Introduction
Prohibitin (PHB) is ubiquitously expressed in cells
and is primarily localized on the inner mitochondrial membrane. PHB
is an evolutionarily, highly conserved protein that serves as a
molecular chaperone in the assembly of the mitochondrial
respiratory chain complex subunit (1,2). PHB
often exists in the form of a complex, consisting of 12–16
heterogenous dimer pairs of PHB1 and PHB2, which are closely
related, that can be anchored to the mitochondrial intima to form a
ring-shaped barrier structure (3,4).
The location of PHB2 proteins determines its
function. For example, the presence of PHB2 on the inner
mitochondrial membrane aids with maintaining the normal morphology
of the mitochondria, as well as anti-oxidative stress and apoptosis
levels, thereby affecting mitochondrial function (5). A previous study reported that
hyperglycemia in patients with type 2 diabetes promoted ATP
production and decreased the mitochondrial membrane potential
(ΔΨm) in platelet mitochondria (6). In addition, another previous study
reported that platelet mitochondrial activity was transiently
enhanced, mitochondrial function was disordered, and the production
of ATP and reactive oxygen species was increased in patients with
diabetes (7).
Platelet activation is closely associated with
mitochondrial dysfunction (8). A
previous study demonstrated that cells primarily participated in
mitochondrial renewal via the selective autophagy pathway (9). The process of selectively removing
mitochondria by autophagy is known as mitophagy (10). Mitophagy maintains a balance in the
number and quality of mitochondria in the cell by scavenging a
portion of the damaged mitochondria. Furthermore, mitophagy not
only removes damaged mitochondria, but also degrades healthy
mitochondria. For example, the exposure of cells to a harsh
environment promotes the synthesis of excess mitochondria to
increase the operational burden of cells, which results in the
degradation of healthy mitochondria to maintain cell survival
(11). The process of autophagy has
been reported to occur in platelets of humans and dogs, wherein
inhibition of the autophagic signaling pathway or autophagic
degradation prevents platelet aggregation and adhesion functions
(12–14). Therefore, platelet autophagy was
suggested to serve an important role in regulating platelet
activation. A previous study demonstrated that mitophagy was
induced in the platelets of patients with diabetes, which was
considered to serve as an intrinsic mechanism that protected
platelets from severe oxidative stress, in addition to preventing
apoptosis and thrombosis (15).
Therefore, the present study aimed to investigate the role and
mechanism of action underlying PHB2 in platelet mitophagy and
activation.
Materials and methods
MEG-01 cell culture
MEG-01 cells (American Type Culture Collection) were
cultured in RPMI-1640 (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.)
at 37°C with 5% CO2. For differentiation into platelets,
MEG-01 cells were treated with 15 ng/ml
phorbol-12-myristate-13-acetate (PMA) for 72 h. To observe cell
morphology, air-dried cell smears were fixed using 0.2% Wright's
staining solution for 2–3 sec at room temperature, followed by
staining with Wright-Giemsa stain (Sigma-Aldrich; Merck KGaA) for
15–25 min at room temperature according to manufacturer's protocol.
Stained cells were visualized using a light microscope (Olympus
Corporation). PE-conjugated anti-CD61 (cat. no. 555754; BD
Biosciences) was used to assess platelet activation via flow
cytometry. Briefly, platelets were fixed with 4% paraformaldehyde
at room temperature for 15 min, rinsed with PBS and stained with
anti-CD61 in the dark for 15–30 min. Subsequently, 1×104
total platelets per sample were acquired and analyzed using a
FACSCalibur™ flow cytometer (BD Biosciences) and FlowJo software
(version 10; FlowJo LLC). CD61 mRNA expression levels were analyzed
via reverse transcription-quantitative PCR (RT-qPCR).
Cell Counting Kit-8 (CCK-8) assay
Following PMA-induced differentiation of MEG-01
cells, platelets were uniformly seeded (100 µl cell suspension;
2×103 cells/well) into 96-well plates and treated with 0
(negative control; NC), 10, 20, 50, 70 or 100 µM carbonyl cyanide
3-chlorophenylhydrazone (CCCP; cat. no. ab141229; Abcam), a
mitochondrial oxidative phosphorylation uncoupler (16,17).
CCCP was diluted using DMSO (Sigma-Aldrich; Merck KGaA). A DMSO
group was used in subsequent experiments to exclude the effects of
DMSO on cells. For each treatment group, 10 replicate wells were
used. Following incubation for 3 h at 37°C, 10 µl CCK-8 solution
(Dojindo Molecular Technologies, Inc.) was added to each well and
incubated for 1 h. In the blank control group, cell-free cell
culture medium was added to the well, followed by CCK-8 solution
according to the aforementioned protocol. Absorbance was measured
at a wavelength of 450 nm using a microplate reader to obtain
optical density values. Cell viability was calculated according to
the following formula: Cell viability (%) = [(experimental
well-blank well)/(NC well-blank well)] ×100.
Measurement of ΔΨm via flow
cytometry
Platelets (5×106) were obtained and
resuspended in 0.5 ml RPMI-1640 supplemented with 10% FBS.
Subsequently, 0.5 ml prepared JC-1 staining working solution (BD
Biosciences) was added to the cell suspension, gently mixed by
pipetting and incubated at 37°C for 20 min. Subsequently, the
suspension was centrifuged at 4°C at 600 × g for 5 min and the
supernatant was discarded. The precipitate was washed with JC-1
staining buffer (1X) as follows: Cells were resuspended in 1 ml
JC-1 staining buffer (1X) and centrifuged at 600 × g at 4°C for 5
min; the supernatant was discarded; and the washing procedure was
repeated three times. Subsequently, cells were resuspended in 500
µl JC-1 Binding Buffer (1X) and analyzed using a FACSCalibur™ flow
cytometer (BD Biosciences). Compensation for FL-1/FL-2 was set up
using FACSComp™ beads (BD Biosciences) and FlowJo software (version
10; FlowJo LLC) was used to set up the flow cytometer.
RT-qPCR
Total RNA was extracted from MEG-01 cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). Total RNA was reverse transcribed into cDNA using random
hexamer primers and a TaqMan reverse transcription kit (Takara Bio,
Inc.) according to the manufacturer's protocol. The temperature
protocol used for RT was 42°C for 2 min, 37°C for 15 min and 85°C
for 5 sec. Subsequently, qPCR was performed using a SYBR Premix Ex
Taq II kit (Takara Bio, Inc.) on a 7900HT Fast Real-Time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.). The following
thermocycling conditions were used for qPCR: Pre-denaturation at
95°C for 30 sec; followed by 40 amplification cycles (denaturation
at 95°C for 5 sec, annealing at 60°C for 34 sec and extension at
95°C for 15 sec). The sequences of the primers used for qPCR are
listed in Table I. mRNA expression
levels were quantified using the 2−ΔΔCq method (18) and normalized to the internal
reference gene GAPDH. the reference gene.
 | Table I.Sequences of primers used for
quantitative PCR. |
Table I.
Sequences of primers used for
quantitative PCR.
| Gene | Sequence
(5′→3′) |
|---|
| Prohibitin 2 | F:
ACACTCGTTCCTCGTAGTCC |
|
| R:
TTGGTTCCAGTACCCCATTA |
| CD61 | F:
TTAGGTTCAGCTTGGGCCTG |
|
| R:
TTGGAGACACGGTGAGCTTC |
| GAPDH | F:
GATTTTGGAGGGATCTCGCT |
|
| R:
CAGGGCTGCTTTTAACTCTGGT |
Western blotting
Total proteins were extracted from platelets using
RIPA lysis buffer (Cell Signaling Technology, Inc.) according to
the manufacturer's protocol. Protein concentrations were quantified
using a bicinchoninic acid protein assay kit (Thermo Fisher
Scientific, Inc.). Subsequently, proteins (30 µg) were separated
via 12.5% SDS-PAGE and electrotransferred onto PVDF membranes.
Following blocking with 3% (w/v) BSA (Sigma-Aldrich; Merck KGaA) in
TBST [150 mM NaCl, 10 mM Tris-HCl (pH 7.5) and 0.1% Tween-20] at
room temperature for 1 h, the membranes were incubated overnight at
4°C with the following primary antibodies: Anti-PHB2 (1:1,000; cat.
no. sc-133094; Santa Cruz Biotechnology, Inc.), anti-LC3B (1:2,000;
cat. no. ab48394; Abcam) and anti-β-tubulin (1:5,000; cat. no.
