Introduction
Transmissible spongiform encephalopathies (TSEs) are
fatal neurodegenerative diseases caused by prions (1). TSEs have common pathognomonic and
histopathological characteristics, including spongiform changes,
glial proliferation, neuronal loss, and the deposition of a
misfolded isoform (PrPsc) (2) of
the cellular prion protein (PrPc). Whether the prion disease at
issue is genetic, transmissible, or an irregular disorder, they all
implicate protein refolding of the normal prion (PrPc), which is
encoded by the Prnp gene (3). PrPsc, a misfolded version of PrPc, was
originally thought to be an accumulated proteinase K-resistant
prion protein. However, patients with PrPc could still incur
disease-related and diagnostically imperative alterations even
without its conversion to the protease-resistant form (4).
Synthetic human prion peptide (PrP) (106–126)
preserves some features of PrPsc's physiological and pathogenic
assets and can initiate apoptosis of hippocampal neurons (5). An investigation of the respective
amino acid sectors showed that the peptide, including amino acids
106–126 of the PrP sequence, was able to replicate several
biological features of PrPsc in vitro, including
amyloidogenesis, as well as its neurotoxic and its gliotrophic
effects (6–8). Moreover, PrP (106–126) covers an
AGAAAAGA arrangement at amino acids 113 to 120, a motif that has
been found in PrP molecules across numerous species (9).
Troglitazone is an antidiabetic medicine used to
treat type 2 diabetes, improve the sensitivity of various tissues
to insulin, and reduce blood glucose levels (10–12).
Troglitazone's antidiabetic effects are closely related to
peroxisome proliferator-activated receptor (PPAR)γ activation,
which suggests that it is the activation of PPAR-responsive genes
that is responsible for the insulin-sensitizing effects of
troglitazone (13–15). PPARγ is a member of the class of
PPARs that are associated with ligand-stimulated nuclear receptors
(16). PPARγ is primarily expressed
in adipose tissue and plays an important role in lipid metabolism
alteration, insulin sensitivity, and adipocyte differentiation
(17,18). Some studies have proposed that PPARγ
activation has a protective effect on neuronal cells and inhibits
neurodegeneration (19–22). With this in mind, we investigated
whether troglitazone protected against prion peptide-mediated
neuronal apoptosis.
Autophagy, also known as programmed cell death-II,
is a homeostatic and catabolic progression by which cellular
constituents and organelles are delivered to lysosomes for
degradation (23,24). Autophagy begins with the development
of double-membrane structures (autophagosomes) that enclose
cellular ingredients and organelles. Once these are combined with
lysosomes, they progress to a maturing system and, ultimately
resulting in the collapse and recycling of the cargo (25,26).
p62 levels correlate with autophagy (27). While p62 is selectively degraded by
autolysosomal degradation, it is not degraded by the
ubiquitin-proteasome system (UPS) (28). p62 levels increase when autophagy is
repressed (29,30).
In our prior research, we uncovered that the
inhibition of autophagy prevented PrP (106–126)-mediated neuronal
cytotoxicity (31). In this study,
we investigate whether troglitazone protects against PrP
(106–126)-mediated neuronal cytotoxicity via autophagy flux and
whether the autophagy flux is controlled by PPARγ.
Materials and methods
Cell culture
Embryonic 18-day ICR mice were purchased from
SAMTAKO (Osan, Korea). No experiments were performed on live
animals. The animals were euthanized by cervical dislocation and
the brain was collected from the pups. Primary murine cortex neuron
culture was performed according to the protocol of Beaudoin et
al (32) and a little modified.
Brain was dissected in Hanks Buffered Saline Solution without
Mg2+ and Ca2+ (HBSS: Gibco; Thermo Fisher
Scientific, Inc.) and digested in 0.25% trypsin containing DNAse I
(2,000 U/mg) (Gibco; Thermo Fisher Scientific, Inc.) for 20 min at
37°C. The obtained cell suspension was diluted in DMEM containing
25 mM glucose and 10% fetal bovine serum (FBS), and then cultured
in tissue culture flasks coated with 50 µg/ml poly-D-lysine at a
density of 3–4×105 cells/cm2. The SK-N-SH
(human neuroblastoma cell line, passage no. 14) was acquired from
the American Type Culture Collection (ATCC). SK-N-SH cells were
cultured in Minimum Essential Medium (Hyclone Laboratories) with
10% fetal bovine serum (FBS, Gibco; Thermo Fisher Scientific, Inc.)
and gentamycin (0.1 mg/ml) at 37°C and 5% CO2.
