Introduction
Short rib-polydactyly syndrome (SRPS) consists of a
group of rare, genetically heterogeneous, autosomal recessive
osteochondrodysplasias, which are characterized by short ribs and
limbs, hypoplastic thorax, polydactyly and multisystem organ
abnormalities (1). An accurate
diagnosis of SRPS is difficult to obtain using prenatal ultrasound
technology alone, due to overlapping phenotypic features that are
characteristic of a large numbers of skeletal dysplasias (2,3).
Indeed, four different types of SRPS have been identified,
including SRPS1/3 (MIM 613091), SRPS2 (MIM 263520), SRPS4 (MIM
269860) and SRPS5 (MIM 614091) (4).
SRPS3 is a perinatal lethal skeletal disorder, with or without
polydactyly, which is characterized by a constricted thoracic cage,
short ribs, shortened tubular bones and a ‘trident’-like appearance
of the acetabular roof.
The present study aimed to further investigate the
pathogenicity of two pairs of compound heterozygotes identified in
two retrospective samples and search for new molecular etiology in
a third SRPS3 fetus. The results revealed novel radiological and
histopathological changes observed in the two retrospective samples
(5,6). The present study also reported the
case of a separate Chinese family, wherein a fetus was diagnosed
with skeletal dysplasia. Targeted next-generation sequencing (NGS)
panels were used to evaluate 463 known skeletal
dysplasia-associated genes in both the parents and fetus.
Hematoxylin and eosin (H&E) staining was also conducted.
Typical and atypical histopathological features, typical
radiological changes and novel dynein cytoplasmic 2 heavy chain 1
gene (DYNC2H1) compound heterozygous variants were
identified. The findings of the present study may facilitate the
clinical and molecular diagnosis of SRPS3.
Materials and methods
Case presentation
Between January 2015 and December 2018, recruitment
of patients and the collection of samples, from two Chinese
families, were described in previous reports (5,6). The
present study also included another two-generation
nonconsanguineous Han Chinese family, focusing on a patient with
SRPS3. The mother, a 30-year-old woman with no history of skeletal
dysplasia, was referred to the Liaoning Centre for Prenatal
Diagnosis (Shenyang, China) at 21 weeks of gestation because
prenatal sonography had identified a fetus with short limbs and a
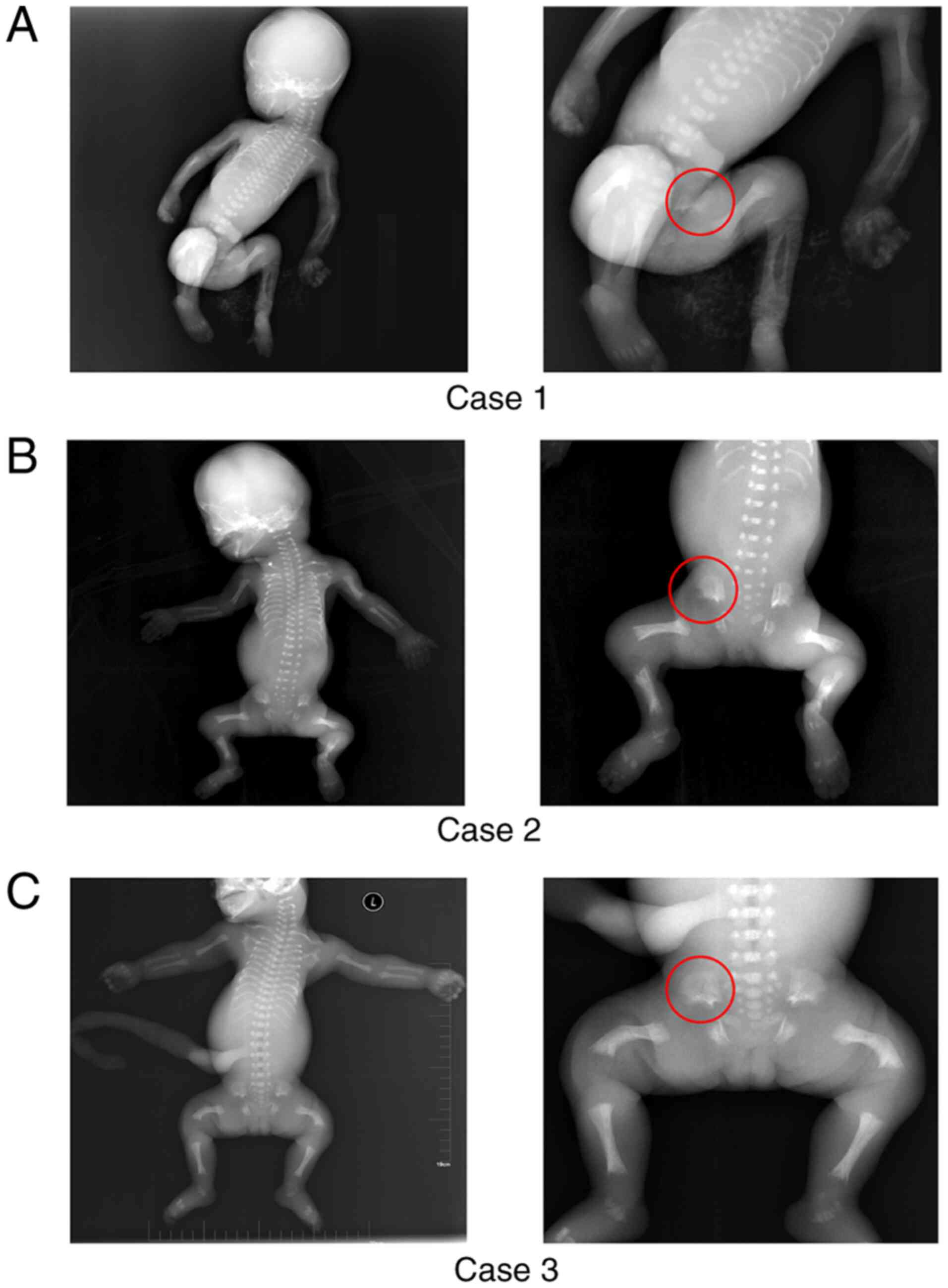
narrow thorax (Fig. 1). Further