10068-1-AP; ProteinTech Group, Inc.). Following the primary
antibody incubation, the membranes were incubated with
HRP-conjugated goat anti-mouse IgG (1:2,000; cat. no. SA00001-1;
ProteinTech Group, Inc.) and goat anti-rabbit IgG (1:2,000; cat.
no. SA00001-2; ProteinTech Group, Inc.) secondary antibodies for 2
h at room temperature. Protein bands were visualized using Pierce™
ECL Western Blotting Substrate (cat. no. 32109; Thermo Fisher
Scientific, Inc.) on a ChemiDoc XRS imager (Bio-Rad Laboratories,
Inc.). Protein expression levels were semi-quantified using Image
Lab software (version 4.0.1; Bio-Rad Laboratories, Inc.) with
β-tubulin as the loading control.
Cell transfection
Short hairpin RNAs (shRNAs/shs) targeting PHB2 were
designed and synthesized by Shanghai GeneChem Co. Ltd. The
recombinant lentivirus (LV) of shRNA targeting PHB2 [PHB2-RNA
interference (RNAi)-LV] and control LV (GFP LV) were commercially
prepared by Shanghai GeneChem Co. Ltd. Briefly, a LV transfer
vector (GV248) was constructed, which contained a puromycin
resistance gene and an enhanced GFP gene. shRNA expression was
driven by a U6 promoter. To package viruses, a 2nd generation
system was used and 293T cells (The Cell Bank of Type Culture
Collection of The Chinese Academy of Sciences).were transiently
transfected with 20 µg GV248 transfer plasmid and three packaging
vectors: i) 20 µg pGC-LV; ii) 15 µg pHelper 1.0; and iii) 10 µg
pHelper 2.0 (all Shanghai GeneChem Co., Ltd.). Following
transfection for 72 h, lentiviral particles were collected,
filtered and concentrated by ultracentrifugation at 12,000 × g for
2 h at 4°C. At 30–50% confluence, MEG-01 cells were transduced with
lentiviral shRNA (100 plaque-forming unit/cell; MOI, 100). At 48 h
post-transduction, transduction efficiencies were evaluated via
RT-qPCR. At 72 h post-transduction, PHB2 protein expression levels
were analyzed via western blotting. The targeting sequences of the
shRNAs were as follows: LV-PHB2-RNAi (57874–1), 5′-AGAATATCTCCAAGACGAT-3′; LV-PHB2-RNAi
(57875–1),
5′-TGAGCTTTAGCCGAGAGTA3′; LV-PHB2-RNAi (57876–2), 5′-TAGCATGTACCAGCGCCTA-3′; and NC-RNAi
(CON077), 5′-TTCTCCGAACGTGTCACGT-3′.
mRNA sequencing (mRNA-seq)
mRNA-seq experiments were performed by Novogene Co.,
Ltd. An mRNA-seq library was prepared for sequencing using standard
Illumina protocols (19). Briefly,
total RNA was extracted from platelets with or without transfection
of shPHB2 treatment using TRIzol reagent (cat. no. 15596026,
Invitrogen; Thermo Fisher Scientific, Inc.). To remove any
contaminating genomic DNA, total RNA was treated with RNase-free
DNase I (cat. no. M0303L; New England BioLabs, Inc.). RIN value
(RNA integrity number) was used as the criterion to determine the
integrity of the sample RNA using the RNA Nano 6000 Assay Kit (cat.
no. 5067-1511; Agilent Technologies, Inc.) and the Bioanalyzer 2100
System (Agilent Technologies, Inc.). mRNA extraction was performed
using Dynabeads oligo(dT) (cat. no. 61002; Invitrogen; Thermo
Fisher Scientific, Inc.). Double-stranded cDNA was synthesized
using Superscript II reverse transcriptase (cat. no. 18-064-022;
Invitrogen; Thermo Fisher Scientific, Inc.) and random hexamer
primers. Subsequently, cDNAs were fragmented by nebulization and
the standard Illumina protocol was followed to generate the
mRNA-seq library using the NEBNext® Ultra™ RNA Library
Prep Kit for Illumina® (cat. no. E7530L; New England
Biolabs, Inc.). The sequencing type we used was PE150 (Hiseq X;
Illumina, Inc.). The inserted fragment length was 250–300 bp and
the sequencing direction was 5′→3′. After the library was
constructed, a Qubit 2.0 Fluorometer (Invitrogen; Thermo Fisher
Scientific, Inc.) was used for preliminary quantification, followed
by dilution and detection using 1.5 ng/µl sample and a CFX96 Touch
Real-Time PCR Detection System (Bio-Rad Laboratories, Inc.). For
data analysis, basecalls were performed using cassava. Reads were
aligned to the genome with the split read aligner TopHat (version
2.0.7, http://tophat.cbcb.umd.edu) and Bowtie2
(version 2.4.2; http://bowtie-bio.sourceforge.net), using default
parameters. HTSeq (version 0.6.0, http://htseq.readthedocs.io) was used to estimate the
abundance of the reads.
Gene Ontology (GO) functional term and
Kyoto Encyclopedia of Genes and Genomes (KEGG) signaling pathway
enrichment analyses
GO functional term enrichment analysis of
differentially expressed genes (DEGs; |log2(FoldChange)|>1 and
adjusted P-value<0.05) was conducted using the GOseq R package
(20). Briefly, GO analysis was
performed by collating the significantly enriched GO terms in the
identified DEGs. Subsequently, DEGs were filtered based on
biological functions and mapped to GO terms in the GO database
(www.geneontology.org). Gene numbers were
calculated for each term using the hypergeometric test to obtain
significantly enriched GO terms for DEGs, which were compared with
sh-NC. GO terms with a corrected P<0.05 were considered as
significantly enriched in DEGs. KEGG Orthology Based Annotation
System software (version no. 4.0.1; bioconductor.org/packages/release/bioc/html/clusterProfiler.html)
was used to perform KEGG pathway enrichment analysis and to
determine the statistical enrichment of the DEGs in KEGG signaling
pathways (www.genome.jp/kegg) (21,22).
The analysis was used to identify the significant enrichment of
genes involved in metabolic or signaling pathways.
Statistical analysis
Statistical analyses were performed using GraphPad
Prism software (version 7.0; GraphPad Software, Inc.). Data are
presented as the mean ± SD. Comparisons between two groups were
analyzed using an unpaired Student's t-test, whereas comparisons
among multiple groups were analyzed using one-way ANOVA followed by
Bonferroni's post hoc test. P<0.05 was considered to indicate a
statistically significant difference. All experiments were repeated
three times.
Results
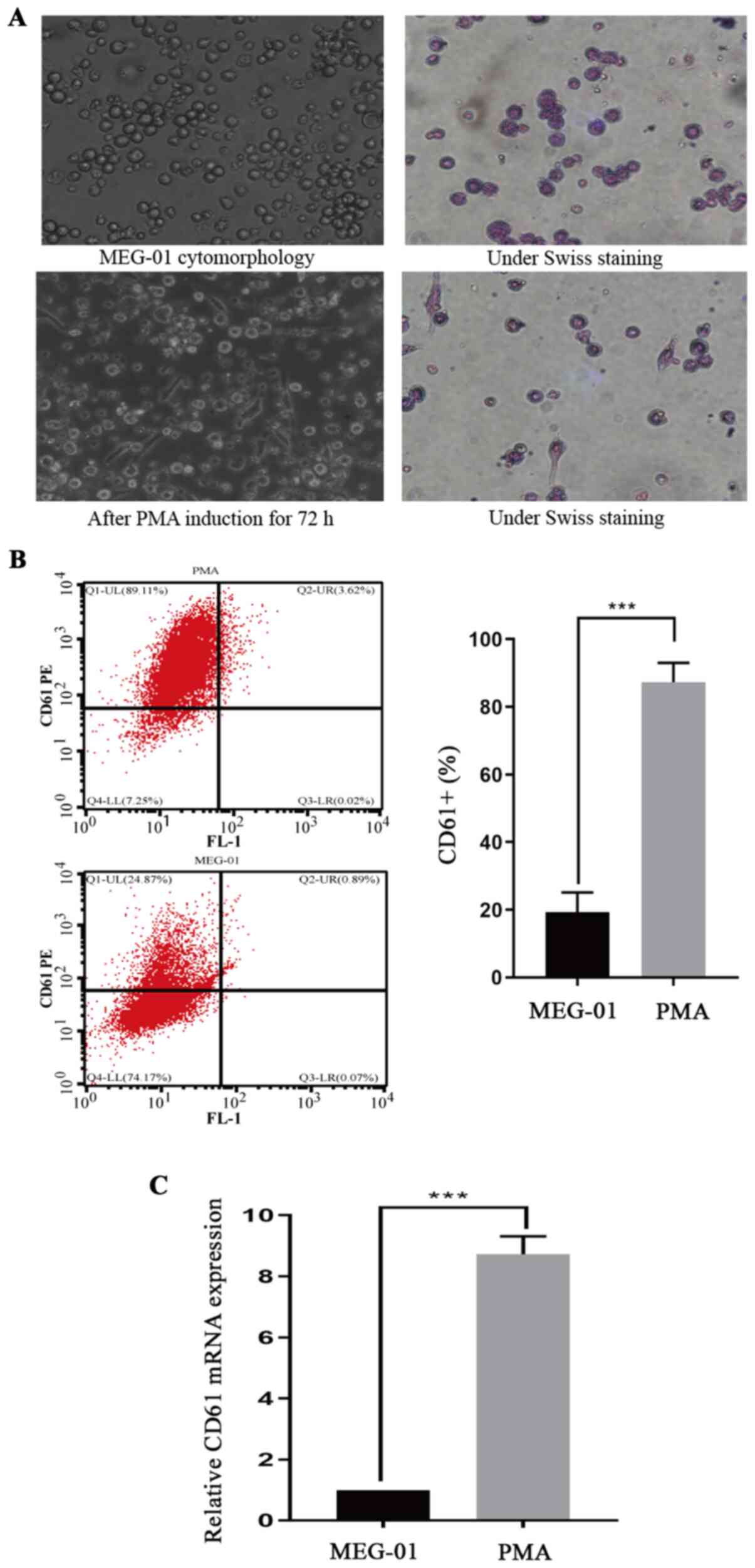
MEG-01 cells are successfully induced
to mature and differentiate into platelets using PMA in vitro
MEG-01 cells are megakaryocyte leukemia cells, which
were cultured in vitro and suspended in RPMI-1640
supplemented with 10% FBS in the present study. Previous studies
have reported that MEG-01 cells were successfully induced to
differentiate into platelet-forming megakaryocytes following
treatment with 15 ng/ml PMA for 72 h (23,24).
