Introduction
Cigarette smoke (CS) exposure is one of the most
important and modifiable risk factors for the development of
atherosclerosis and associated cardiovascular and cerebrovascular
diseases (1,2), representing 31% of all global deaths
in 2015 (3). Epidemiological
studies of various populations have shown that CS exposure is a
preventable risk factor for dyslipidemia (4–6).
However, the cessation of cigarette smoking increases serum levels
of high-density lipoprotein cholesterol (HDL-C) (5). In addition, exposure to CS for various
time periods has been shown to cause dyslipidemia in various
experimental animals (7–10). Dyslipidemia, particularly high
levels of low-density lipoprotein cholesterol (LDL-C), is a key
mechanism by which CS induces atherosclerosis (3). Previous studies have indicated that CS
exposure raises total cholesterol and circulating LDL-C levels
(4,9,10). The
accumulation of LDL-C in the subendothelial matrix is a primary
event in atherosclerosis, and elevated levels of LDL-C in the
circulation accelerate this process (3,11). Our
previous study demonstrated that CS exposure reduced the expression
of the LDL receptor (LDLR) in mouse hepatocytes and HepG2 cells
(10). However, the underlying
mechanisms by which CS mediated the change in LDLR expression
remain unclear.
Proprotein convertase subtilisin/kexin type 9
(PCSK9), which is mainly synthesized and secreted by hepatocytes,
binds to the epidermal growth factor-like repeat homology domain of
the LDLR on the surface of hepatocytes (12). The resulting complex is transported
to the lysosome where it is degraded, increasing circulating LDL-C
levels (12,13). PCSK9 loss-of-function
mutations have been shown to be associated with low circulating
levels of LDL-C and a reduced risk of coronary artery disease
(14,15). Monoclonal antibodies against PCSK9
have been used to treat patients with hyperlipidemia who are
statin-intolerant or have familial hypercholesterolemia (16). In addition to increasing the
circulating levels of LDL-C, PCSK9 plays an important role in
inflammation (17).
Lipopolysaccharide (LPS), tumor necrosis factor α (TNF-α) and
reactive oxygen species (ROS) markedly increase the expression of
PCSK9 in vivo and in vitro (17). However, the effect of CS on
PCSK9 expression has not yet been elucidated.
Melatonin has a variety of physiological functions,
including a regulatory effect of circadian rhythm, as well as
anti-inflammatory and antioxidant activity (18). Our previous study showed that
melatonin increased the expression of LDLR in mouse hepatocytes and
HepG2 cells (10). Other studies
have demonstrated that melatonin exerts anti-inflammatory and
antioxidant effects via the upregulation of sirtuin 1 (SIRT1)
activity and expression (19–21).
However, whether melatonin and SIRT1 mediate the CSE-induced
regulation of PCSK9 and LDLR expression remains unclear.
We hypothesize that CS and PCSK9/LDLR expression may
be linked. To examine this postulation, HepG2 cells were treated
with CS extract (CSE) and then the mRNA and protein expression
levels of PCSK9 and LDLR were evaluated via reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) and
western blotting, respectively. In addition, the role of
ROS/nuclear factor κB (NF-κB) signaling in the regulation of PCSK9
and LDLR expression by CSE was studied.
Materials and methods
Cell culture and treatment
HepG2 cells were purchased from the American Type
Culture Collection, and cultured in Dulbecco's modified Eagle's
medium (DMEM, HyClone; Cytiva) supplemented with 10% fetal bovine
serum (FBS; HyClone; Cytiva) and 1% (v/v) penicillin/streptomycin
at 37°C with 5% CO2. The cells were stimulated with 0,
1.25, 2.5 and 5% CSE for 24 h or with 5% CSE for 6, 12 and 24 h at
37°C. The concentrations of CSE were selected according to our
previous study (10). The medium
was changed to DMEM containing 1% FBS during treatment with CSE
and/or 5 mM ROS inhibitor [N-acetyl-L-cysteine (NAC);
Sigma-Aldrich; Merck KGaA], 10 µM NF-κB inhibitor (BAY11-7082;
Sigma-Aldrich, Merck KGaA), 100 µM melatonin (Sigma-Aldrich; Merck
KGaA) and 2 µM SIRT1 inhibitor (Inauhzin; Sigma-Aldrich; Merck
KGaA). In most experiments, HepG2 cells were pretreated with or
without BAY11-7082, melatonin or Inauhzin for 1 h prior to
stimulation with 5% CSE for 24 h at 37°C. However, the expression
level of phosphorylated protein was measured after treatment with
CSE for only 30 min, as it would be degraded if the treatment was
prolonged. All experiments were performed at least three times and
representative results are shown.
Preparation of CSE
CSE was obtained from ordinary filtered cigarettes
(each containing 0.90 mg nicotine and 10.0 mg tar) for all
experiments. One cigarette was bubbled into 15 ml DMEM, which was
then adjusted to pH 7.4 and filtered through a 0.22-mm filter
(Roche Diagnostics). This solution was defined as 100% CSE. The CSE
used in all experiments was freshly prepared.
Western blot analysis
Total protein from HepG2 cells was prepared using
lysis buffer (50 mM Tris, 150 mM NaCl, 1% NP-40, 0.5% sodium
deoxycholate, 0.1% SDS; pH 7.4) with 1 mM phenylmethylsulfonyl
fluoride (Beyotime Institute of Biotechnology) on ice; when
detecting phosphorylated protein, 1% (v/v) phosphatase inhibitor
(Shanghai Yeasen Biotechnology Co., Ltd.) was added to the lysis
solution. The lysates were centrifuged at 13,000 × g for 20 min at
4°C to remove cell debris, and the total protein concentration was
determined using a bicinchoninic acid protein assay kit (Beyotime
Institute of Biotechnology) according to the manufacturer's
instructions. Protein (20 µg/lane) was separated by 4–15% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (Applygen
Technologies, Inc.) and then electroblotted onto polyvinylidene
fluoride membranes. After blocking with 5% skimmed milk or 3%
bovine serum albumin (Shanghai Yeasen Biotechnology Co., Ltd.) in
Tris-buffered saline containing 0.1% Tween-20 (TBST) for 60 min at
room temperature, the membranes were incubated with primary
antibodies against the following: PCSK9 (1:1,000; cat. no. 85813S;
Cell Signaling Technology, Inc.), LDLR (1:1,000; cat. no.