ultrasonography revealed a normal amount of amniotic fluid, a
thoracic circumference of 10.6 cm (<5th percentile), a femur
length of 1.6 cm (<5th percentile) and a humerus length of 1.9
cm (<5th percentile). The biparietal diameter and head
circumference of the fetus measured 4.9 and 18 cm, respectively,
which was normal for the gestational age. Abnormalities were not
detected in the liver and pancreas, but the kidneys were
polycystic. The present study was approved by the ethics committee
of Medical Scientific Research and New Technology of Shengjing
Hospital of China Medical University (approval no. 2016PS159K;
Shenyang, China). Informed consent was obtained for each
participant included in the study.
H&E staining
Sections from each paraffin block of the distal
femur growth plates, taken from the previously reported cases, the
newly identified case and aborted fetuses without skeletal
malformations were processed and stained with H&E according to
routine protocols. In brief, the distal femur growth plates were
fixed in 4% paraformaldehyde for 24 h at 4°C, followed by
decalcification with a decalcifying solution. Subsequently, the
samples were washed in tap water for 24 h to clean the tissues, and
then dehydrated in serially graded ethanol solutions and embedded
in paraffin (Tissue Tek processor and Leica embedder; Leica
Microsystems, Inc.). The sections (5 µm) were sagittally sectioned
at a thickness of 5 µm, and deparaffinized in xylene, rehydrated in
descending concentrations of alcohol, and stained with H&E
(Thermo Fisher Scientific, Inc.) using routine protocols. The
images were acquired under at ×10 and ×40 magnification using a
light microscope (Nikon ECLIPSE Ci; Nikon Corporation).
NGS and Sanger sequencing
Peripheral blood was collected from the parents and
amniotic fluid was collected from the fetus. Targeted capturing,
NGS and data analysis were conducted as previously described
(6). In brief, genomic DNA (case 3
I-1, I-2 and II-1) sequences were captured by a customized capture
array (NimbleGen; Roche Applied Science), which was designed to
capture all exons and splicing areas of 463 genes known to be
associated with genetic skeletal disorders, including SRPS. The
library was sequenced on an Illumina HiSeq 2000 (Illumina, Inc.).
NGS produced >200 times the average sequencing depth and
>98.95% overall coverage. The human reference sequence
GRCh37.-hg19 was used to validate the experimental sequencing data.