Morphological observations and Wright-Giemsa staining demonstrated
that cells were larger in size with irregular edges and coarse
granular chromatin structures, and were tightly arranged with
unclear nucleoli prior to induction (Fig. 1A). After induction, cavitation
occurred and pseudopods were generated. Following induction, the
flow cytometry results demonstrated that CD61 expression on MEG-01
cells was significantly increased compared with untreated MEG-01
cells (P<0.05; Fig. 1B).
Similarly, the RT-qPCR results also demonstrated that CD61 mRNA
expression levels were significantly upregulated following
induction compared with untreated MEG-01 cells (P<0.05; Fig. 1C).
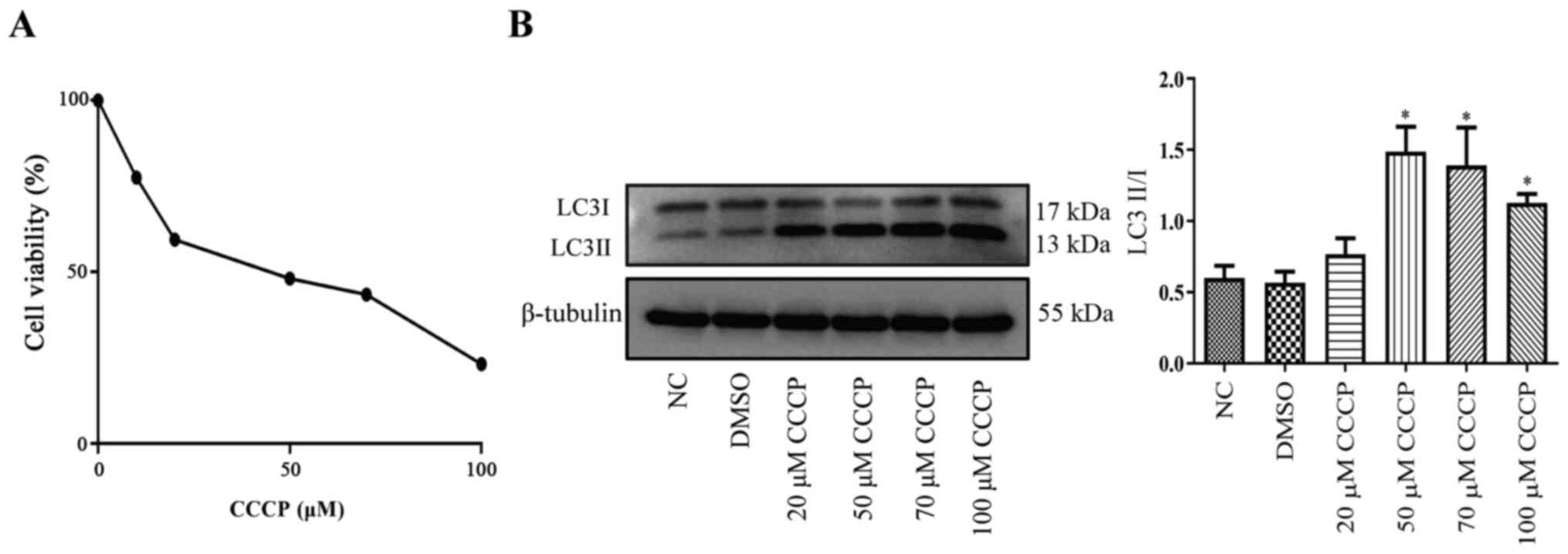
Effect of CCCP on platelet
activity
The effect of CCCP on platelet activity was analyzed
by performing CCK-8 assays. Compared with the control group (100%),
the survival rates of megakaryocytes/platelets following treatment
with 10, 20, 50, 70 or 100 µM CCCP for 3 h were 77.57, 59.42,
48.13, 43.43 or 23.25%, respectively (Fig. 2A). The results indicated that CCCP
treatment decreased cell survival rates in a
concentration-dependent manner. In addition, the expression levels
of the autophagy-related protein, LC3-II/LC3-I, were significantly
upregulated by CCCP treatment (≥50 µM) compared with the NC group
(Fig. 2B). Among the CCCP treatment
groups, the highest expression levels of LC3-II/I were observed
following treatment with 50 µM CCCP for 3 h. Therefore, treatment
with 50 µM CCCP for 3 h was selected as the optimal condition for
the induction of mitochondrial autophagy in platelets.
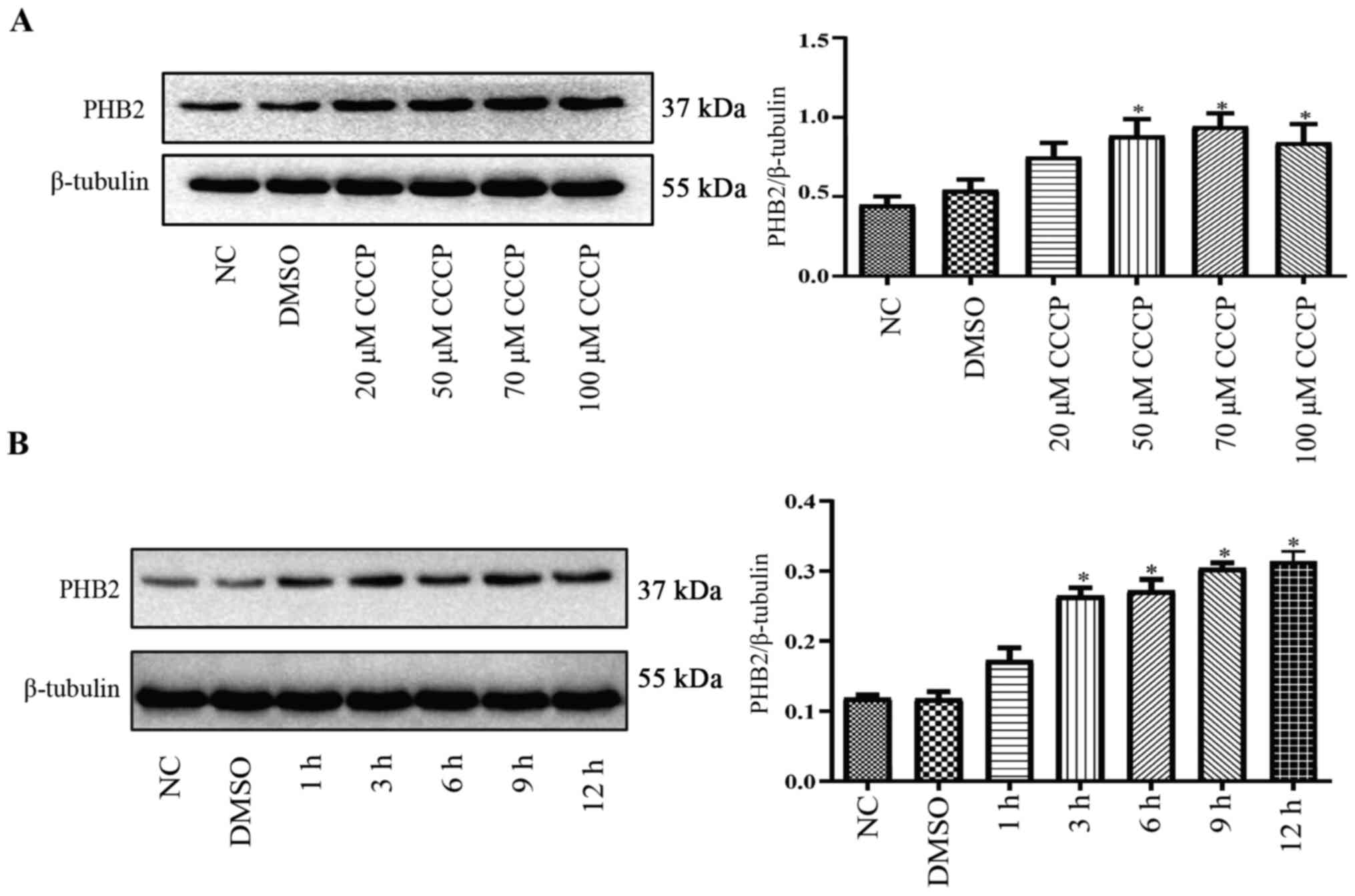
PHB2 expression levels in platelets
during mitophagy
Compared with the NC group, PHB2 protein expression
levels were significantly upregulated by CCCP (≥50 µM) treatment
for 3 h in a dose-dependent manner in platelets (Fig. 3A). Following treatment with 50 µM
CCCP for 0, 1, 3, 6, 9 or 12 h, PHB2 protein expression levels were
notably increased in a time-dependent manner in platelets compared
with the NC group (Fig. 3B).