Chemical and PrP (106–126)
treatment
Synthetic prion peptides PrP (106–126)
(Lys-Thr-Asn-Met-Lys-His-Met-Ala-Gly-Ala-Ala-Ala-Ala-Gly-Ala-Val-Val-Gly-Gly-Leu-Gly)
were manufactured by Peptron. The peptides were dissolved in
sterile dimethyl sulfoxide (DMSO) at a stock concentration of 10 mM
and stored at −20°C.
The stock solution of troglitazone (8 mM;
Sigma-Aldrich; Merck KGaA) was dissolved in DMSO. The cells were
pre-incubated with 20, 40, or 80 µM troglitazone with/without PrP
(100 µM) for 12 h. For the inhibitor treatments, the cultures were
pretreated with a 10 µM CQ (autophagy inhibitor; Sigma-Aldrich) or
10 µM GW9662 (PPARγ antagonist; Sigma-Aldrich; Merck KGaA).
Trypan blue exclusion assay
Cell viability was evaluated by trypan blue
exclusion assay using a hemocytometer. Untreated cells were used as
the control group and cell viability was compared with the control.
Each treatment was performed in triplicate.
Annexin V/PI assay
Cells in the logarithmic phase were collected and
cultured in 24-well plates at 4×104 cells/well. Cell
survival was evaluated using an Annexin V/PI Assay Kit (Santa Cruz
Biotechnology) following the manufacturer's procedure. Briefly,
cells were treated for 24 h and then harvested, washed in cold
phosphate-buffered saline (PBS) twice and then stained with
fluorescein isothiocyanate (FITC)-conjugated Annexin V and PI dyes.
The externalization of phosphatidylserine and the permeability to
PI were evaluated using a flow cytometer. Data from 5,000 gated
events per sample were collected. Cells in early stages of
apoptosis were positively stained with Annexin V, whereas cells in
late apoptosis were positively stained with both Annexin V and PI.
The fluorescence was determined at 488 nm excitation and 525/30
emission using a Guava EasyCyte HT System (Millipore).
Terminal deoxynucleotidyl transferase
dUTP nick end-labeling (TUNEL) assay
Cells in the logarithmic phase were collected and
cultured in 6-well plates at 3×105 cells/well. Neuronal
apoptosis was assessed after treatment using an ApoBrdU DNA
Fragmentation Assay Kit (BioVision), following the manufacturer's
instructions. The nuclei were counterstained with PI.
RNA interference
SK-N-SH cells were transfected with ATG5
small-interfering RNA (siRNA; oligoID HSS114104; Invitrogen; Thermo
Fisher Scientific, Inc.) using Lipofectamine 2000 according to the
manufacturer's instructions. After a 48-h culture, knockdown
efficiency was measured at the protein level by immunoblot
analysis. Nonspecific siRNA (oligoID 12935-300; Invitrogen; Thermo
Fisher Scientific, Inc.) was used as the negative control.
BacMam transduction
GFP-LC3B puncta assay was evaluated in neuronal
cells using the virus from the Premo Autophagy Sensor LC3B-GFP
(BacMam 2.0) kit (Life Technologies, P36235). LC3B-FP and LC3B
(G120A)-FP viral vectors (MOI, 30) were employed to monitor
autophagosome dynamics by fluorescence microscopy analysis.
Immunocytochemistry
Cells in the logarithmic phase were collected and
cultured on 1% gelatin-coated coverslips (12 mm; Nalge Nunc
International) in 24-well plates at 4×104 cells/well.
After treatment, the cells were fixed with 4% PFA in PBS (1X) at pH
7.4 for 20 min at room temperature (RT). The cells were washed in
sterile Tris-buffered saline TBS (1M), 3 g Tris (24.8 mM), 8 g NaCl
(137 mM), and 0.2 g KCl (2.7 mM) in 1 liter distilled water at pH
7.4 with 0.1% Tween-20 (TBST) for 10 min. They were then blocked
for 15 min in TBST with 5% FBS, and incubated for 3 h at RT with
the primary antibodies (anti-PPARγ diluted 1:1,000; sc-7273; Santa
Cruz Biotechnology), which had themselves been diluted in TBST with
5% FBS. Alexa Fluor 488-labeled donkey anti-rabbit IgG antibody
(A21206; Molecular Probes) diluted 1:1,000 was employed to
visualize the channel expression using fluorescence microscopy
(Nikon Eclipse 80i). The images were evaluated using NIS-Elements F
Ver4.60 imaging software.