10785-1-AP; ProteinTech Group, Inc.), phosphorylated (p)-p65
(1:1,000; cat. no. sc-136548; Santa Cruz Biotechnology, Inc.),
total p65 (1:1,000; cat. no. sc-8008; Santa Cruz Biotechnology,
Inc.) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH;
1:10,000; cat. no. 10494-1-AP; ProteinTech Group, Ltd.) overnight
at 4°C in a shaker. Then, the membranes were washed with TBST
(three times for 10 min each) before incubating with the secondary
anti-rabbit horseradish peroxidase-conjugated antibody (1:5,000;
cat. no. ZB-2301; Beijing Zhongshan Jinqiao Biotechnology Co.,
Ltd.) for 60 min at room temperature. The antibody-antigen
complexes were detected using enhanced electrochemiluminescence
reagents (Shanghai Yeasen Biotechnology Co., Ltd.) and
densitometrically analyzed with ImageJ software (version 1.46;
National Institutes of Health).
RT-qPCR
Total RNA was extracted from cells using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.) and
quantified with a NanoDrop™ 2000 Spectrophotometer (Thermo Fisher
Scientific, Inc.). Complementary DNA (cDNA) was synthesized with 1
µg total RNA using a High-Capacity cDNA Reverse Transcription Kit
(Takara Bio, Inc.), the temperature protocol was as follows: 42°C
for 60 min, 70°C for 15 min, and then 4°CC for 5 min. qPCR was then
performed in a CFX96 Touch Real-Time PCR Detection System (Bio-Rad
Laboratories, Inc.) using SYBR Premix (Shanghai Yeasen
Biotechnology Co., Ltd.), the thermocycling conditions were as
follows: Predenaturation step at 95°C for 3 min, followed by 40
cycles of 95°C for 10 sec and 60°C for 40 sec. Each sample was
performed in triplicate. Table I
lists the primer sequences used. The relative mRNA expression
levels were normalized to GAPDH expression using the
2−ΔΔCq method (22).
 | Table I.Sequences of primers. |
Table I.
Sequences of primers.
| Gene | Forward
(5′→3′) | Reverse
(5′→3′) |
|---|
| PCSK9 |
CCTGCGCGTGTCAACT |
GCTGGCTTTTCCGAAACTC |
| LDLR | GTGTCACAGCGGCG | CGCACTCTTTGATG |
| GAPDH |
GAAGGTGAAGGTCGGAGTC |
GAAGATGGTGATGGGATTTC |
Measurement of ROS production
Total intracellular ROS generation was measured
using an ROS Assay Kit (Beyotime Institute of Biotechnology)
according to the manufacturer's instructions. HepG2 cells were