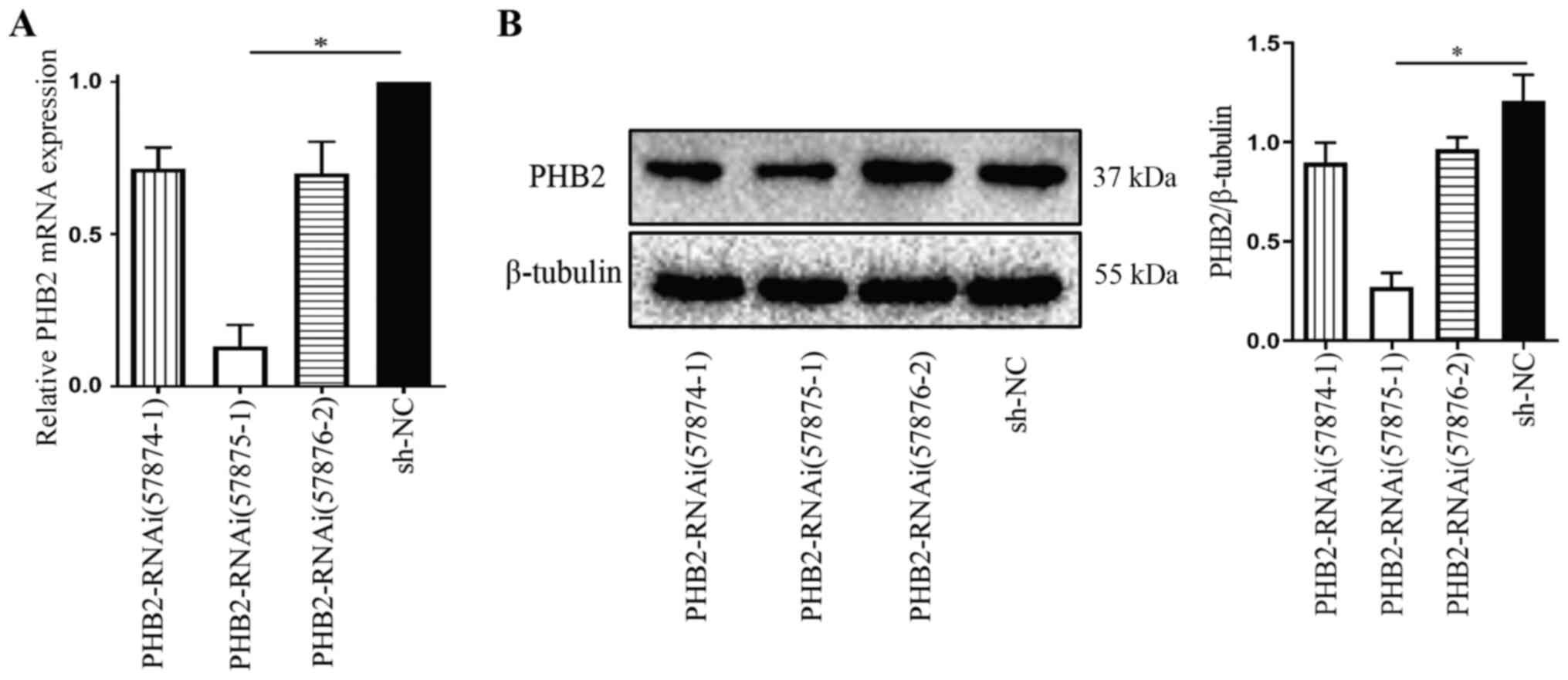
Effect of PHB2 knockdown on platelet
mitophagy
Under the optimal established LV transfection
conditions, the interfering fragments that effectively silenced
PHB2 were screened. Platelets were divided into the following four
groups: i) LV-PHB2-RNAi (57874–1);
ii) LV-PHB2-RNAi (57875–1); iii)
LV-PHB2-RNAi (57876–2); and iv)
NC-RNAi (CON077). At 72 h post-transduction, transduction
efficiency was assessed via RT-qPCR and western blotting.
Subsequently, mRNA and protein were extracted from platelets in
each group to measure PHB2 expression levels. PHB2 mRNA expression
levels in the LV-PHB2-RNAi (57875–1) group were significantly downregulated
compared with the NC-RNAi (CON077) group (P<0.05; Fig. 4A). Similarly, PHB2 protein
expression levels were also significantly downregulated in the
LV-PHB2-RNAi (57875–1) group
compared with the NC-RNAi (CON077) group (P<0.05: Fig. 4B). PHB2 mRNA and protein expression
levels among the other groups were not significantly different
(Fig. 4). Therefore, LV-PHB2-RNAi
(57875–1) was identified as the
most effective interfering fragment of PHB2.