Confocal microscopy
After being subjected to either immunocytochemistry
or the GFP-LC3B puncta assay, the coverslips were placed in
mounting medium and imaged through a 63× oil objective on a Zeiss
LSM710 microscope equipped with a standard set of lasers. The
excitation wavelengths were 488, 543, and 633 nm. The bandpass
filters were set at 500–550 (AlexaFluor488), 560–615 nm (Cy3,
AlexaFluor568), and 650–750 nm (AlexaFluor647).
Western blot analysis
Cells in the logarithmic phase were collected and
cultured in 6-well plates at 3×105 cells/well. After the
treatments, the cells were washed with PBS and lysed in lysis
buffer [25 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic
acid) at pH 7.4, 100 mM NaCl, 1 mM ethylenediaminetetraacetic acid
(EDTA), 5 mM MgCl2, 0.1 mM dithiothreitol (DTT), and a
protease inhibitor mixture]. Equal quantities of cellular proteins
(15–30 µg/µl) were electrophoretically resolved on a 10% sodium
dodecyl sulfate (SDS) polyacrylamide gel and transferred to a
nitrocellulose membrane. Immunoreactivity was detected through
consecutive incubations with a blocking solution containing 5% skim
milk and primary antibodies followed by the corresponding
horseradish peroxidase-conjugated secondary antibodies, and finally
developed using enhanced chemiluminescence substances in the
WESTSAVE Gold Detection Kit (LF-QC0103; AbFrontier Inc.). The
primary antibodies, anti-PPARγ diluted 1:1,000 (sc-7273; Santa Cruz
Biotechnology), anti-LC3B diluted 1:1,000 (#4108; Cell Signaling
Technology), anti-P62 diluted 1:1,000 (#5114; Cell Signaling
Technology) and anti-β-actin diluted 1:5,000 (A5441; Sigma-Aldrich;
Merck KGaA), in the antibody solution (1% skim milk in TBST) were
used for immunoblotting. The images were inspected using a Fusion
FX7 imaging system (Vilber Lourmat, Torcy Z.I. Sud). The density of
the signal bands was evaluated using Bio-1D software (Vilber
Lourmat).
Transmission electron microscopy (TEM)
analysis
The cells for TEM were fixed in 2% glutaraldehyde
[Electron Microscopy Sciences (EMS)] and 2% paraformaldehyde (EMS)
in 0.05 M sodium cacodylate (pH 7.2; EMS) for 2 h at 4°C. The
samples were fixed in 1% osmium tetroxide (EMS) for 1 h at 4°C,
dehydrated with a graded ethanol series (25, 50, 70, 90 and 100%)
for 5 min each, and embedded in epoxy resin (Embed 812; EMS) for 48
h at 60°C following to the manufacturer's instructions. Ultrathin
sections (60 nm) were cut using an LKB-III Ultratome (Leica
Microsystems GmbH) and stained with 0.5% uranyl acetate (EMS) for
20 min and 0.1% lead citrate (EMS) for 7 min at RT. The fluorescent
images were recorded on a Hitachi H7650 electron microscope
(Hitachi, Ltd.; magnification, ×10,000) installed at the Center for
University-Wide Research Facilities (CURF) at Jeonbuk National
University.
Statistical analysis
Results are expressed as the means ± standard
deviations (SD) from at least three independent replicates. All
experiments were analyzed by the one-way analysis of variance.
Comparisons of three or more groups were made using Tukey's post
hoc test. All statistical analyses were implemented with GraphPad
Prism version 5.0 software. P<0.05 was considered to indicate a
statistically significant difference.
