated with 100 mg/l ox-LDL for
24 h, followed by transfection with inhibitor-NC + si-NC,
miR-541-3p inhibitor + si-NC, miR-541-3p inhibitor + si-STIM1 or
inhibitor-NC + si-STIM1. (A) Expression levels of miR-541-3p and
STIM1 were analyzed using reverse transcription-quantitative PCR.
(B) Protein expression levels of STIM1 were analyzed using western
blotting. (C) Cell Counting Kit-8 assay was used to analyze cell
viability. (D) Transwell assay was used to determine cell
migration. Magnification, ×200. (E) Expression levels of VEGF,
MMP9, p-PI3K p85, PI3K p85, p-AKT1 and AKT1 were analyzed using
western blotting. n=3; *P<0.05 vs. inhibitor-NC + si-NC;
#P<0.05 vs. miR-541-3p inhibitor + si-NC (one-way
ANOVA and Bonferroni post hoc test). miR, microRNA; STIM1, stromal
interaction molecular 1; VSMCs, vascular smooth muscle cells;
ox-LDL, oxidized low-density lipoprotein; NC, negative control; si,
small interfering RNA; p-, phosphorylated; MMP9, matrix
metalloproteinase 9. |
In addition, the knockdown of miR-541-3p in the
miR-541-3p inhibitor + si-NC group significantly upregulated the
expression levels of VEGF and MMP9, and the p-AKT1:AKT1 and
p-PI3K:PI3K ratios as compared with the inhibitor-NC + si-NC group.
However, transfection with si-STIM1 in the inhibitor-NC + si-STIM1
group induced the opposite results and rescued the effect of the
miR-541-3p inhibitor (miR-541-3p inhibitor + si-STIM1 group vs.
miR-541-3p inhibitor + si-NC group) (Fig. 4E). These results suggested that the
miR-541-3p/STIM1 axis may modulate VSMC viability and migration via
PI3K/AKT signaling.
Discussion
The enhanced proliferation of VSMCs has been
identified as the main mechanism of aberrant neointima formation in
vascular diseases (17). During
atherogenesis, VSMCs respond to several factors, including
thrombin, platelet derived growth factor-BB, IFN-γ, endothelin-1
and IL-1, resulting in the migration of VSMCs to the intima
(18,19). The migration of VSMCs leads to
plaque formation and also facilitates the atherosclerosis hardening
process (20). Thus, inhibition of
VSMC proliferation and migration is a potential strategy for the
prevention or treatment of atherosclerosis (21). The present study aimed to determine
the role of the miR-541-3p/STIM1 axis in the viability and
migration of VSMCs. The results illustrated that miR-541-3p
suppressed ox-LDL-mediated VSMC viability and migration by
targeting STIM1.
To date, numerous miRNAs have been reported to play
an important role in the pathogenesis of atherosclerosis by
modulating VSMC proliferation and migration (22). For example, miR-146a expression was
revealed to be upregulated in proliferating VSMCs, and knockdown of
miR-146a expression weakened the proliferative and migratory
capacities of VSMCs in vitro (23). In addition, miR-92a expression was
upregulated in the atherosclerotic plaques of mice and the
overexpression of miR-92a significantly increased the proliferation
of VSMCs (24). Xu et al
(25) also reported that miR-647
expression was upregulated in the serum samples of patients with
atherosclerosis and ox-LDL-treated VSMCs, and the upregulation of
miR-647 expression levels promoted the proliferation and migration
of ox-LDL-treated VSMCs via targeting PTEN. miR-541-3p expression
levels were also shown to be downregulated in non-small cell lung
cancer (NSCLC) tissues and plasma, and the overexpression
subsequently suppressed NSCLC cell growth and metastasis (26). To the best of our knowledge, the
present study was the first to demonstrate that miR-541-3p
expression levels were downregulated in ox-LDL-treated VSMCs, and
that the overexpression of miR-541-3p significantly suppressed
ox-LDL-induced increases in VSMC viability and migration.