Effect of PHB2 expression on platelet
mitophagy
Following the successful screening of the
interference fragment of PHB2, cells were divided into the
following groups: i) control (Con); ii) small interfering RNA
(sh)-NC; and iii) sh-PHB2. Following successful lentiviral
transduction, MEG-01 cells in each group were treated with PMA to
induce the maturation and differentiation of the cells, and then
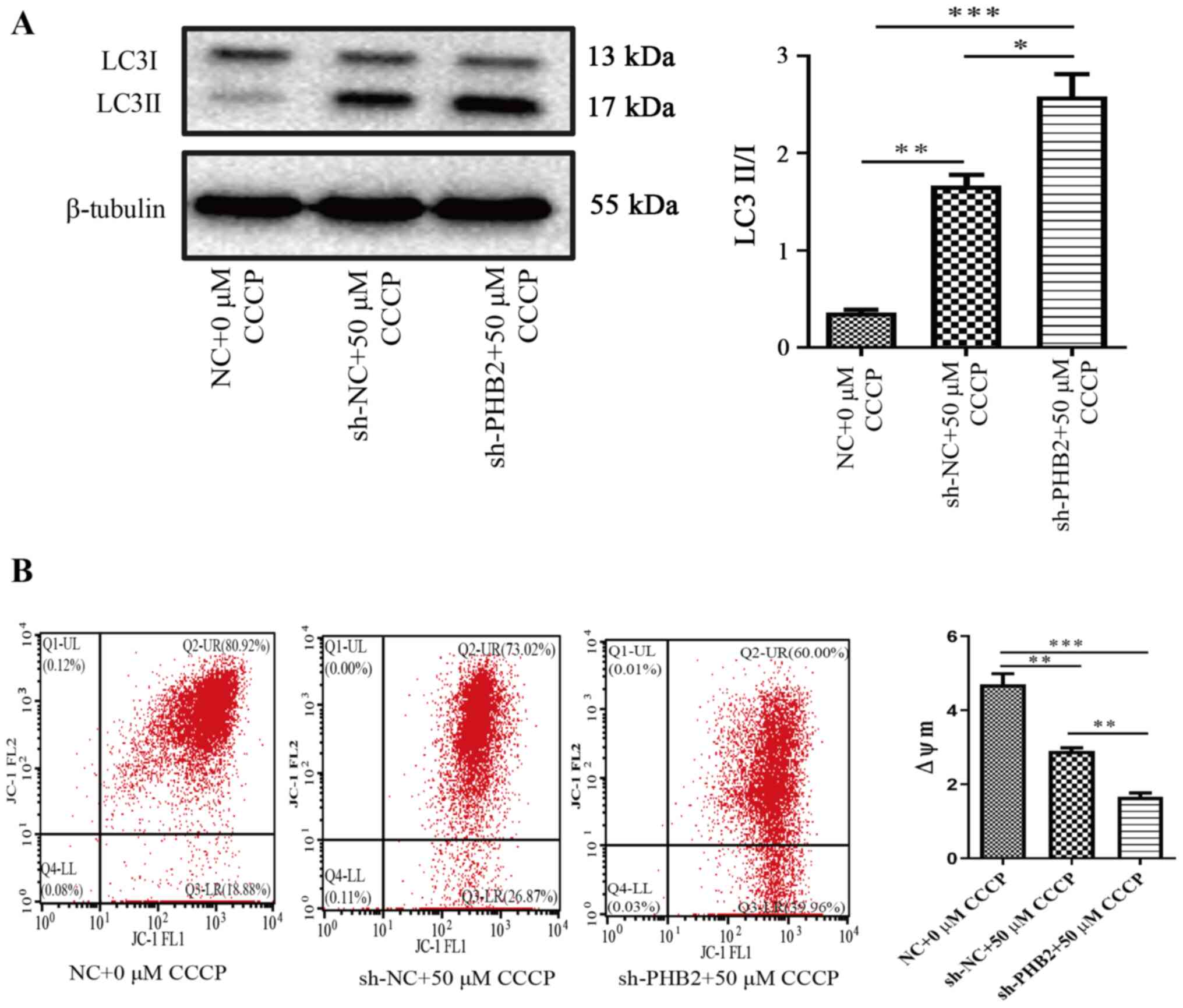
treated with 0 or 50 µM CCCP for 3 h. The western blotting results
indicated that the expression levels of the autophagy-associated
protein, LC3-II/LC3-I, were significantly upregulated in the sh-NC
+ 50 µM CCCP group compared with the NC + 0 µM CCCP group
(P<0.01; Fig. 5A). Furthermore,
the LC3-II/LC3-I ratio was also significantly upregulated in the
sh-PHB2 + 50 µM CCCP group compared with the NC + 0 µM CCCP group
(P<0.001). The LC3-II/LC3-I ratio was significantly higher in
the sh-PHB2 + 50 µM CCCP group compared with the sh-NC + 50 µM CCCP
group (P<0.05), which suggested that PHB2 knockdown may increase
the level of autophagy in platelets. In addition, the
ΔΨm was analyzed via flow cytometry. The results
revealed that the ΔΨm was significantly decreased
in the sh-NC + 50 µM CCCP group compared with the NC + 0 µM CCCP
group (P<0.01; Fig. 5B).
Similarly, the ΔΨm level in the sh-PHB2 + 50 µM
CCCP group was also significantly decreased compared with the NC +
0 µM CCCP group (P<0.001). Moreover, the ΔΨm
level was significantly reduced in the sh-PHB2 + 50 µM CCCP group
compared with the sh-NC + 50 µM CCCP group (P<0.01).
Transcriptome sequencing analysis of
platelet activation following PHB2 knockdown
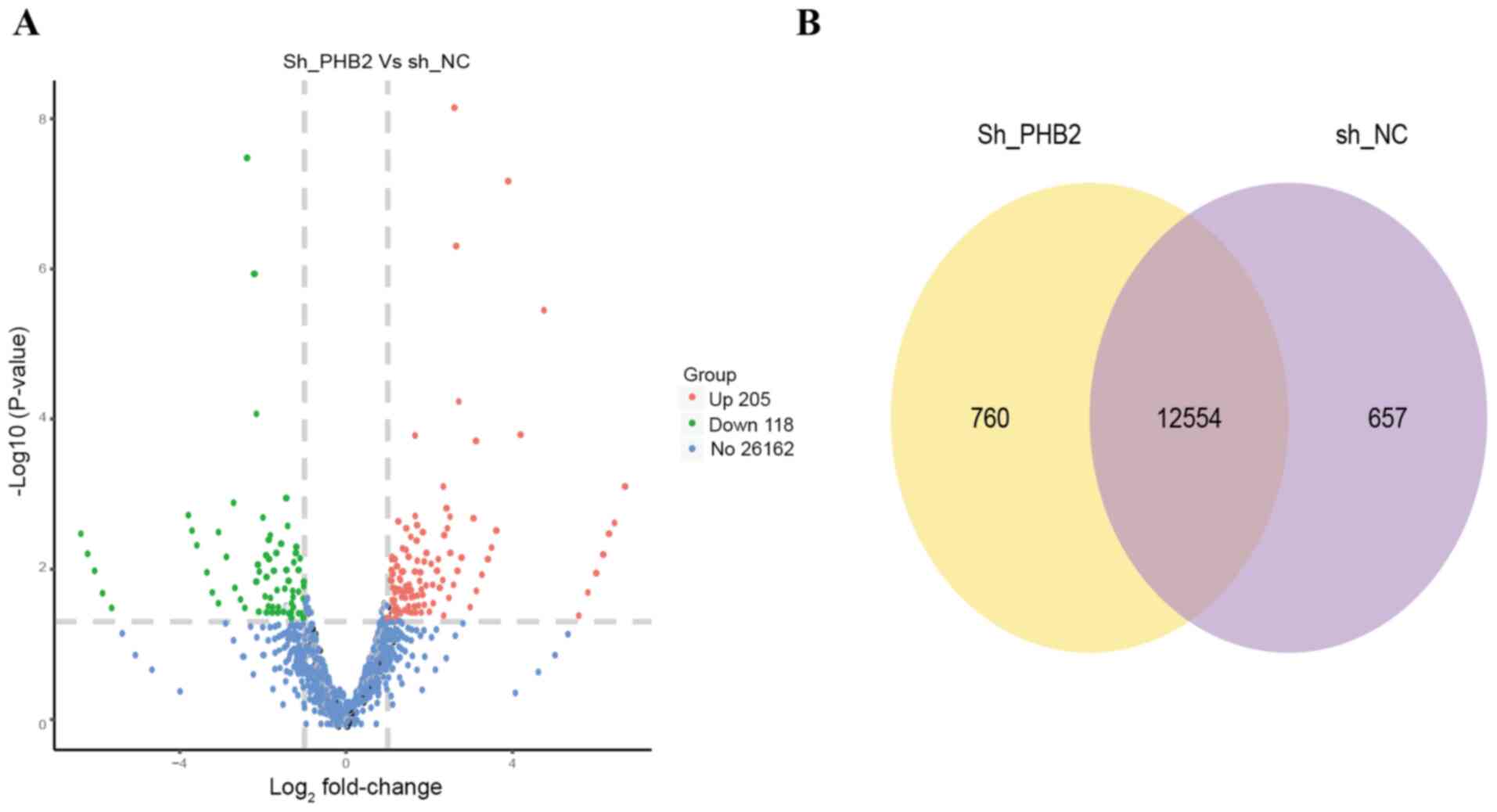
After mRNA-seq, 2.4 million clean reads were
obtained, of which 85.96% were aligned to the exon region, whereas
the remaining reads were distributed in the intronic and intergenic
regions. Compared with the sh-NC group, 323 differentially
expressed transcripts in the sh-PHB2 group were screened (Fig. 6A). A total of 12,554 DEGs were
screened from the NC and sh-PHB2 groups by differential gene
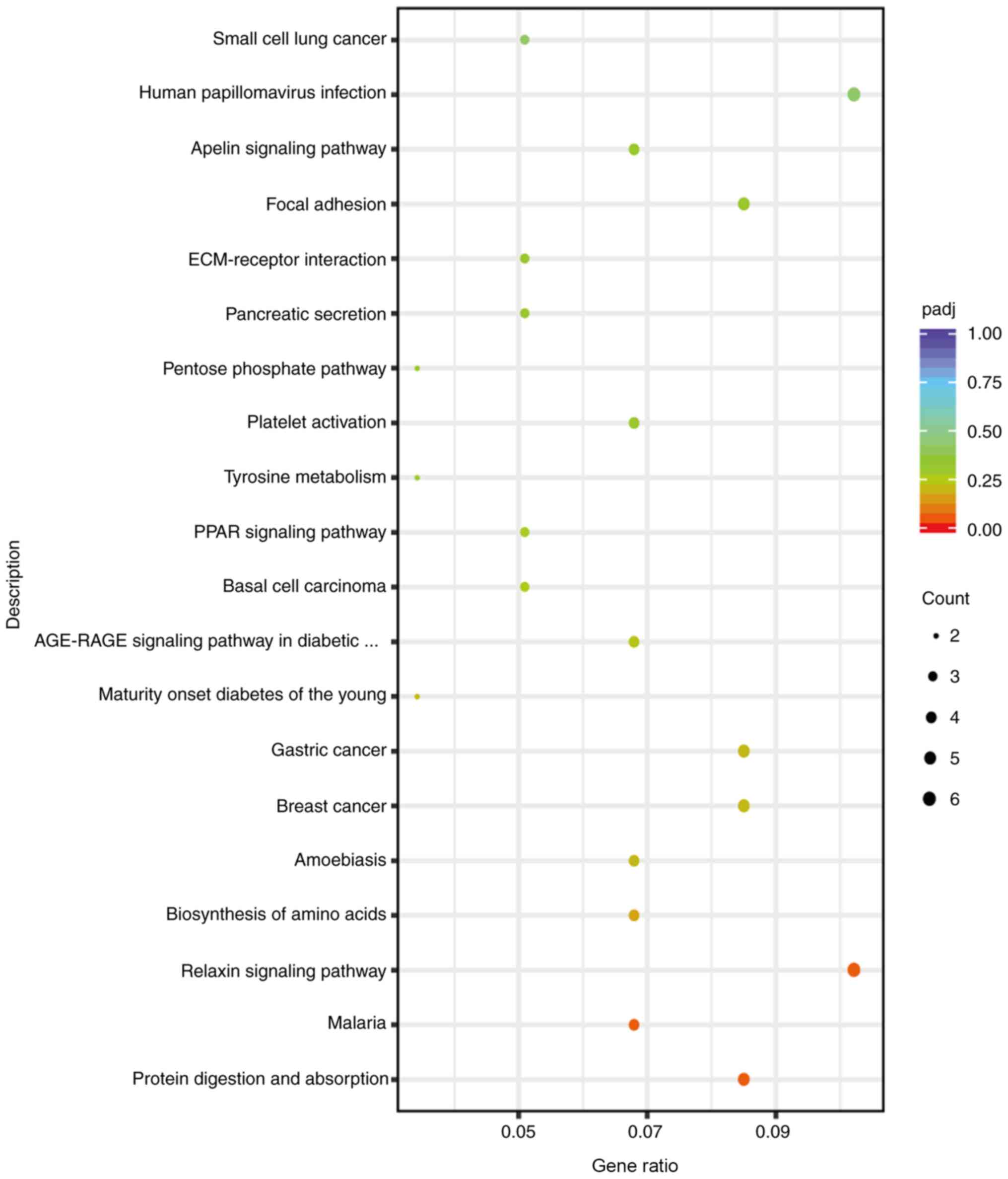
screening (Fig. 6B). KEGG signaling
pathway enrichment analysis of the DEGs revealed that 285 DEGs were
enriched in 183 KEGG pathways. Statistical analysis identified 20
significant KEGG signaling pathways, including ‘Focal adhesion’,
‘ECM-receptor interaction’ and ‘Apelin signaling pathway’ (adjusted
P<0.05; Fig. 7). Among these,
genes that were associated with the platelet activation pathway
included collagen type 1 α 2 chain (COL1A2), talin 2 (TLN2), nitric
oxide synthase 3 (NOS3) and collagen type III α 1 chain (COL3A1)
(Table II).
| Table II.Analysis of platelet-associated
activation gene signaling pathways. |
Table II.
Analysis of platelet-associated
activation gene signaling pathways.
| Gene | shPHB2 | NC |
log2-FoldChange | P-value |
|---|
| COL1A2 | 13.78 | 3.05 | 2.13 | <0.05 |
| TLN2 | 61.04 | 27.42 | 1.15 | <0.05 |
| NOS3 | 51.19 | 23.36 | 1.13 | <0.05 |
| COL3A1 | 34.46 | 14.22 | 1.27 | <0.05 |
Discussion
Previous studies have reported that the inner
mitochondrial membrane protein PHB2 is a key mitophagy receptor
(25,26). Mitophagy is a specific selection
process regulated by various factors, including ischemia, other
cardiac stress or heart disease conditions, that regulates the
number of mitochondria that adapt to the cell survival environment
(27). To the best of our
knowledge, the regulation of PHB2 in the process of platelet
mitophagy has not been previously reported; thus, for the first
time, transcriptomics sequencing in the present study discovered
PHB2-mediated regulation of downstream platelet activation related
genes. However, the precise mechanisms underlying mitophagy are not
completely understood.
PHB2 is a highly conserved inner mitochondrial
membrane protein that has been reported to regulate mitochondrial
assembly and function due to its unique localization on the
mitochondria membrane (5,28). Wei et al (25) previously reported that mitochondrial
receptors were involved in the mitochondrial degradation of PHB2,
and mitophagy was inhibited following PHB2 knockdown, which was a