Results
Protective effect of troglitazone on
prion protein-mediated neuronal cell death
Consistent with the relevant literature (33,34)
and our preliminary experiments, we used different concentrations
of troglitazone for efficacy testing. We selected 80 µM
troglitazone as highest concentration because it significantly
increased cell viability (at 100 µM PrP). Once our preliminary
experiments were complete, we proceeded to examine the effect of
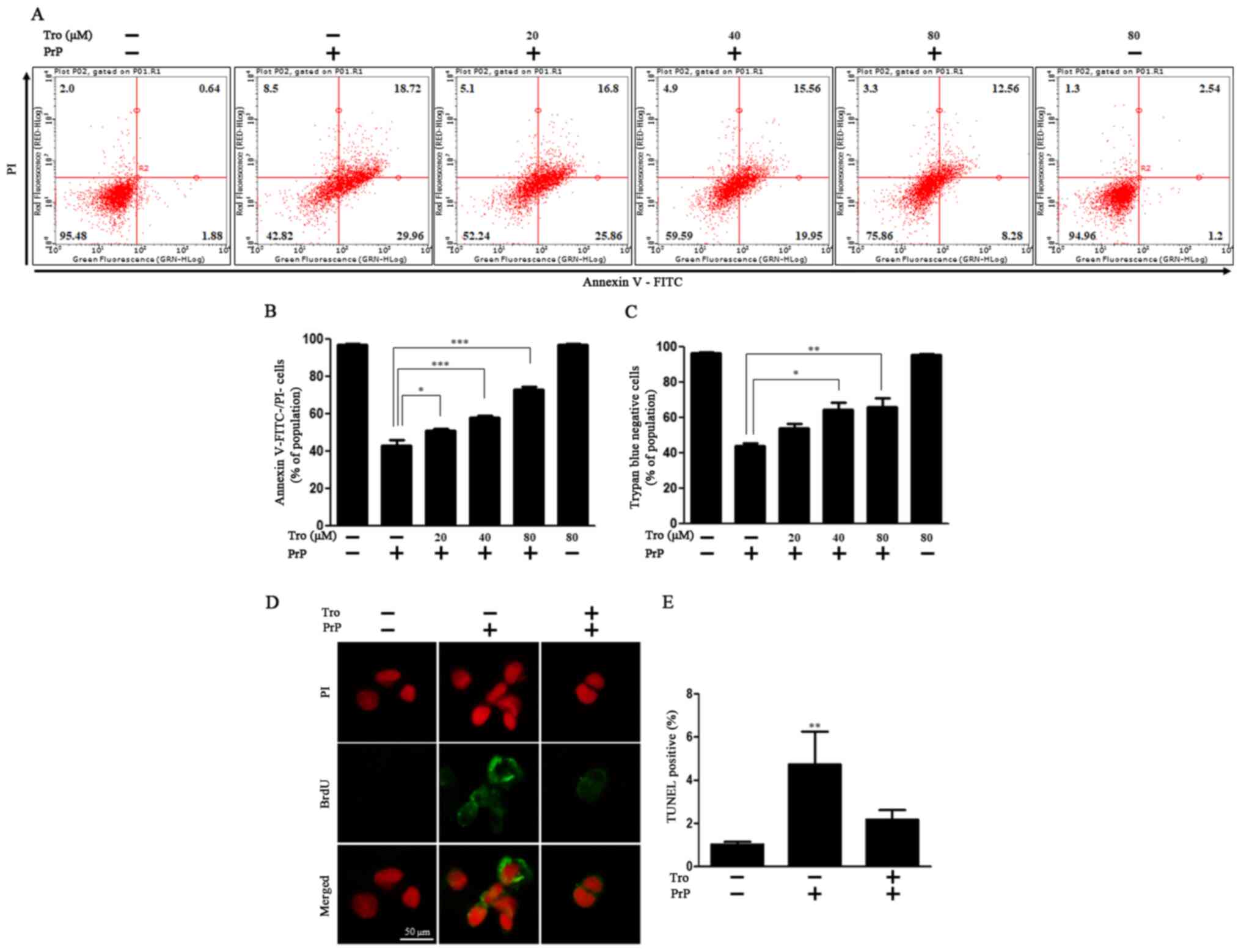
troglitazone on PrP (106–126)-mediated neurotoxicity by an Annexin
V/PI assay in primary neuronal cells. Cells were treated with PrP
in combination with troglitazone. PrP treatment led to a 2.25-fold
decrease in cell viability (Annexin V−/PI−)
compared to the control. In detail, PrP treatment increased early
apoptosis (Annexin V+/PI−) from about 2% to
about 30% and late apoptosis (Annexin V+/PI+)
from about 1% to about 19%. We observed that pretreatment with
troglitazone dose-dependently protected neurons against PrP
(106–126)-induced neurotoxicity. Treatment of the cells with 20,
40, and 80 µM troglitazone increased cell viability by 1.18±1.20
(SD), 1.34±1.19 (SD), and 1.70±1.23 (SD)-fold, respectively,
compared to PrP-treated cells (Fig. 1A
and B). In addition, we confirmed the protective effect of 80
µM troglitazone in SK-N-SH cells (data not shown). Consistent with
this, in the trypan blue assay, troglitazone repressed PrP-mediated
cell death dose-dependently (Fig.
1C). Troglitazone also decreased DNA strand damage triggered by
PrP treatment (Fig. 1D and E).
These results indicate that troglitazone attenuated PrP
(106–126)-mediated neuronal cell death.