The pathogenicity of variants was annotated by referring to the
ClinVar dataset (https://www.ncbi.nlm.nih.gov/clinvar/), Human Gene
Mutation Database (http://www.hgmd.cf.ac.uk/) and the Standards and
Guidelines for the Interpretation of Sequence Variants of American
College of Medical Genetics and Genomics (7). Variants with minor allele frequencies
(<0.01) in any of the following databases were selected: gnomAD
(https://gnomad.broadinstitute.org/),
Exome Aggregation Consortium (https://gnomad.broadinstitute.org/), 1000 Genomes
Project (https://www.internationalgenome.org/) and an in-house
database. The M-Cap tool (M-CAP version 1.4; http://bejerano.stanford.edu/MCAP/) was used to
predict the variant pathogenicity. As an autosomal recessive
inheritance model should be applied according to the pedigree
chart, variants of compound heterozygotes or homozygotes should be
relevant first. The DYNC2H1 variants were amplified by
general PCR and sequenced using two DYNC2H1-specific primer
pairs, as follows: Primer7883 forward,
5′-GCCAATATATTTATCCAGGATTACC-3′ and Primer7883 reverse,
5′-AAACCAAATAAAGCAAAAGAGA GTG-3′; and Primer6591_6593 forward,
5′-TGAGTTTAAAAATGGTTCTTGAAAAGG-3′ and Primer6591_6593 reverse,
5′-CAAATCATTGTGTTTTGGCAGTTAAG-3′.
Results
Radiographic presentation of SRPS3
cases
Radiographs of all three cases demonstrated similar
skeletal characteristics, including the typical phenotypic trident
appearance of the acetabulum; a long, narrow thorax with short
ribs; shortened long bones; shortened femurs and remarkable spurs
at the metaphysis of the long bones (Fig. 1A-C). In addition, case 3 showed
obvious congenital bowing of both femurs (Fig. 1C and Table I).
 | Table I.Clinical features of cases with
SRPS3. |
Table I.
Clinical features of cases with
SRPS3.
|
| Cases |
|---|
|
|
|
|---|
| Clinical