similar effect to that observed following autophagy related 7
(ATG7) knockdown. In addition, mitochondrial respiratory
inhibitor-induced mitophagy significantly upregulated PHB2
expression levels, which were subsequently blocked following
transfection with si-ATG7, indicating that PHB2 may serve as a
marker of mitophagy (29). Xiao
et al (26) also reported
that PHB2 was required for cholestasis-induced mitophagy, whereby
PHB2 transferred LC3 to damaged mitochondria by interacting with
sequestosome 1 and LC3, which subsequently maintained homeostasis.
The aforementioned results suggested that PHB2 may also serve an
important role in the development of platelet mitophagy.
The present study aimed to investigate the role of
PHB2 in the induction of a differentiated platelet model. CCCP was
used as an inducer of mitochondrial autophagy, as it has been
reported to directly act on mitochondria, uncouple mitochondria and
damage the membrane potential (15). Previous literature indicated that
CCCP should be used at a concentration range of 10–100 µM;
therefore, the present study analyzed the most effective
concentration by performing CCK-8 assays and western blotting. In
the present study, 50 µM CCCP was used to induce platelet
mitophagy. As platelets derived from the differentiation of the
MEG-01 cell line were used in the present study, the platelets may
have been more tolerable and relatively insensitive to stimulation
compared with platelets directly isolated from humans or animals.
The aforementioned hypothesis may provide an explanation for the
higher concentration of CCCP used in the present study compared
with previous studies (16,17). Following treatment with 70 or 100 µM
CCCP, platelet viability was notably inhibited compared with the NC
group in the present study. Autophagy is a double-edged sword that
displays a protective effect at an appropriate levels, but the
level of autophagy can be increased. At high concentrations,
excessive autophagy activation occurs, which presents as decreased
levels of autophagy protein molecule LC3II/I (30). The results of the present study
demonstrated that PHB2 was involved in platelet mitophagy, and its
expression levels were altered according to the concentration and
duration of CCCP treatment. Moreover, the results indicated that
PHB2 protein expression levels were upregulated in a dose- and
time-dependent manner. Therefore, the present study suggested that
PHB2 may serve as a mitophagy receptor, maintaining the stability
of the mitochondrial structure in platelets.
Since PHB2 is widely expressed in biological cells,
distinguishing its role in mitochondria compared with other
organelles or cytoplasm is important. Lee et al (15) demonstrated that CCCP could induce
mitophagy in individual platelets. Therefore, CCCP, a proton
carrier (H+ ionophore), is a powerful uncoupling agent
for mitochondrial oxidative phosphorylation. CCCP enhances the
permeability of the mitochondrial inner membrane to H+,
resulting in the loss of the membrane potential on both sides of
the mitochondrial inner membrane, which ultimately leads to
mitophagy (15,31). Although CCCP-induced PHB2-mediated
mitophagy cannot directly reflect the precise positioning and
function of PHB2 on the mitochondria, it can be partially
speculated that it is caused by the changes of PHB2 following
CCCP-induced mitophagy.
PHB2 has been previously reported to serve an
important role in platelets. For example, PHB2 participated in
Prader Willi/Angelman region RNA 1-mediated platelet aggregation,
and regulated thrombocyte activation and related thrombin-induced
signaling pathways (32).
Therefore, it was hypothesized that PHB2 may serve an important
role in regulating mitochondrial function and platelet activation.
Previous studies have reported the contribution of several
mitochondrial functions in platelet activation, including increased
mitochondrial reactive oxygen species (mtROS) generation (33–35), a
decrease in the ΔΨm (36)
and the induction of mitochondrial dysfunction (31). In type II diabetes mellitus, ROS
production, an increased mitochondrial mass, higher ΔΨm and
elevated levels of mitochondrial respiration were reported be
associated with platelet activation (37,38).