pretreated with NAC (5 mM), BAY11-7082 (10 µM) or melatonin (100
µM) for 1 h, and then stimulated with 5% CSE for 24 h at 37°C.
After that, the medium was replaced with serum-free culture medium
containing 10 µM 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA)
for 20 min at 37°C, and then the cells were washed three times with
serum-free medium. The green fluorescence was measured using a
fluorescence microscope using an excitation of 488 nm and an
emission of 525 nm.
Statistical analysis
All data are presented as the mean ± standard
deviation of three different experiments in triplicate. The data
were analyzed using one-way analysis of variance with Bonferroni
test (GraphPad Prism Version 7.0; GraphPad Software, Inc.). All
data were obtained from at least three independent experiments.
P<0.05 was considered to indicate a statistically significant
result.
Results
Effects of CSE on PCSK9 and LDLR
expression in HepG2 cells
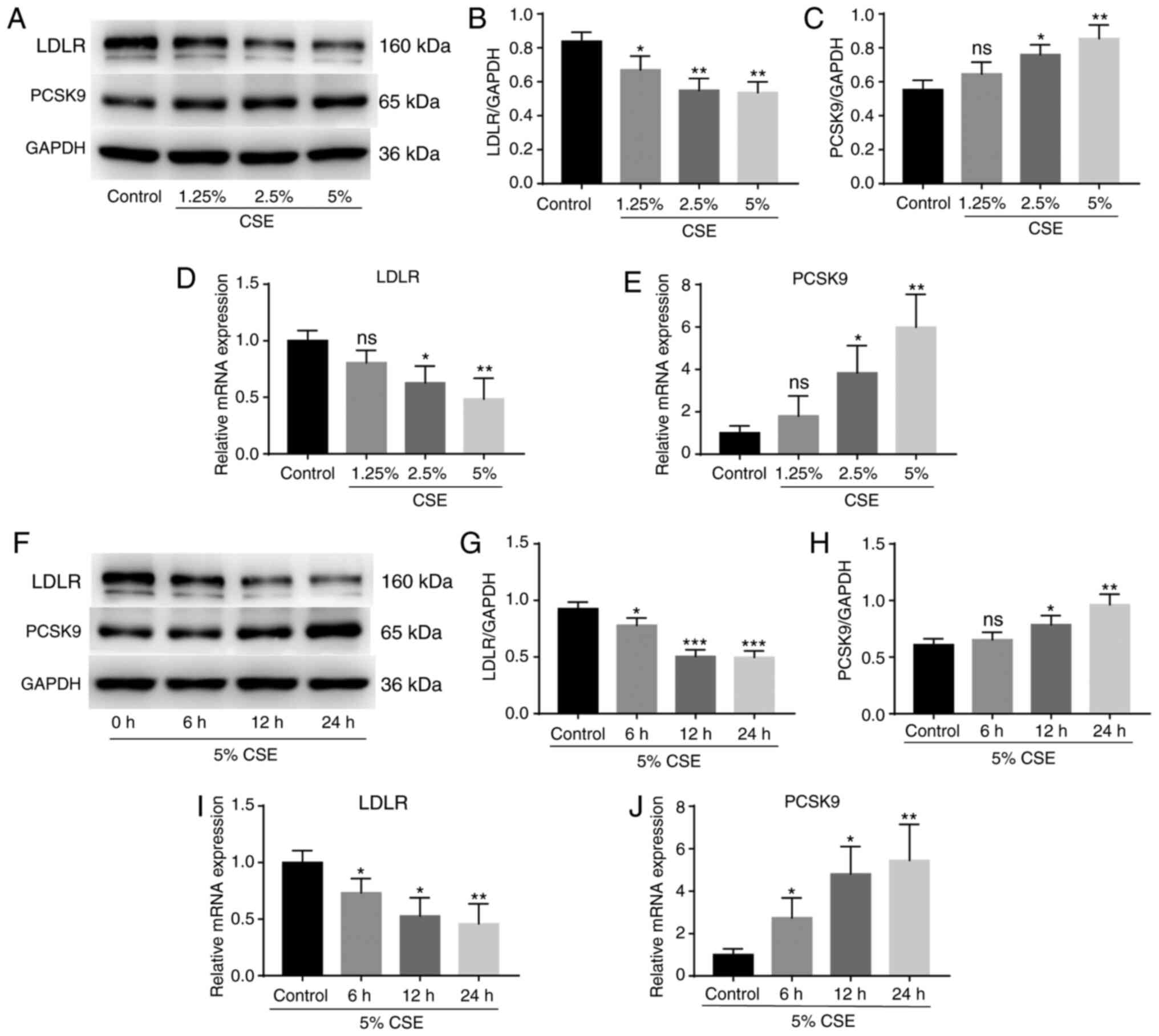
To investigate whether CSE regulates PCSK9 and LDLR
expression, HepG2 cells were treated with 0, 1.25, 2.5 and 5% CSE
for 24 h, and the expression levels of PCSK9 and LDLR were
determined using western blotting and RT-qPCR. CSE reduced the
expression of LDLR protein (Fig. 1A and
B) and mRNA (Fig. 1D) and
induced the expression of PCSK9 protein (Fig. 1A and C) and mRNA (Fig. 1E) in a concentration-dependent
manner. Then, HepG2 cells were stimulated with 5% CSE for 6, 12 and
24 h. The results revealed that CSE decreased the production of
LDLR protein (Fig. 1F and G) and
mRNA (Fig. 1I) and induced the
production of PCSK9 protein (Fig. 1F
and H) and mRNA (Fig. 1J) in a
time-dependent manner.
Effects of ROS on PCSK9 and LDLR
expression in CSE-stimulated HepG2 cells
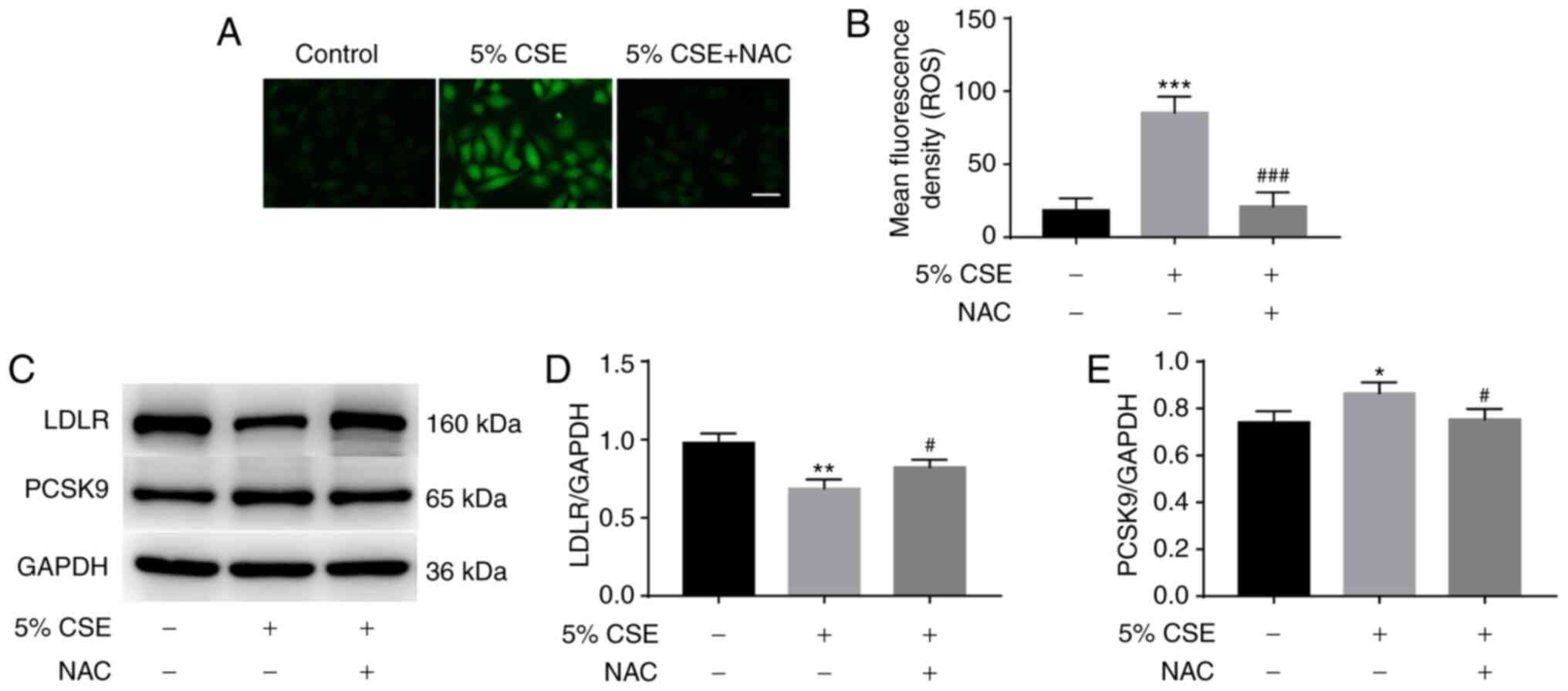
To investigate whether ROS contribute to the effect
of CSE on PCSK9 and LDLR expression, HepG2 cells were incubated
with or without 5 mM NAC for 1 h prior to stimulation with 5% CSE
for 24 h, and then total intracellular ROS generation was measured
using DCFH-DA. CSE treatment significantly increased ROS
production; however, pretreatment with NAC significantly inhibited
the CSE-induced production of ROS (Fig.