Using TargetScan and miRDB, the current study
predicted that miR-541-5p was a regulator of STIM1, which has been
previously identified to promote VSMC proliferation (14,15).
Subsequently, dual luciferase gene reporter assays were used to
verify that STIM1 was a target gene of miR-541-3p in VSMCs. Further
in vitro experiments revealed that the genetic knockdown of
STIM1 significantly weakened the miR-541-3p knockdown-induced
stimulation of VSMC viability and migration, indicating that
miR-541-3p may inhibit ox-LDL-induced VSMC viability and migration
by targeting STIM1. miR-541-5p was found to negatively regulate
STIM1 expression by binding to the 3′-UTR of STIM1 mRNA, leading to
the inhibition of VSMC viability. The present results showed that
STIM1 may serve a role in regulating cell viability, which was
consistent with previous studies (27–30),
which reported that the dysregulation of STIM1 caused a
deregulation in cell viability.
The PI3K/AKT signaling pathway, one of the most
important intracellular pathways in the body, is involved in a
variety of cellular processes, including cell proliferation,
survival, differentiation and migration (31). Notably, PI3K/AKT signaling was also
discovered to play a crucial role in the pathogenesis of
atherosclerosis (32). It has
previously been reported that the selective inhibition of PI3K/AKT
signaling could potently suppress the progression of
atherosclerosis (33,34), whereas the activation of PI3K/AKT
signaling enhanced the proliferation, survival and migration of
VSMCs and human umbilical vein endothelial cells (35). These findings indicated that
PI3K/AKT signaling may be an important target for the treatment of
atherosclerosis. Therefore, the present study investigated the
effect of the miR-541-3p/STIM1 axis on the activation of PI3K/AKT
signaling. The results revealed that the knockdown of miR-541-3p
significantly increased the phosphorylation levels of PI3K and AKT;
however, this trend was abolished following the silencing of STIM1
in ox-LDL-treated VSMCs. Consistent with these findings, a previous
study reported that the knockdown of STIM1 inactivated PI3K/AKT
signaling in human prostate cancer cells (36). These aforementioned results
suggested that PI3K/AKT signaling may serve a role in the
miR-541-5p/STIM1 axis-mediated inhibition of VSMC viability and
migration.
There are two main limitations of the present study.
Firstly, the specific role of the PI3K/AKT signaling pathway in the
miR-541-5p/STIM1 axis-mediated inhibition of VSMC viability and
migration was not clarified. Secondly, the role of the
miR-541-3p/STIM1 axis was only investigated in vitro. Thus,
in vivo experiments should be performed in future studies to
determine the role of the miR-541-3p/STIM1 axis in the pathogenesis
of atherosclerosis.
In conclusion, the findings of the present study
suggested that miR-541-3p may efficiently suppress ox-LDL-induced
increases in VSMC viability and migration by targeting STIM1, which
may be associated with the suppression of PI3K/AKT signaling. These
results indicated that the miR-541-3p/STIM1 axis may represent a
potential target to modulate VSMC viability and migration.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZL, DY, CZ and YX designed the study, analyzed the
data and interpreted the results. ZL, DY, YX and HH performed
experiments. ZL, DY, CZ and YX wrote the manuscript and prepared
the figures. ZF and XL analyzed data, and reviewed and edited the
manuscript. XL coordinated and supervised the study. ZL and XL
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Libby P, Ridker PM and Hansson GK:
Progress and challenges in translating the biology of
atherosclerosis. Nature. 473:317–325. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tabas I, García-Cardeña G and Owens GK:
Recent insights into the cellular biology of atherosclerosis. J
Cell Biol. 209:13–22. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kolodgie FD, Burke AP, Farb A, Gold HK,
Yuan J, Narula J, Finn AV and Virmani R: The thin-cap
fibroatheroma: A type of vulnerable plaque: The major precursor
lesion to acute coronary syndromes. Curr Opin Cardiol. 16:285–292.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pal AS and Kasinski AL: Animal models to
study MicroRNA function. Adv Cancer Res. 135:53–118. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bernardo BC, Ooi JY, Lin RC and McMullen
JR: miRNA therapeutics: A new class of drugs with potential
therapeutic applications in the heart. Future Med Chem.