Therefore, it was hypothesized that similar processes, including
increased mtROS generation, a higher ΔΨm and an elevated
mitochondrial mass, may also occur in platelets during mitophagy.
By using JC-1 as the main indicator for evaluating platelet
mitochondrial function, the results of the present study revealed
that the ∆Ψm was significantly decreased in the sh-NC + 50
µM CCCP and sh-PHB2 + 50 µM CCCP groups compared with the NC + 0 µM
CCCP group. ∆Ψm refers to the difference in
transmembrane potential produced by the different ion
concentrations on both sides of the inner mitochondrial membrane,
which is the driving force for ATP synthesis and release (36). Thus, the ΔΨm is a sensitive
indicator for evaluating mitochondrial function. The results of the
present study also demonstrated that PHB2 knockdown significantly
decreased the ∆Ψm in 50 µM CCCP-treated cells,
which also resulted in upregulation of the LC3-II/LC3-I ratio. A
potential mechanism underlying PHB2 knockdown-mediated effects is
that PHB2 served as a specific inner mitochondrial membrane
receptor. As a result of autophagy inhibition due to PHB2
knockdown, damaged or redundant mitochondria were not removed in
time and the stability of the structure and function of
mitochondria was not maintained in vivo, leading to a
decrease in the ∆Ψm and the accumulation of the
autophagy-related protein, LC3.
Zhang et al (39) reported for the first time that FUN14
domain containing 1 (FUNDC1)-mediated mitophagy in platelets was
crucial for ischemic adaptation in response to hypoxia, which
serves a crucial role in platelet activation. Furthermore, the
aforementioned study also identified BCL2 interacting protein 3
like as a receptor that mediated platelet mitophagy by regulating
the mitophagy flux, as a housekeeping process, to maintain
mitochondrial quality in platelets (40). The aforementioned results suggested
the existence of an association between receptor-mediated platelet
mitophagy and platelet activation. Therefore, further investigation
into PHB2-mediated platelet mitophagy may identify an important
mechanism for the prevention and treatment of cardiovascular
diseases. Increasing evidence has suggested that hypoxic
preconditioning in the clinic reduces ischemia-reperfusion
(I/R)-induced-heart injury (41,42).
Furthermore, hypoxic preconditioning was reported to induce
mitophagy in platelets and aid with determining the outcome of I/R
injury (43). Of particular
interest, FUNDC1-mediated mitophagy in cardiomyocytes and platelets
was identified as a major mechanism underlying cardioprotection
against I/R-induced heart injury. Under normal physiological
conditions, damaged toxic platelet mitochondria are removed to
maintain mitochondrial quality and platelet activation. Platelets
adhere to the site of injury to reduce blood oxygen levels, which
promotes myocardial infarction. Subsequently, the occlusion of the
blood vessels creates a hypoxic environment that triggers platelet
mitophagy to lower platelet activity, thereby preventing I/R damage
from worsening (44). It was
hypothesized that PHB2 may also be involved in I/R-induced heart
injury; however, the underlying mechanism requires further
investigation.
In the present study, the transcriptome sequencing
analysis revealed that PHB2 knockdown altered the expression of
platelet-associated activation genes, including COL1A2, TLN2, NOS3
and COL3A1. Type I and III collagen encoded by the COL1A2 and
COL3A1 genes, respectively, were previously reported to serve an
important role in activating platelets, forming thrombi and
maintaining the elasticity of the arterial wall (45–47).
Another previous study reported that NOS3 was an
endothelial-induced NOS that was involved in endothelial NOS
signaling transduction and regulation of nitric oxide (NO) in
vivo (48,49). The synthesis of NO not only altered
endothelial function (50), but
also affected platelet activity. For example, the release of NO
inhibited platelet aggregation and adhesion functions to a certain
degree (51). However, the role of
TLN2 in the context of platelet activation has not been reported
and requires further investigation. The present study aimed to
investigate platelet activation and mitophagy. However, a key
limitation of the present study was that other platelet functions
were not investigated.
In conclusion, the present study demonstrated that
PHB2, as an inner mitochondrial membrane receptor, may be involved
in platelet mitophagy and inhibit platelet activation by
downregulating the expression of platelet activation genes.
However, the related functions and mechanisms underlying PHB2
require further investigation. It was hypothesized that PHB2 may
influence the activation of platelets by altering the function of
mitochondria; therefore, PHB2 may serve as a novel therapeutic
target for thrombosis-related diseases due to its unique
localization on the mitochondrial membrane.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81960081 and
81660063) and the Natural Science Foundation of Jiangxi (grant no.
20171BAB205041).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request. The sequencing data were submitted to Sequence Read
Archive (accession no. PRJNA692313; www.ncbi.nlm.nih.gov/bioproject/PRJNA692313).
Authors' contributions
LLH and KZ conceived and designed the study,
analyzed the data and wrote the manuscript. LLH, KZ, YC, LJW, JC,
XYX, LW and XSC performed the experiments, and analyzed and
interpreted the data. LLH and KZ confirm the authenticity of all
the raw data. QZX and RQY designed the study and critically revised
the manuscript. RQY guaranteed the work, had full access to all the
data in the study and takes responsibility for the integrity of the
data and the accuracy of the data analysis. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Snedden WA and Fromm H: Characterization
of the plant homologue of prohibitin, a gene associated with
antiproliferative activity in mammalian cells. Plant Mol Biol.
33:753–756. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Loyer X, Potteaux S, Vion AC, Guerin CL,
Boulkroun S, Rautou PE, Ramkhelawon B, Esposito B, Dalloz M, Paul
JL, et al: Inhibition of microRNA-92a prevents endothelial
dysfunction and atherosclerosis in mice. Circ Res. 114:434–443.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Merkwirth C and Langer T: Prohibitin
function within mitochondria: Essential roles for cell
proliferation and cristae morphogenesis. Biochim Biophys Acta.
1793:27–32. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Osman C, Merkwirth C and Langer T:
Prohibitins and the functional compartmentalization of
mitochondrial membranes. J Cell Sci. 122:3823–3830. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Artal-Sanz M and Tavernarakis N: Opposing
function of mitochondrial prohibitin in aging. Aging (Albany NY).
2:1004–1011. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Guo X, Wu J, Du J, Ran J and Xu J:
Platelets of type 2 diabetic patients are characterized by high ATP
content and low mitochondrial membrane potential. Platelets.
20:588–593. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Avila C, Huang RJ, Stevens MV, Aponte AM,
Tripodi D, Kim KY and Sack MN: Platelet mitochondrial dysfunction
is evident in type 2 diabetes in association with modifications of
mitochondrial anti-oxidant stress proteins. Exp Clin Endocrinol
Diabetes. 120:248–251. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fuentes E, Araya-Maturana R and Urra FA:
Regulation of mitochondrial function as a promising target in
platelet activation-related diseases. Free Radic Biol Med.
136:172–182. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xue L, Fletcher GC and Tolkovsky AM:
Mitochondria are selectively eliminated from eukaryotic cells after
blockade of caspases during apoptosis. Curr Biol. 11:361–365. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lemasters JJ: Selective mitochondrial
autophagy, or mitophagy, as a targeted defense against oxidative
stress, mitochondrial dysfunction, and aging. Rejuvenation Res.
8:3–5. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bhatia-Kissova I and Camougrand N:
Mitophagy: A process that adapts to the cell physiology. Int J
Biochem Cell Biol. 45:30–33. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pieczarka EM, Yamaguchi M, Wellman ML and
Judith Radin M: Platelet vacuoles in a dog with severe
nonregenerative anemia: Evidence of platelet autophagy. Vet Clin
Pathol. 43:326–329. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Feng W, Chang C, Luo D, Su H, Yu S, Hua W,
Chen Z, Hu H and Liu W: Dissection of autophagy in human platelets.
Autophagy. 10:642–651. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cao Y, Cai J, Zhang S, Yuan N, Li X, Fang
Y, Song L, Shang M, Liu S, Zhao W, et al: Loss of autophagy leads
to failure in megakaryopoiesis, megakaryocyte differentiation, and
thrombopoiesis in mice. Exp Hematol. 43:488–494. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee SH, Du J, Stitham J, Atteya G, Lee S,
Xiang Y, Wang D, Jin Y, Leslie KL, Spollett G, et al: Inducing
mitophagy in diabetic platelets protects against severe oxidative
stress. EMBO Mol Med. 8:779–795. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Griffith JM, Basting PJ, Bischof KM, Wrona
EP, Kunka KS, Tancredi AC, Moore JP, Hyman MRL and Slonczewski JL:
Experimental evolution of Escherichia coli K-12 in the
presence of proton motive force (PMF) uncoupler carbonyl cyanide
m-chlorophenylhydrazone selects for mutations affecting PMF-driven
drug efflux pumps. Appl Environ Microbiol. 85:e02792–e02718.