2A and B). In addition, the effects of NAC on LDLR and PCSK9
expression were measured using western blotting. The pretreatment
of HepG2 cells with NAC attenuated the inhibitory effect of CSE on
LDLR expression (Fig. 2C and D) and
the stimulatory effect of CSE on PCSK9 expression (Fig. 2C and E).
Effects of NF-κB activation on PCSK9
and LDLR expression in CSE-stimulated HepG2 cells
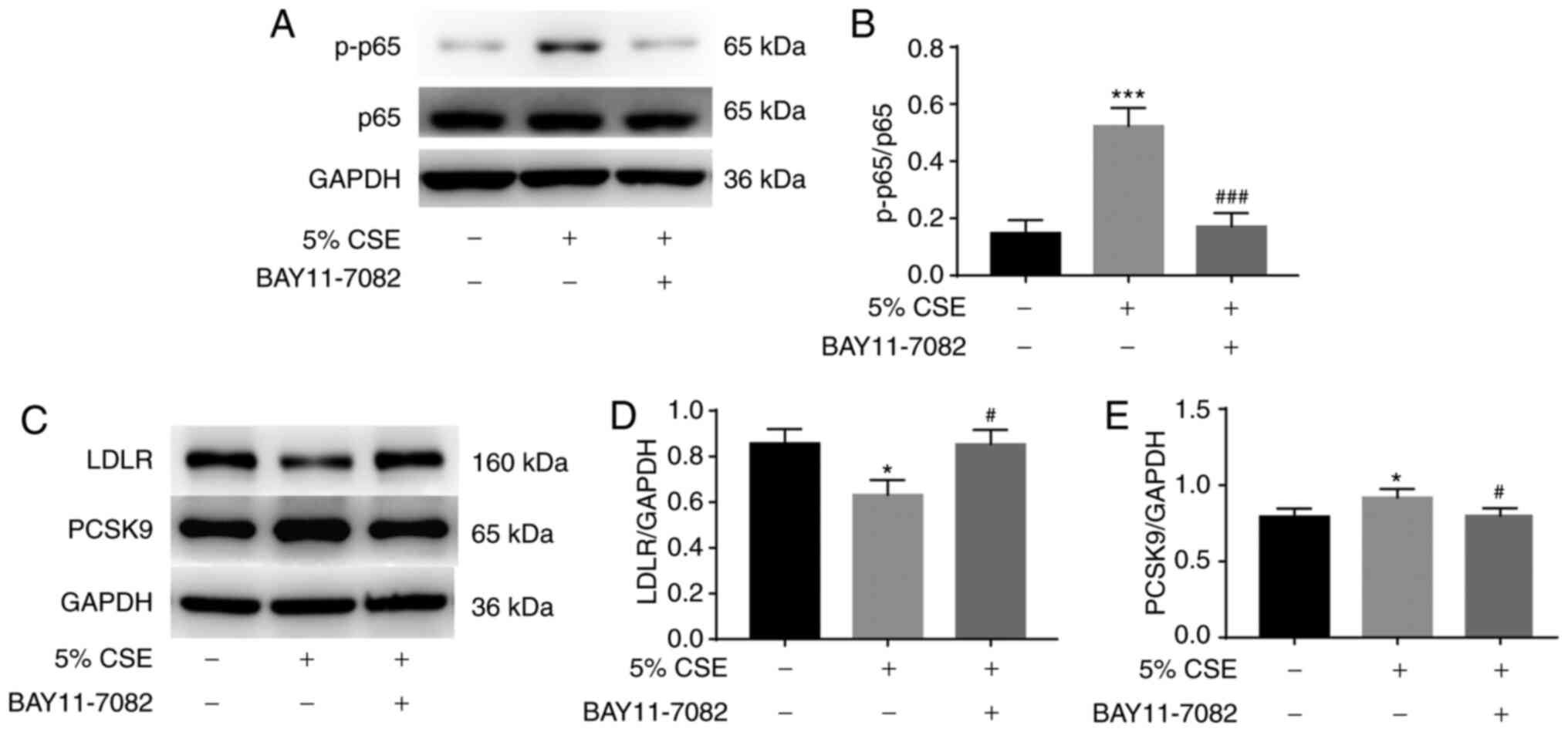
Next, whether NF-κB activation contributes to the
effect of CSE on PCSK9 and LDLR expression was investigated. HepG2
cells were incubated with or without 10 µM BAY11-7082 for 1 h, and
then stimulated with 5% CSE. Stimulation with 5% CSE for 30 min
induced the phosphorylation of p65 significantly; however, this
increase was attenuated by BAY11-7082 pretreatment (Fig. 3A and B). Furthermore, BAY11-7082
pretreatment also reversed the inhibition of LDLR expression and
increase in PCSK9 expression induced by treatment with 5% for 24 h
(Fig. 3C-E).