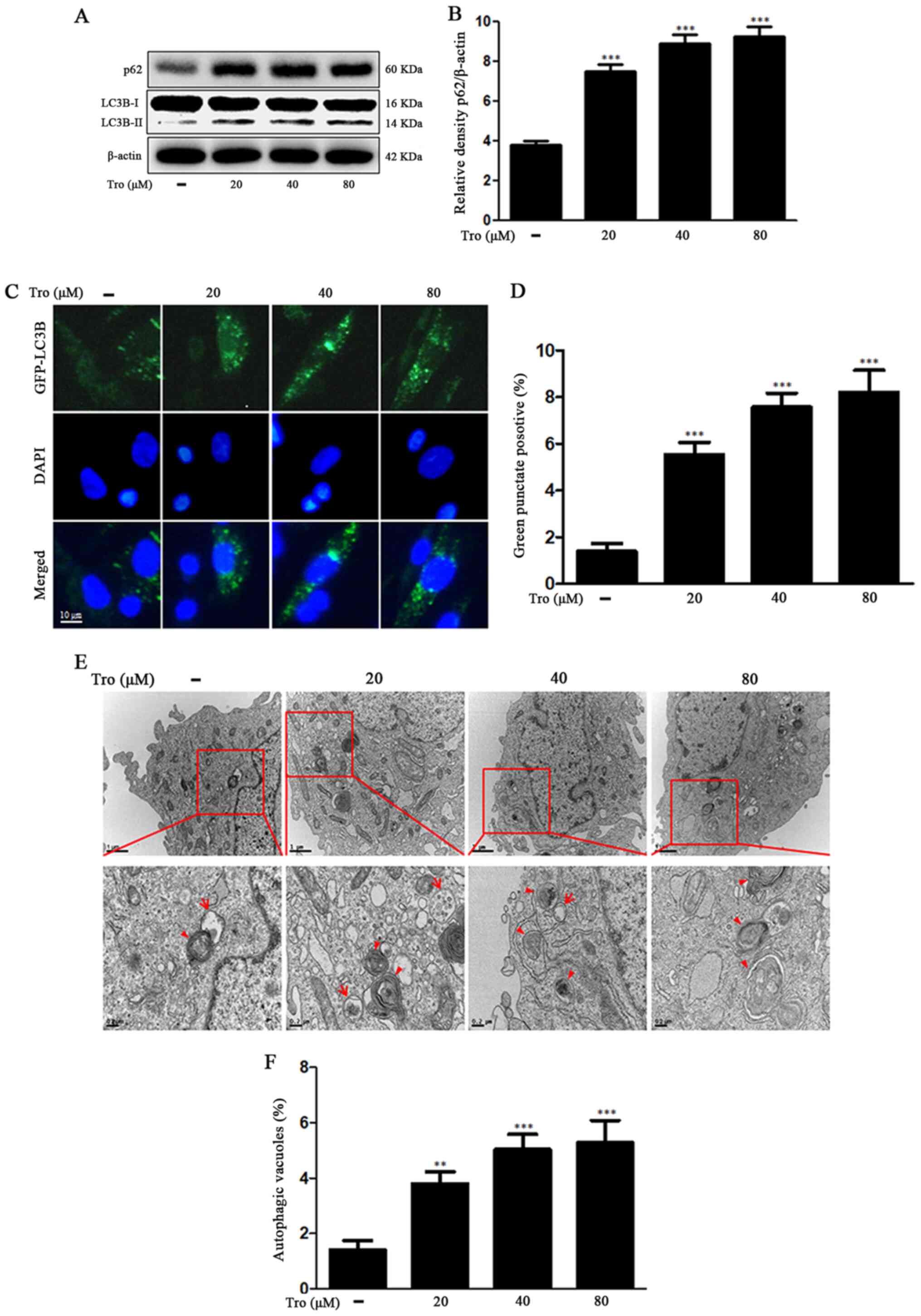
Troglitazone restrains autophagy flux
in neuronal cells
It has traditionally been thought that LC3 and
SQSTM1/p62 are consumed by lysosomal degradation via the process of
autophagy flux (28). We found that
LC3-II and SQSTM1/p62 protein levels were increased in the
troglitazone-treated groups. Treatment of the cells with 80 µM
troglitazone increased SQSTM1/p62 protein level by 2.44±0.50-fold
compared to the control (Fig. 2A and
B). The BacMam 2.0 system was employed to observe the
construction of autophagosomes in the neuroblastoma cells.