7:1771–1792. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Karnati HK, Panigrahi MK, Gutti RK, Greig
NH and Tamargo IA: miRNAs: Key players in neurodegenerative
disorders and epilepsy. J Alzheimers Dis. 48:563–580. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mehrgou A and Akouchekian M: Therapeutic
impacts of microRNAs in breast cancer by their roles in regulating
processes involved in this disease. J Res Med Sci. 22:1302017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang SC, Wang M, Wu WB, Wang R, Cui J, Li
W, Li ZL, Li W and Wang SM: Mir-22-3p inhibits arterial smooth
muscle cell proliferation and migration and neointimal hyperplasia
by targeting HMGB1 in arteriosclerosis obliterans. Cell Physiol
Biochem. 42:2492–2506. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen Q, Yang F, Guo M, Wen G, Zhang C,
Luong le A, Zhu J, Xiao Q and Zhang L: miRNA-34a reduces neointima
formation through inhibiting smooth muscle cell proliferation and
migration. J Mol Cell Cardiol. 89:75–86. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhu J, Liu B, Wang Z, Wang D, Ni H, Zhang
L and Wang Y: Exosomes from nicotine-stimulated macrophages
accelerate atherosclerosis through miR-21-3p/PTEN-mediated VSMC
migration and proliferation. Theranostics. 9:6901–6919. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang H, Jiang M, Xu Z, Huang H, Gong P,
Zhu H and Ruan C: miR-146b-5p promotes VSMC proliferation and
migration. Int J Clin Exp Pathol. 8:12901–12907. 2015.PubMed/NCBI
|
|
13
|
Parekh AB and Putney JW Jr: Store-operated
calcium channels. Physiol Rev. 85:757–810. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Takahashi Y, Watanabe H, Murakami M, Ono
K, Munehisa Y, Koyama T, Nobori K, Iijima T and Ito H: Functional
role of stromal interaction molecule 1 (STIM1) in vascular smooth
muscle cells. Biochem Biophys Res Commun. 361:934–940. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Potier M, Gonzalez JC, Motiani RK,
Abdullaev IF, Bisaillon JM, Singer HA and Trebak M: Evidence for
STIM1- and Orai1-dependent store-operated calcium influx through
ICRAC in vascular smooth muscle cells: Role in proliferation and
migration. FASEB J. 23:2425–2437. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Giachini FR, Lima VV, Hannan JL, Carneiro
FS, Webb RC and Tostes RC: STIM1/Orai1-mediated store-operated
Ca2+ entry: The tip of the iceberg. Braz J Med Biol Res.
44:1080–1087. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lopes J, Adiguzel E, Gu S, Liu SL, Hou G,
Heximer S, Assoian RK and Bendeck MP: Type VIII collagen mediates
vessel wall remodeling after arterial injury and fibrous cap
formation in atherosclerosis. Am J Pathol. 182:2241–2253. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rudijanto A: The role of vascular smooth
muscle cells on the pathogenesis of atherosclerosis. Acta Med
Indones. 39:86–93. 2007.PubMed/NCBI
|
|
20
|
Pan J, Lu L, Wang X, Liu D, Tian J, Liu H,
Zhang M, Xu F and An F: AIM2 regulates vascular smooth muscle cell
migration in atherosclerosis. Biochem Biophys Res Commun.