Effects of melatonin on CSE-regulated
PCSK9 and LDLR production
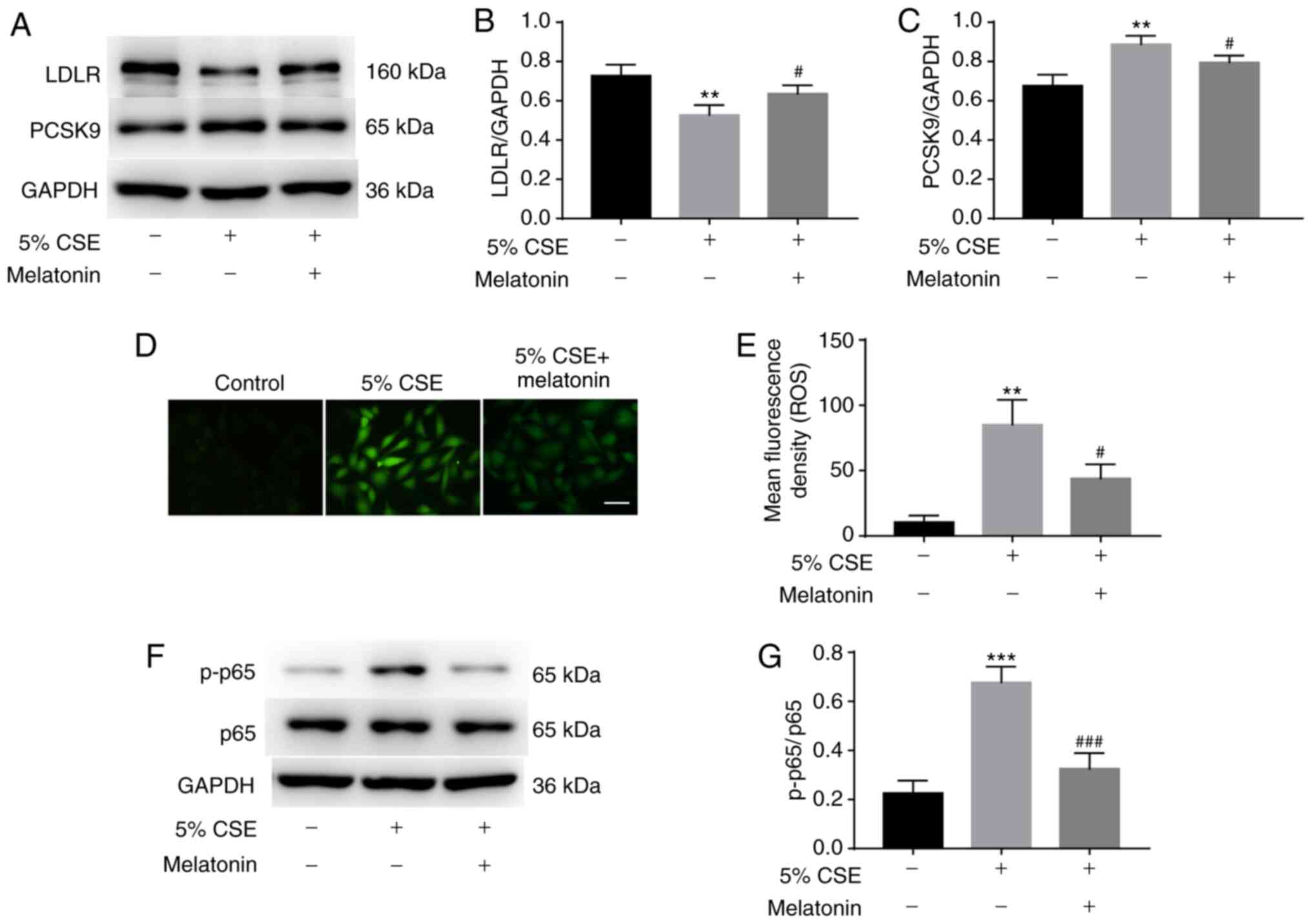
To assess whether melatonin inhibited CSE-regulated
PCSK9 and LDLR production, HepG2 cells were pretreated with or
without 100 µM melatonin for 1 h and then stimulated with 5% CSE
for 24 h. The inhibitory effect of CSE treatment on LDLR expression
was reversed by melatonin, and the CSE-induced increase in PCSK9
expression was also attenuated by melatonin (Fig. 4A-C).
Whether ROS and/or NF-κB are involved in the
mechanism by which melatonin regulates CSE-induced changes in PCSK9
and LDLR expression was then examined. The levels of ROS were
measured using DCFH-DA, and p-p65 levels were measured by western
blotting. Pretreatment with melatonin significantly decreased the
production of ROS (Fig. 4D and E)
and the phosphorylation of p65 (Fig. 4F
and G) in the CSE-stimulated HepG2 cells.
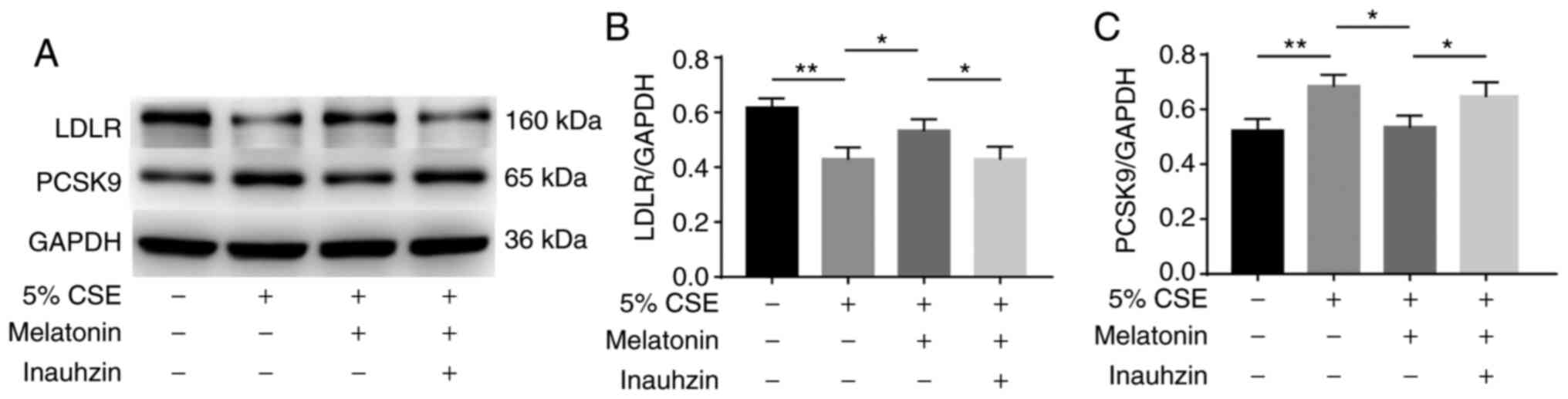
Effects of SIRT1 on the regulation of
PCSK9 and LDLR expression by melatonin in CSE-stimulated HepG2
cells
The role of SIRT1 in the regulatory effect of
melatonin on PCSK9 and LDLR production was also investigated in
CSE-stimulated cells. HepG2 cells were treated with 100 µM
melatonin alone or with 2 µM Inauhzin for 1 h, and then stimulated
with 5% CSE for 24 h. The CSE-induced reduction in LDLR expression
was attenuated by melatonin, and Inauhzin blocked the effect of
melatonin (Fig. 5A and B). In
addition, the CSE-induced increase in PCSK9 expression was
suppressed by melatonin, and Inauhzin blocked the inhibitory effect
of melatonin (Fig. 5A and C).