Troglitazone led to an increase in punctate fluorescence in SK-N-SH
cells (Fig. 2C and D). The observed
punctate fluorescence distribution pattern suggested that the
LC3B-FP protein was collected in the autophagosomes. Using
transmission electron microscopy (TEM), we observed that
troglitazone treatment improved phagophore accumulation that was
not degraded by the lysosomes (Fig. 2E
and F), suggesting that troglitazone inhibited autophagy flux
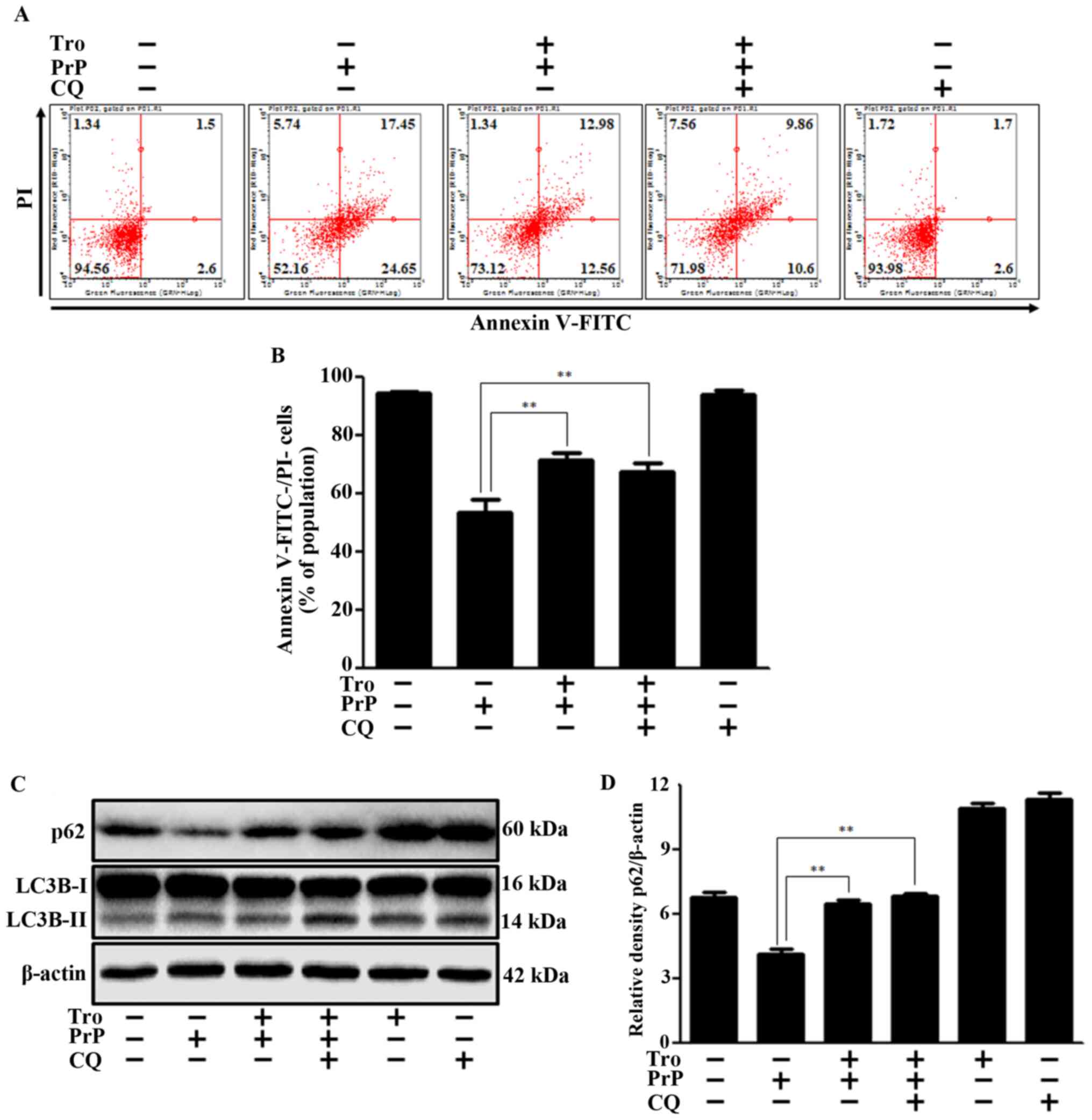
in neuronal cells. To demonstrate the effect of autophagy
inhibition on PrP-mediated neuronal cell death, we performed cell
viability assays using Annexin V/propidium iodide (PI) with/without
chloroquine (CQ). We chose the CQ concentration on the basis of
other studies (35,36). We confirmed that, like the CQ
autophagy inhibitor, troglitazone had neuroprotective effects via
autophagy inhibition (Fig. 3A and
B). We also found that troglitazone and CQ increased levels of
p62 and LC3B-II protein (Fig. 3C and
D), suggesting an inhibition of autophagy flux. On the basis of
the observation that p62 protein level and cell viability did not
increase by more when troglitazone and CQ were treated together, we
propose that troglitazone and CQ play the same role in inhibiting
autophagy flux.