features | Case 1 | Case 2 | Case 3 |
|---|
| CS | No | No | No |
| Week of diagnosis,
wg | 25 | 28 | 21 |
| Diagnosis | SRPS3 | SRPS3 | SRPS3 |
| Chest circumference,
cm | NA | 11.2 | 10.6 |
| Abdominal
circumference, cm | 19.9 | 13.8 | 14.4 |
| Femur length, cm | 3.4 | 2.1 | 1.6 |
| Humerus length,
cm | 3 | 2.1 | 1.9 |
| Polydactyly | No | No | No |
| Kidney anomaly | No | No | Polycystic
kidney |
| Liver/pancreas
microscopic changes | No | No | No |
| Trident appearance of
acetabulum | Yes | Yes | Yes |
| Short ribs | Yes | Yes | Yes |
| Short long bones | Yes | Yes | Yes |
| Long bones,
metaphysis spurs | Yes | Yes | Yes |
| Congenitally bowed
femurs | No | No | Yes |
| Histopathological
changes | Irregularly organized
proliferation and hypertrophy zones | Irregularly organized
proliferation and hypertrophy zones | NA |
| Other features | NA | NA | NA |
Cartilage growth plate abnormalities
due to DYNC2H1 variants
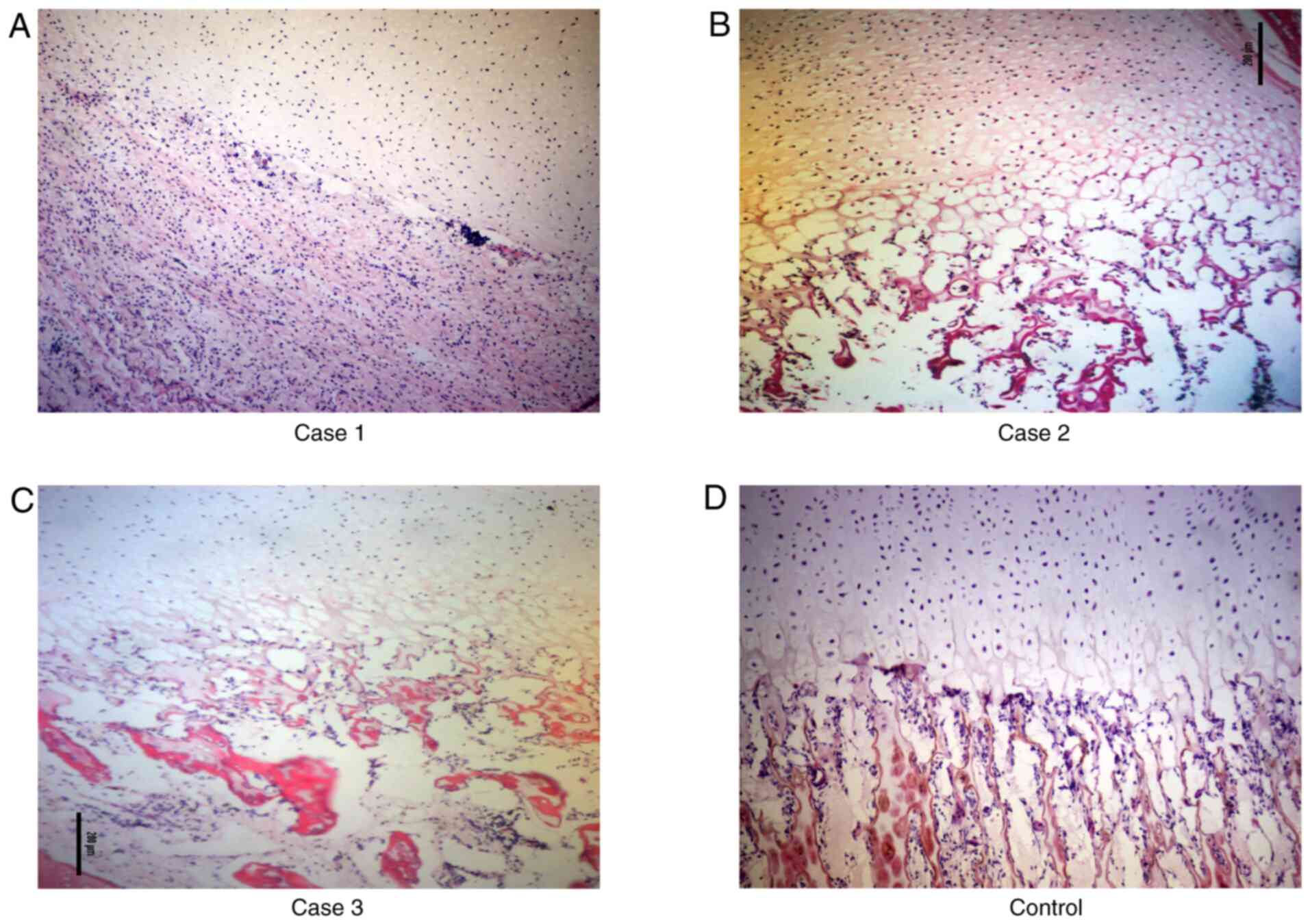
Histological sections of the distal femur growth
plates obtained from the three cases were stained with H&E
(Fig. 2). As shown in Fig. 2D, the growth plate of the femur bone
from an aborted fetus without skeletal malformations had regular
organized zones of proliferation and hypertrophy. However, in case
1, zones of proliferation and hypertrophy could not be visualized;
thus, the chondrocytes did not appear to be proliferating. Within
the region of osteogenesis, the primary spongiosum showed an
abundance of retaining cartilage (Fig.
2A). By contrast, in cases 2 and 3, although proliferation was
evident, irregular columnar formations and a lack of normal
chondrocytes were observed; indeed, the chondrocytes appeared to be
increasingly hypertrophic. Disorganization and increased
vacuolization were evident in the region of osteogenesis (Fig. 2B and C).