2019.PubMed/NCBI
|
|
17
|
Rodriguez C, Simon V, Conget P and Vega
IA: Both quiescent and proliferating cells circulate in the blood
of the invasive apple snail Pomacea canaliculata. Fish
Shellfish Immunol. 107:95–103. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Trapnell C, Williams BA, Pertea G,
Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ and Pachter
L: Transcript assembly and quantification by RNA-Seq reveals
unannotated transcripts and isoform switching during cell
differentiation. Nat Biotechnol. 28:511–515. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Young MD, Wakefield MJ, Smyth GK and
Oshlack A: Gene ontology analysis for RNA-seq: Accounting for
selection bias. Genome Biol. 11:R142010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kanehisa M, Araki M, Goto S, Hattori M,
Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T
and Yamanishi Y: KEGG for linking genomes to life and the
environment. Nucleic Acids Res. 36:D480–D484. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mao X, Cai T, Olyarchuk JG and Wei L:
Automated genome annotation and pathway identification using the
KEGG orthology (KO) as a controlled vocabulary. Bioinformatics.
21:3787–3793. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ogura M, Morishima Y, Okumura M, Hotta T,
Takamoto S, Ohno R, Hirabayashi N, Nagura H and Saito H: Functional
and morphological differentiation induction of a human
megakaryoblastic leukemia cell line (MEG-01s) by phorbol diesters.
Blood. 72:49–60. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Takeuchi K, Satoh M, Kuno H, Yoshida T,
Kondo H and Takeuchi M: Platelet-like particle formation in the
human megakaryoblastic leukaemia cell lines, MEG-01 and MEG-01s. Br
J Haematol. 100:436–444. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wei Y, Chiang WC, Sumpter R Jr, Mishra P
and Levine B: Prohibitin 2 is an inner mitochondrial membrane
mitophagy receptor. Cell. 168:224–238.e10. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xiao Y, Zhou Y, Lu Y, Zhou K and Cai W:
PHB2 interacts with LC3 and SQSTM1 is required for bile
acids-induced mitophagy in cholestatic liver. Cell Death Dis.
9:1602018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Galluzzi L, Bravo-San Pedro JM and Kroemer
G: Mitophagy: Permitted by prohibitin. Curr Biol. 27:R73–R76. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bavelloni A, Piazzi M, Raffini M, Faenza I
and Blalock WL: Prohibitin 2: At a communications crossroads. IUBMB
Life. 67:239–254. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lahiri V and Klionsky DJ: PHB2/prohibitin
2: An inner membrane mitophagy receptor. Cell Res. 27:311–312.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schiattarella GG and Hill JA: Therapeutic
targeting of autophagy in cardiovascular disease. J Mol Cell
Cardiol. 95:86–93. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang Y, Nartiss Y, Steipe B, McQuibban GA
and Kim PK: ROS-induced mitochondrial depolarization initiates
PARK2/PARKIN-dependent mitochondrial degradation by autophagy.
Autophagy. 8:1462–1476. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang Y, Wang Y, Xiang Y, Lee W and Zhang
Y: Prohibitins are involved in protease-activated receptor
1-mediated platelet aggregation. J Thromb Haemost. 10:411–418.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lopez JJ, Salido GM, Gómez-Arteta E,
Rosado JA and Pariente JA: Thrombin induces apoptotic events
through the generation of reactive oxygen species in human
platelets. J Thromb Haemost. 5:1283–1291. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yamagishi SI, Edelstein D, Du XL and
Brownlee M: Hyperglycemia potentiates collagen-induced platelet
activation through mitochondrial superoxide overproduction.
Diabetes. 50:1491–1494. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Choo HJ, Saafir TB, Mkumba L, Wagner MB
and Jobe SM: Mitochondrial calcium and reactive oxygen species
regulate agonist-initiated platelet phosphatidylserine exposure.
Arterioscler Thromb Vasc Biol. 32:2946–2955. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jobe SM, Wilson KM, Leo L, Raimondi A,
Molkentin JD, Lentz SR and Di Paola J: Critical role for the
mitochondrial permeability transition pore and cyclophilin D in
platelet activation and thrombosis. Blood. 111:1257–1265. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Siewiera K, Kassassir H, Talar M, Wieteska
L and Watala C: Higher mitochondrial potential and elevated
mitochondrial respiration are associated with excessive activation
of blood platelets in diabetic rats. Life Sci. 148:293–304. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wu F, Liu Y, Luo L, Lu Y, Yew D, Xu J and
Guo K: Platelet mitochondrial dysfunction of DM rats and DM
patients. Int J Clin Exp Med. 8:6937–6946. 2015.PubMed/NCBI
|
|
39
|
Zhang W, Ren H, Xu C, Zhu C, Wu H, Liu D,
Wang J, Liu L, Li W, Ma Q, et al: Hypoxic mitophagy regulates
mitochondrial quality and platelet activation and determines
severity of I/R heart injury. Elife. 5:e214072016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang W, Ma Q, Siraj S, Ney PA, Liu J,
Liao X, Yuan Y, Li W, Liu L and Chen Q: Nix-mediated mitophagy
regulates platelet activation and life span. Blood Adv.
3:2342–2354. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Eitel I, Stiermaier T, Rommel KP, Fuernau
G, Sandri M, Mangner N, Linke A, Erbs S, Lurz P, Boudriot E, et al:
Cardioprotection by combined intrahospital remote ischaemic
perconditioning and postconditioning in ST-elevation myocardial
infarction: The randomized LIPSIA CONDITIONING trial. Eur Heart J.
36:3049–3057. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hausenloy DJ, Barrabes JA, Bøtker HE,
Davidson SM, Di Lisa F, Downey J, Engstrom T, Ferdinandy P,
Carbrera-Fuentes HA, Heusch G, et al: Ischaemic conditioning and
targeting reperfusion injury: A 30 year voyage of discovery. Basic
Res Cardiol. 111:702016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang W, Siraj S, Zhang R and Chen Q:
Mitophagy receptor FUNDC1 regulates mitochondrial homeostasis and
protects the heart from I/R injury. Autophagy. 13:1080–1081. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang W, Chen C, Wang J, Liu L, He Y and
Chen Q: Mitophagy in cardiomyocytes and in platelets: A major
mechanism of cardioprotection against ischemia/reperfusion injury.
Physiology (Bethesda). 33:86–98. 2018.PubMed/NCBI
|
|
45
|
Penz S, Reininger AJ, Brandl R, Goyal P,
Rabie T, Bernlochner I, Bernlochner I, Rother E, Goetz C, Engelmann
B, et al: Human atheromatous plaques stimulate thrombus formation
by activating platelet glycoprotein VI. FASEB J. 19:898–909. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lv W, Lin Y, Song W, Sun K, Yu H, Zhang Y,
Zhang C, Li L, Suo M, Hui R and Chen J: Variants of COL3A1 are
associated with the risk of stroke recurrence and prognosis in the
Chinese population: A prospective study. J Mol Neurosci.
53:196–203. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Buxhofer-Ausch V, Steurer M, Sormann S,
Schloegl E, Schimetta W, Gisslinger B, Ruckser R, Gastl G and
Gisslinger H: Influence of platelet and white blood cell counts on
major thrombosis-analysis from a patient registry in essential
thrombocythemia. Eur J Haematol. 97:511–516. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fleming I: Molecular mechanisms underlying
the activation of eNOS. Pflugers Arch. 459:793–806. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Xia N, Forstermann U and Li H: Resveratrol
and endothelial nitric oxide. Molecules. 19:16102–16121. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Khan M, Meduru S, Gogna R, Madan E, Citro
L, Kuppusamy ML, Sayyid M, Mostafa M, Hamlin RL and Kuppusamy P:
Oxygen cycling in conjunction with stem cell transplantation
induces NOS3 expression leading to attenuation of fibrosis and
improved cardiac function. Cardiovasc Res. 93:89–99. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bharath LP, Mueller R, Li Y, Ruan T, Kunz
D, Goodrich R, Mills T, Deeter L, Sargsyan A, Babu PVA, et al:
Impairment of autophagy in endothelial cells prevents
shear-stress-induced increases in nitric oxide bioavailability. Can
J Physiol Pharmacol. 92:605–612. 2014. View Article : Google Scholar : PubMed/NCBI
|