Discussion
The present study demonstrated that CSE induced
PCSK9 expression and decreased LDLR expression in HepG2 cells,
while melatonin attenuated those effects. Furthermore, CSE induced
ROS production and NF-κB activation, and pretreatment with a ROS
scavenger or NF-κB inhibitor suppressed the CSE-mediated regulation
of PCSK9 and LDLR expression in the cells. These data indicate that
CSE induced PCSK9 expression and inhibited LDLR expression via ROS
and NF-κB signaling pathways in the HepG2 cells.
Epidemiological studies have shown that dyslipidemia
is associated with CS exposure (23–26);
smokers have higher serum levels of cholesterol (24), higher plasma triglyceride
concentrations (25) and lower
concentrations of HDL-C (26) than
non-smokers, and smoking cessation for >6 years can reduce the
risk of dyslipidemia (23). Animal
experiments have also shown that CS exposure can cause dyslipidemia
(7,27). LDL-C is an indubitable causal factor
in atherosclerosis (3,28). Long-term exposure to CS has been
shown to increase serum LDL-C levels in experimental animals
(9), and our previous study
(10) indicated that the reduced
expression of the LDLR on hepatocytes caused by CS exposure may
underlie the increase in serum LDL-C levels. The present study
demonstrated that CSE stimulated the expression of PCSK9 mRNA and
protein, which may explain the mechanism by which CSE inhibits LDLR
expression.
PCSK9 serves a critical role in the regulation of
cholesterol homeostasis (13,29).
It binds the LDLR at the surface of hepatocytes, activating the
endosomal and lysosomal degradation of LDLR in the liver, resulting
in increased serum LDL-C levels (30,31).
In the present study, CSE induced PCSK9 expression in HepG2 cells
in a time- and concentration-dependent manner, consistent with the
decreased LDLR expression. However, in addition to decreasing LDLR
at the surface of hepatocytes, another important role of PCSK9 is
the regulation of inflammation (17). Pro-inflammatory factors such as LPS,
TNF-α and oxidized LDL (ox-LDL) have been shown to upregulate PCSK9
expression (17). Moreover,
compared with wild-type mice, PCSK9 knockout mice display a
decreased response to LPS stimulation, manifested by the reduced
production of inflammatory factors such as TNF-α, interleukin
(IL)-6, monocyte chemotactic protein 1 and IL-1β (32,33),
which suggests that PCSK9 may participate in the process of
inflammation. The present study demonstrated that CSE induced PCSK9
expression in HepG2 cells, and suggests that PCSK9 may play a role
in CSE-induced inflammation.
CSE simulates the various harmful substances
contained in real CS, including high concentrations of oxidants,
which can induce ROS production and are important in the
pathogenesis of atherosclerosis (1,2). In
our previous study, we reported that CSE-induced pyroptosis in
human umbilical vein endothelial cells (ECs) required ROS, and that
CS increased the production of ROS in rat carotid arteries
(34). Notably, Ding et al
(35) reported that hemodynamic
shear stress modulated PCSK9 expression in human primary aortic ECs
and smooth muscle cells (SMCs) via ROS production. Furthermore,
another study demonstrated PCSK9 expression was enhanced by the
induction of mitochondrial ROS and reduced by their inhibition
(33). In the present study, the
increased PCSK9 expression induced by CSE was inhibited by NAC, a
ROS scavenger, which indicates that CSE induced PCSK9 expression
via ROS.
The transcription factor NF-κB is known as the
master regulator of inflammation and immune homeostasis (36), and CS can induce its activation
(37). Inhibiting the activity of
NF-κB has been shown to alleviate the progression of
atherosclerosis (38,39). By contrast, the autophagy of SMCs
induced by nicotine, one of the main components in CS, accelerates
atherosclerosis via ROS/NF-κB signaling (40). Furthermore, LPS, ox-LDL and TNF-α
have been demonstrated to regulate PCSK9 expression via the NF-κB
signaling pathway (33). NF-κB
downstream signaling, including IL-1β, IL-6 and TNF-α, has also
been shown to significantly induce PCSK9 expression (41). The present study demonstrated that
the inhibition of NF-κB activation significantly suppressed
CSE-induced PCSK9 expression, suggesting that CSE induced PCSK9
expression via NF-κB signaling. Notably, there is complex crosstalk
between ROS and the NF-κB signaling pathway; NF-κB regulatory genes
are important in regulating the amount of ROS in cells, and ROS
have inhibitory or stimulatory effects on NF-κB signal transduction
(42). However, this relationship
was not investigated further as it was not the focus of the present
study.
A meta-analysis of 12 randomized controlled trials
found that melatonin ameliorates dyslipidemia and reduces LDL-C and
triglyceride levels (43). In
addition, studies have shown that treatment with melatonin
regulates dyslipidemia in rats (44–46)
and can reprogram the gut microbiota to improve lipid dysmetabolism
in mice fed a high-fat diet (47).
In the present study, melatonin attenuated the CSE-induced increase
in PCSK9 expression and reduction in LDLR expression, which
indicates that melatonin regulated CSE-induced dyslipidemia by
downregulating PCSK9 expression and upregulating LDLR expression.
In addition, the present study demonstrated that melatonin
significantly decreased ROS production and p65 phosphorylation in
CSE-stimulated HepG2 cells, suggesting that melatonin regulated
PCSK9 and LDLR expression by blocking the ROS and NF-κB signaling
pathways in HepG2 cells.
Multiple studies have shown that melatonin exerts
anti-inflammatory and antioxidant effects by upregulating SIRT1
activity and expression (19–21),
and that SIRT1 activation inhibits NF-κB signaling (48,49),
thereby protecting the cells from ROS (50). The results of the present study
indicate that the inhibition of SIRT1 reversed the regulatory
effect of melatonin on CSE-induced PCSK9 and LDLR expression. The
absence of in vivo data to confirm the results is a
potential limitation of the present study, and such experiments are
planned in the future.