Novel variants identified in SRPS3
case 3
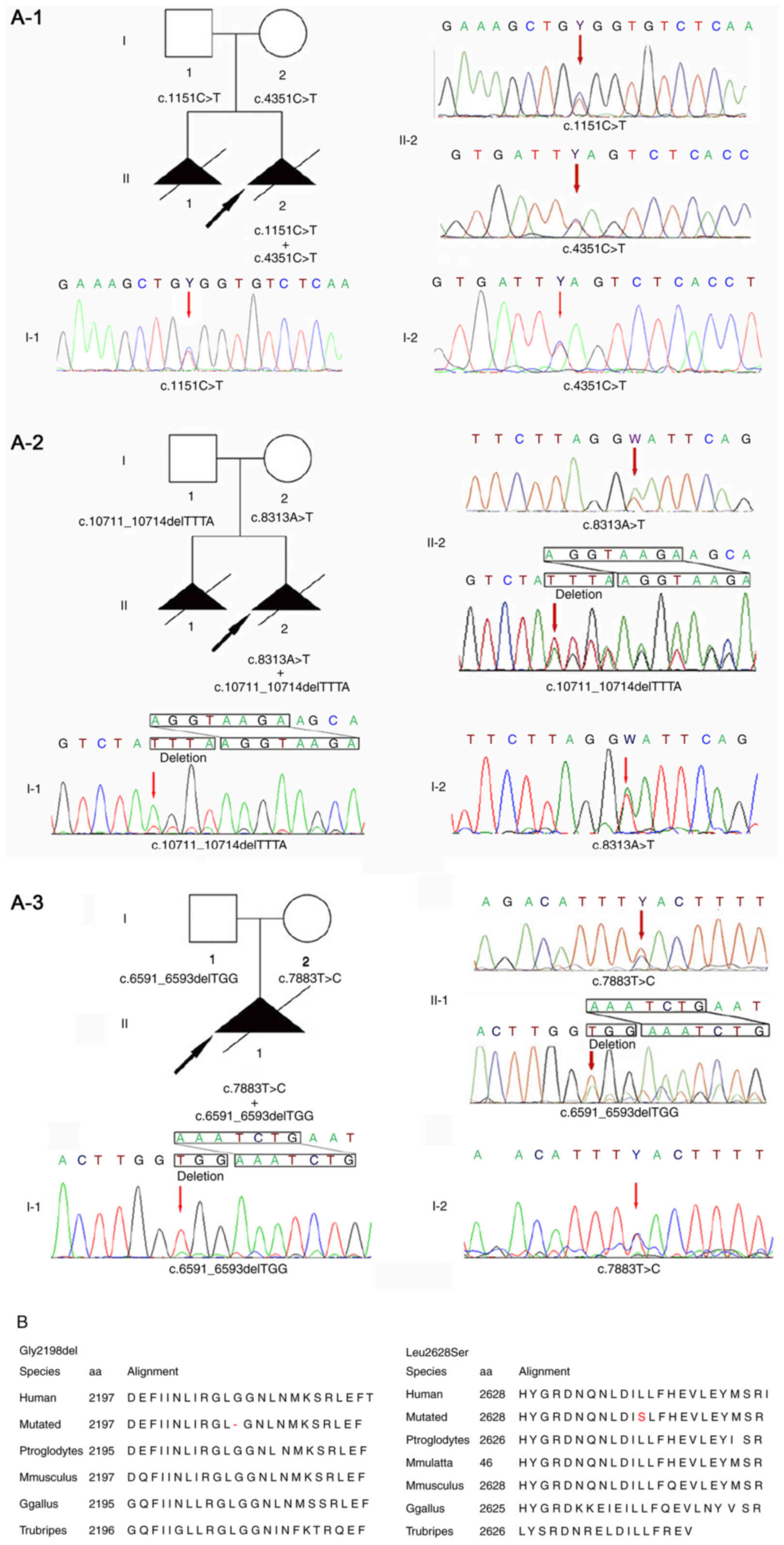
Targeted exon sequencing of 463 known skeletal
disorder-related genes was performed in the newly identified
proband and parents (case 3, Fig.
3A-3). Together, two compound heterozygous maternal and
paternal variants were identified in the DYNC2H1 gene:
NM_001080463.1, c.6591_6593delTGG (chr11:103055738-103055740) and
NM_001080463.1, c.7883T>C (chr11:103070000), respectively. These
results were further confirmed by Sanger sequencing. The two
variants were highly conserved upon comparison of the sequences
over a range of species (Fig.
3B).
Discussion
All three fetuses considered in this report showed
prenatal sonographic findings that were consistent with SRPS and
all parents agreed to induced labor. Radiography showed similar
phenotypes for the three fetuses (Fig.