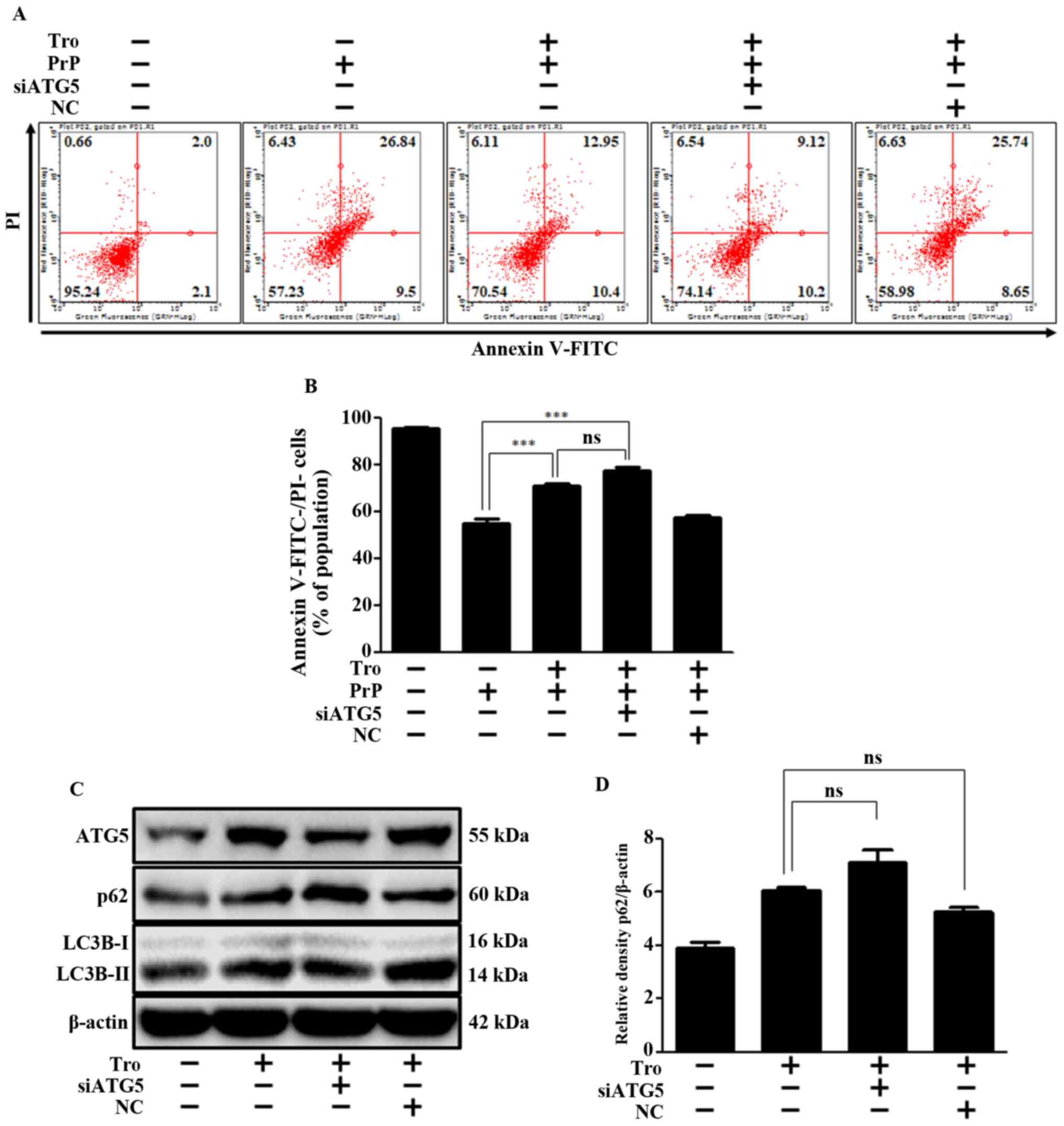
Moreover, the Atg5 knockdown had little impact on
troglitazone-mediated neuroprotection (Fig. 4A and B) and inhibition of autophagy
flux (Fig. 4C and D). Specifically,
troglitazone repressed autophagy flux and PrP-mediated neuronal
apoptosis by inhibiting fusion between the autophagosome and the
lysosome, not by reduction of Atg5. We confirmed the successful
siRNA transfection of ATG5 through additional experiments; the
results revealed that Atg5 siRNA reduced the expression levels of
Atg5 and LC3B, and increased p62 (Fig.