497:401–409. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang L, Cheng H, Yue Y, Li S, Zhang D and
He R: H19 knockdown suppresses proliferation and induces apoptosis
by regulating miR-148b/WNT/beta-catenin in ox-LDL-stimulated
vascular smooth muscle cells. J Biomed Sci. 25:112018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li L, Li Y and Tang C: The role of
microRNAs in the involvement of vascular smooth muscle cells in the
development of atherosclerosis. Cell Biol Int. 43:1102–1112. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dong S, Xiong W, Yuan J, Li J, Liu J and
Xu X: miRNA-146a regulates the maturation and differentiation of
vascular smooth muscle cells by targeting NF-κB expression. Mol Med
Rep. 8:407–412. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang J, Zhang C, Li C, Zhao D, Li S, Ma L,
Cui Y, Wei X, Zhao Y and Gao Y: MicroRNA-92a promotes vascular
smooth muscle cell proliferation and migration through the
ROCK/MLCK signalling pathway. J Cell Mol Med. 23:3696–3710. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xu CX, Xu L, Peng FZ, Cai YL and Wang YG:
miR-647 promotes proliferation and migration of ox-LDL-treated
vascular smooth muscle cells through regulating PTEN/PI3K/AKT
pathway. Eur Rev Med Pharmacol Sci. 23:7110–7119. 2019.PubMed/NCBI
|
|
26
|
Lu YJ, Liu RY, Hu K and Wang Y: miR-541-3p
reverses cancer progression by directly targeting TGIF2 in
non-small cell lung cancer. Tumour Biol. 37:12685–12695. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mao YY, Wang JQ, Guo XX, Bi Y and Wang CX:
Circ-SATB2 upregulates STIM1 expression and regulates vascular
smooth muscle cell proliferation and differentiation through
miR-939. Biochem Biophys Res Commun. 505:119–125. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ge C, Zeng B, Li R, Li Z, Fu Q, Wang W,
Wang Z, Dong S, Lai Z, Wang Y, et al: Knockdown of STIM1 expression
inhibits non-small-cell lung cancer cell proliferation in vitro and
in nude mouse xenografts. Bioengineered. 10:425–436. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cheng H, Wang S and Feng R: STIM1 plays an
important role in TGF-β-induced suppression of breast cancer cell
proliferation. Oncotarget. 7:16866–16878. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim JH, Lkhagvadorj S, Lee MR, Hwang KH,
Chung HC, Jung JH, Cha SK and Eom M: Orai1 and STIM1 are critical
for cell migration and proliferation of clear cell renal cell
carcinoma. Biochem Biophys Res Commun. 448:76–82. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Aoki M and Fujishita T: Oncogenic roles of
the PI3K/AKT/mTOR axis. Curr Top Microbiol Immunol. 407:153–189.
2017.PubMed/NCBI
|
|
32
|
Linton MF, Moslehi JJ and Babaev VR: Akt
signaling in macrophage polarization, survival, and
atherosclerosis. Int J Mol Sci. 20:27032019. View Article : Google Scholar
|
|
33
|
Zhai C, Cheng J, Mujahid H, Wang H, Kong
J, Yin Y, Li J, Zhang Y, Ji X and Chen W: Selective inhibition of
PI3K/Akt/mTOR signaling pathway regulates autophagy of macrophage
and vulnerability of atherosclerotic plaque. PLoS One.
9:e905632014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhou M, Ren P, Zhang Y, Li S, Li M, Li P,
Shang J, Liu W and Liu H: Shen-Yuan-Dan capsule attenuates
atherosclerosis and foam cell formation by enhancing autophagy and
inhibiting the PI3K/Akt/mTORC1 signaling pathway. Front Pharmacol.
10:6032019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yao X, Yan C, Zhang L, Li Y and Wan Q:
LncRNA ENST00113 promotes proliferation, survival, and migration by
activating PI3K/Akt/mTOR signaling pathway in atherosclerosis.
Medicine (Baltimore). 97:e04732018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhou Y, Gu P, Li J, Li F, Zhu J, Gao P,
Zang Y, Wang Y, Shan Y and Yang D: Suppression of STIM1 inhibits
the migration and invasion of human prostate cancer cells and is
associated with PI3K/Akt signaling inactivation. Oncol Rep.
38:2629–2636. 2017. View Article : Google Scholar : PubMed/NCBI
|