1). These findings were consistent with previous reports
regarding the radiographic presentation of SRPS3 cases (5,6).
Although other multisystem anomalies are often associated with SRPS
(8), only a polycystic kidney was
found in case 3 (Table I).
A number of histopathological changes have been
found in the growth plates of patients with SRPS. Such phenomena
have been observed in several skeletal dysplasia-associated genes,
including intraflagellar transport protein 43 homolog
(IFT43), IFT80 and IFT52 (9–11).
However, few histopathological changes have been reported in the
growth plates of cases involving DYNC2H1 variant-induced
SRPS (7). To confirm the
involvement of DYNC2H1 variants in endochondral
ossification, the histological sections of femur growth plates were
stained. Case 1 was atypical, wherein proliferation was absent and
an abundance of retaining cartilage was observed in the primary
spongiosum (Fig. 2A). Phenotypes
similar to those reported in the literature were observed in cases
2 and 3 (Fig. 2B and C).
Variants in DYNC2H1 can inactivate the
retrograde ciliary motor, causing skeletal dysplasia (12). A number of the morphological
abnormalities observed in human ciliopathies are probably caused by
disruption of the hedgehog signaling pathway, including variation
in the DYNC2H1 gene (13,14).
Although different histopathological changes were observed in the
three cases evaluated in the present study, they all showed an
abnormal pattern of proliferation and differentiation at the femur
growth plates. This may be due to disruption of the hedgehog
signaling pathway caused by a DYNC2H1 variant.
One of the novel DYNC2H1 variants found in
case 3 (Fig. 3A-3), namely
c.7883T>C(p.Leu2628Ser) (chr11:103070000), is located in exon 49
within an ATP binding and hydrolysis domain (AAA_4 domain). The
M-Cap tool predicted this variant to be ‘deleterious’. Regardless
of this prediction, this variant occurs in the conserved AAA_4
domain, which should affect ATP binding and hydrolysis. In
addition, another variant, reported to cause SRPS, has been located
within close proximity to c.7883T>C (15).
The other variant identified in this study in case
3, c.6591_6593delTGG(Gly2198del) (chr11:103055738-103055740;
Fig. 3A-3), might hinder ATP
hydrolysis and thus, the generation of adequate cellular energy for
the movement of microtubules. An R2205H substitution close to the
novel variant has also been reported to cause SRPS (16). Neither p.Leu2628Ser nor Gly2198del
were detected in the control database, indicating that these two
variants were pathogenic in nature, rather than polymorphisms.
The present study identified atypical
histopathological changes and confirmed the radiological features
of three SRPS3 cases. In addition, it successfully diagnosed SRPS3
in a Chinese fetus and identified two novel compound heterozygous
DYNC2H1 variants. In conclusion, three pairs of compound
heterozygous DYNC2H1 variants were identified as pathogenic.
The findings of the present study expand the current understanding
of histopathological changes associated with DYNC2H1
variants and provide further confirmation of the radiological
features of SRPS3. Collectively, the findings should promote more
efficient imaging, as well as histopathological and molecular
diagnoses of SRPS3.
Acknowledgements
Not applicable.
Funding
The present study was jointly supported by the
Natural Science Foundation of China (grant no. 81701462), the
National Key R&D Program of China (grant no. 2018YFC1002900)
and the 345 Talent Project.
Availability of data and materials
The datasets generated and/or analyzed during the
current study are not publicly available due to the participants
not wanting to share all their sequencing data on a public database
except for the pathogenic variants, but are available from the
corresponding author on reasonable request.
Authors' contributions
CLX, SQX, XY and YL conceived and designed the
experiments. CXL, HKJ, JLL and YL assisted in the recruitment of
the patients and acquisition of experimental data. CLX, SQX, XY and
HQ performed the experiments. SQX, XY, YL and HQ helped in genetic
analysis. CLX, SQX, XY, YL and HQ wrote the manuscript. CLX and YL
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All procedures performed in studies involving human
participants were in accordance with the ethical standards of the
institutional and/or national research committee and in accordance
with the 1964 Helsinki declaration and its later amendments or
comparable ethical standards. This study was approved by the ethics
committee of Medical Scientific Research and New Technology of
Shengjing Hospital of China Medical University (approval no.