In conclusion, the present study showed that CSE
increased PCSK9 production and inhibited LDLR expression in HepG2
cells. Additionally, it demonstrated that ROS and NF-κB signaling
pathways are mediators of CSE-regulated PCSK9 and LDLR expression,
which may contribute to the lipid metabolism disorders caused by
CS. Melatonin regulated PCSK9 and LDLR expression via SIRT1, which
blocked the ROS/NF-κB signaling in HepG2 cells. However, additional
experiments are required to clarify the results.
Acknowledgements
The authors would like to thank Professor Jing Li
and Professor Shihua Wang of the Medical Science Research Center,
Chinese Academy of Medical Sciences and Peking Inion Medical
college for technical assistance.
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 81970417).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
BTM and CWL conceived and designed the study. BTM,
XBW, RZ, and SN performed the experiments. BTM, XBW and QH
interpreted the results, analyzed the data and wrote the paper. LN,
XD and ZHR analyzed the data and designed the figures and table.
CWL and QH reviewed and edited the manuscript. BTM and CWL confirm
the authenticity of all the raw data. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests
References
|
1
|
Messner B and Bernhard D: Smoking and
cardiovascular disease: Mechanisms of endothelial dysfunction and
early atherogenesis. Arterioscler Thromb Vasc Biol. 34:509–515.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siasos G, Tsigkou V, Kokkou E, Oikonomou
E, Vavuranakis M, Vlachopoulos C, Verveniotis A, Limperi M,
Genimata V, Papavassiliou AG, et al: Smoking and atherosclerosis:
Mechanisms of disease and new therapeutic approaches. Curr Med
Chem. 21:3936–3948. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Libby P, Buring JE, Badimon L, Hansson GK,
Deanfield J, Bittencourt MS, Tokgözoğlu L and Lewis EF:
Atherosclerosis. Nat Rev Dis Primers. 5:562019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Craig WY, Palomaki GE and Haddow JE:
Cigarette smoking and serum lipid and lipoprotein concentrations:
An analysis of published data. BMJ. 298:784–788. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Maeda K, Noguchi Y and Fukui T: The
effects of cessation from cigarette smoking on the lipid and
lipoprotein profiles: A meta-analysis. Prev Med. 37:283–290. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Selya AS and Hesse ND: Time to first
cigarette and serum cholesterol levels. Soc Sci Med. 174:213–219.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Latha MS, Vijayammal PL and Kurup PA:
Effect of exposure of rats to cigarette smoke on the metabolism of
lipids. Atherosclerosis. 70:225–231. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lietz M, Berges A, Lebrun S, Meurrens K,
Steffen Y, Stolle K, Schueller J, Boue S, Vuillaume G,
Vanscheeuwijck P, et al: Cigarette-smoke-induced atherogenic lipid
profiles in plasma and vascular tissue of apolipoprotein
E-deficient mice are attenuated by smoking cessation.
Atherosclerosis. 229:86–93. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zong C, Song G, Yao S, Guo S, Yu Y, Yang
N, Guo Z and Qin S: Cigarette smoke exposure impairs reverse
cholesterol transport which can be minimized by treatment of
hydrogen-saturated saline. Lipids Health Dis. 14:1592015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ma B, Chen Y, Wang X, Zhang R, Niu S, Ni
L, Di X, Han Q and Liu C: Cigarette smoke exposure impairs lipid
metabolism by decreasing low-density lipoprotein receptor
expression in hepatocytes. Lipids Health Dis. 19:882020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lusis AJ: Atherosclerosis. Nature.
407:233–241. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Poirier S, Mayer G, Poupon V, McPherson
PS, Desjardins R, Ly K, Asselin MC, Day R, Duclos FJ, Witmer M, et
al: Dissection of the endogenous cellular pathways of PCSK9-induced
low density lipoprotein receptor degradation: Evidence for an
intracellular route. J Biol Chem. 284:28856–28864. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Urban D, Pöss J, Böhm M and Laufs U:
Targeting the proprotein convertase subtilisin/kexin type 9 for the
treatment of dyslipidemia and atherosclerosis. J Am Coll Cardiol.
62:1401–1408. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cohen JC, Boerwinkle E, Mosley TH Jr and
Hobbs HH: Sequence variations in PCSK9, low LDL, and protection
against coronary heart disease. N Engl J Med. 354:1264–1272. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kathiresan S; Myocardial Infarction
Genetics C; Myocardial Infarction Genetics Consortium, : A PCSK9
missense variant associated with a reduced risk of early-onset
myocardial infarction. N Engl J Med. 358:2299–2300. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sabatine MS: PCSK9 inhibitors: Clinical
evidence and implementation. Nat Rev Cardiol. 16:155–165. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tang ZH, Li TH, Peng J, Zheng J, Li TT,
Liu LS, Jiang ZS and Zheng XL: PCSK9: A novel inflammation
modulator in atherosclerosis? J Cell Physiol. 234:2345–2355. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cipolla-Neto J and Amaral FGD: Melatonin
as a hormone: New physiological and clinical insights. Endocr Rev.
39:990–1028. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mayo JC, Sainz RM, Gonzalez Menendez P,
Cepas V, Tan DX and Reiter RJ: Melatonin and sirtuins: A ‘not-so
unexpected’ relationship. J Pineal Res. 62:e123912017. View Article : Google Scholar
|
|
20
|
Arioz BI, Tastan B, Tarakcioglu E, Tufekci
KU, Olcum M, Ersoy N, Bagriyanik A, Genc K and Genc S: Melatonin
attenuates LPS-induced acute depressive-like behaviors and
microglial NLRP3 inflammasome activation through the SIRT1/Nrf2
pathway. Front Immunol. 10:15112019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hardeland R: Melatonin and inflammation -
Story of a double-edged blade. J Pineal Res. 65:e125252018.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shi J, Bai Y, Qiu S, Li Y, Kou C, Tao Y,
Zhen Q, Gu Y, Yu Y, Zhang K, et al: Classified status of smoking
and quitting has different associations with dyslipidemia in
residents in northeast China. Clin Chim Acta. 486:209–213. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Muscat JE, Harris RE, Haley NJ and Wynder
EL: Cigarette smoking and plasma cholesterol. Am Heart J.