2016PS159K; Shenyang, China). Informed consent was obtained for
each participant included in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Huber C and Cormier-Daire V: Ciliary
disorder of the skeleton. Am J Med Genet C Semin Med Genet.
160C:165–174. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Krakow D and Rimoin DL: The skeletal
dysplasias. Genet Med. 12:327–341. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mäkitie O: Molecular defects causing
skeletal dysplasias. Endocr Dev. 21:78–84. 2011. View Article : Google Scholar
|
|
4
|
Bonafe L, Cormier-Daire V, Hall C, Lachman
R, Mortier G, Mundlos S, Nishimura G, Sangiorgi L, Savarirayan R,
Sillence D, et al: Nosology and classification of genetic skeletal
disorders: 2015 revision. Am J Med Genet A. 167A:A2869–A2892. 2015.
View Article : Google Scholar
|
|
5
|
Chen LS, Shi SJ, Zou PS, Ma M, Chen XH and
Cao DH: Identification of novel DYNC2H1 mutations associated with
short rib-polydactyly syndrome type III using next-generation panel
sequencing. Genetics and molecular research. Genet Mol Med.
15:gmr81342016.
|
|
6
|
Mei L, Huang Y, Pan Q, Su W, Quan Y, Liang
D and Wu L: Targeted next-generation sequencing identifies novel
compound heterozygous mutations of DYNC2H1 in a fetus with short
rib-polydactyly syndrome, type III. Clin Chim Acta. 447:47–51.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al ACMG
Laboratory Quality Assurance Committee, : Standards and guidelines
for the interpretation of sequence variants: A joint consensus
recommendation of the American College of Medical Genetics and
Genomics and the Association for Molecular Pathology. Genet Med.
17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Elçioglu NH and Hall CM: Diagnostic
dilemmas in the short rib-polydactyly syndrome group. Am J Med
Genet. 111:392–400. 2002. View Article : Google Scholar
|
|
9
|
Zhang W, Taylor SP, Nevarez L, Lachman RS,
Nickerson DA, Bamshad M, Krakow D and Cohn DH; University of
Washington Center for Mendelian Genomics Consortium, : IFT52
mutations destabilize anterograde complex assembly, disrupt
ciliogenesis and result in short rib polydactyly syndrome. Hum Mol
Genet. 25:4012–4020. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cavalcanti DP, Huber C, Sang KH, Baujat G,
Collins F, Delezoide AL, Dagoneau N, Le Merrer M, Martinovic J,
Mello MF, et al: Mutation in IFT80 in a fetus with the phenotype of
Verma-Naumoff provides molecular evidence for Jeune-Verma-Naumoff
dysplasia spectrum. J Med Genet. 48:88–92. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Duran I, Taylor SP, Zhang W, Martin J,
Qureshi F, Jacques SM, Wallerstein R, Lachman RS, Nickerson DA,
Bamshad M, et al: Mutations in IFT-A satellite core component genes
IFT43and IFT121 produce short rib polydactyly syndrome with
distinctive campomelia. Cilia. 6:72017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ocbina PJ, Eggenschwiler JT, Moskowitz I
and Anderson KV: Complex interactions between genes controlling
trafficking in primary cilia. Nat Genet. 43:547–553. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Weatherbee SD, Niswander LA and Anderson
KV: A mouse model for Meckel syndrome reveals Mks1 is required for
ciliogenesis and Hedgehog signaling. Hum Mol Genet. 18:4565–4575.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Goetz SC and Anderson KV: The primary
cilium: A signalling centre during vertebrate development. Nat Rev
Genet. 11:331–344. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
El Hokayem J, Huber C, Couvé A, Aziza J,
Baujat G, Bouvier R, Cavalcanti DP, Collins FA, Cordier MP,
Delezoide AL, et al: NEK1 and DYNC2H1 are both involved in short
rib polydactyly Majewski type but not in Beemer Langer cases. J Med
Genet. 49:227–233. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Merrill AE, Merriman B, Farrington-Rock C,
Camacho N, Sebald ET, Funari VA, Schibler MJ, Firestein MH, Cohn
ZA, Priore MA, et al: Ciliary abnormalities due to defects in the
retrograde transport protein DYNC2H1 in short-rib polydactyly
syndrome. Am J Hum Genet. 84:542–549. 2009. View Article : Google Scholar : PubMed/NCBI
|