121:141–147. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kuzuya M, Ando F, Iguchi A and Shimokata
H: Effect of smoking habit on age-related changes in serum lipids:
A cross-sectional and longitudinal analysis in a large Japanese
cohort. Atherosclerosis. 185:183–190. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Merianos AL, Jandarov RA, Khoury JC and
Mahabee-Gittens EM: Tobacco smoke exposure association with lipid
profiles and adiposity among U.S. adolescents. J Adolesc Health.
62:463–470. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Han SG, Howatt DA, Daugherty A and Gairola
CG: Atherogenic and pulmonary responses of ApoE- and LDL
receptor-deficient mice to sidestream cigarette smoke. Toxicology.
299:133–138. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ridker PM: LDL cholesterol: Controversies
and future therapeutic directions. Lancet. 384:607–617. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Seidah NG and Prat A: The proprotein
convertases are potential targets in the treatment of dyslipidemia.
J Mol Med (Berl). 85:685–696. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Seidah NG, Awan Z, Chrétien M and Mbikay
M: PCSK9: A key modulator of cardiovascular health. Circ Res.
114:1022–1036. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Park SW, Moon YA and Horton JD:
Post-transcriptional regulation of low density lipoprotein receptor
protein by proprotein convertase subtilisin/kexin type 9a in mouse
liver. J Biol Chem. 279:50630–50638. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Walley KR, Thain KR, Russell JA, Reilly
MP, Meyer NJ, Ferguson JF, Christie JD, Nakada TA, Fjell CD, Thair
SA, et al: PCSK9 is a critical regulator of the innate immune
response and septic shock outcome. Sci Transl Med. 6:258ra1432014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ding Z, Liu S, Wang X, Deng X, Fan Y,
Shahanawaz J, Shmookler Reis RJ, Varughese KI, Sawamura T and Mehta
JL: Cross-talk between LOX-1 and PCSK9 in vascular tissues.
Cardiovasc Res. 107:556–567. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang X, Bian Y, Zhang R, Liu X, Ni L, Ma
B, Zeng R, Zhao Z, Song X and Liu C: Melatonin alleviates cigarette
smoke-induced endothelial cell pyroptosis through inhibiting
ROS/NLRP3 axis. Biochem Biophys Res Commun. 519:402–408. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ding Z, Liu S, Wang X, Deng X, Fan Y, Sun
C, Wang Y and Mehta JL: Hemodynamic shear stress via ROS modulates
PCSK9 expression in human vascular endothelial and smooth muscle
cells and along the mouse aorta. Antioxid Redox Signal. 22:760–771.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mitchell JP and Carmody RJ: NF-κB and the
transcriptional control of inflammation. Int Rev Cell Mol Biol.
335:41–84. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ahn KS and Aggarwal BB: Transcription
factor NF-kappaB: A sensor for smoke and stress signals. Ann N Y
Acad Sci. 1056:218–233. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ben J, Jiang B, Wang D, Liu Q, Zhang Y, Qi
Y, Tong X, Chen L, Liu X, Zhang Y, et al: Major vault protein
suppresses obesity and atherosclerosis through inhibiting IKK-NF-κB
signaling mediated inflammation. Nat Commun. 10:18012019.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wu Y, Wang F, Fan L, Zhang W, Wang T, Du Y
and Bai X: Baicalin alleviates atherosclerosis by relieving
oxidative stress and inflammatory responses via inactivating the
NF-κB and p38 MAPK signaling pathways. Biomed Pharmacother.
97:1673–1679. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang Z, Liu B, Zhu J, Wang D and Wang Y:
Nicotine-mediated autophagy of vascular smooth muscle cell
accelerates atherosclerosis via nAChRs/ROS/NF-κB signaling pathway.
Atherosclerosis. 284:1–10. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu S, Deng X, Zhang P, Wang X, Fan Y,
Zhou S, Mu S, Mehta JL and Ding Z: Blood flow patterns regulate
PCSK9 secretion via MyD88-mediated pro-inflammatory cytokines.
Cardiovasc Res. 116:1721–1732. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Morgan MJ and Liu ZG: Crosstalk of
reactive oxygen species and NF-κB signaling. Cell Res. 21:103–115.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Loloei S, Sepidarkish M, Heydarian A,
Tahvilian N, Khazdouz M, Heshmati J and Pouraram H: The effect of
melatonin supplementation on lipid profile and anthropometric
indices: A systematic review and meta-analysis of clinical trials.
Diabetes Metab Syndr. 13:1901–1910. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Agil A, Navarro-Alarcón M, Ruiz R,
Abuhamadah S, El-Mir MY and Vázquez GF: Beneficial effects of
melatonin on obesity and lipid profile in young Zucker diabetic
fatty rats. J Pineal Res. 50:207–212. 2011.PubMed/NCBI
|
|
45
|
Hussain SA: Effect of melatonin on
cholesterol absorption in rats. J Pineal Res. 42:267–271. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hoyos M, Guerrero JM, Perez-Cano R, Olivan
J, Fabiani F, Garcia-Pergañeda A and Osuna C: Serum cholesterol and
lipid peroxidation are decreased by melatonin in diet-induced
hypercholesterolemic rats. J Pineal Res. 28:150–155. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yin J, Li Y, Han H, Chen S, Gao J, Liu G,
Wu X, Deng J, Yu Q, Huang X, et al: Melatonin reprogramming of gut
microbiota improves lipid dysmetabolism in high-fat diet-fed mice.
J Pineal Res. 65:e125242018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kauppinen A, Suuronen T, Ojala J,
Kaarniranta K and Salminen A: Antagonistic crosstalk between NF-κB
and SIRT1 in the regulation of inflammation and metabolic
disorders. Cell Signal. 25:1939–1948. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Xu F, Xu J, Xiong X and Deng Y:
Salidroside inhibits MAPK, NF-κB, and STAT3 pathways in
psoriasis-associated oxidative stress via SIRT1 activation. Redox
Rep. 24:70–74. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Singh CK, Chhabra G, Ndiaye MA,
Garcia-Peterson LM, Mack NJ and Ahmad N: The role of sirtuins in
antioxidant and redox signaling. Antioxid Redox Signal. 28:643–661.
2018. View Article : Google Scholar : PubMed/NCBI
|