S1). These experimental results indicate that troglitazone or
Atg5 knockdown result in an inhibition of autophagy flux, and
thereby exert a neuroprotective effect in an in vitro prion
model.
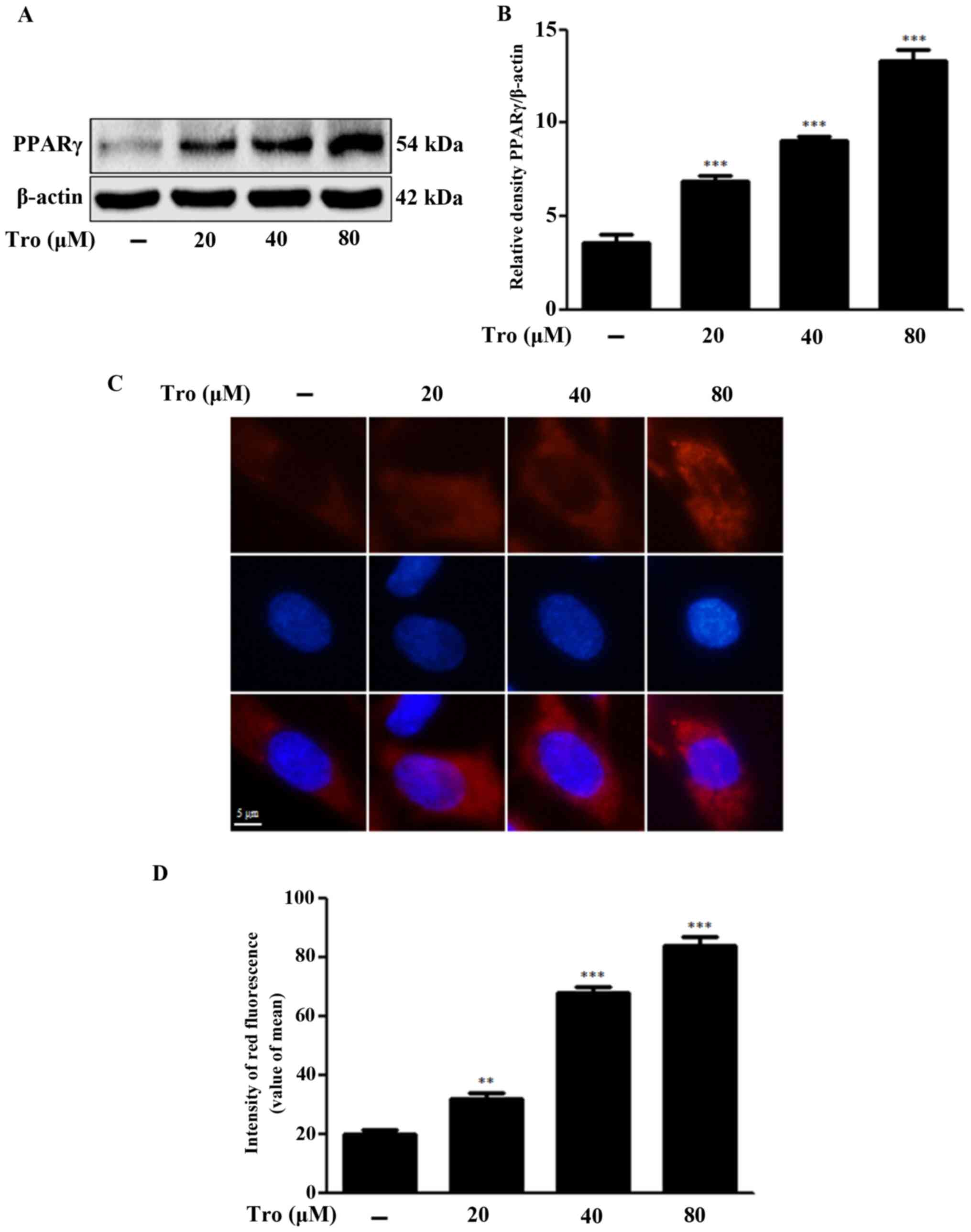
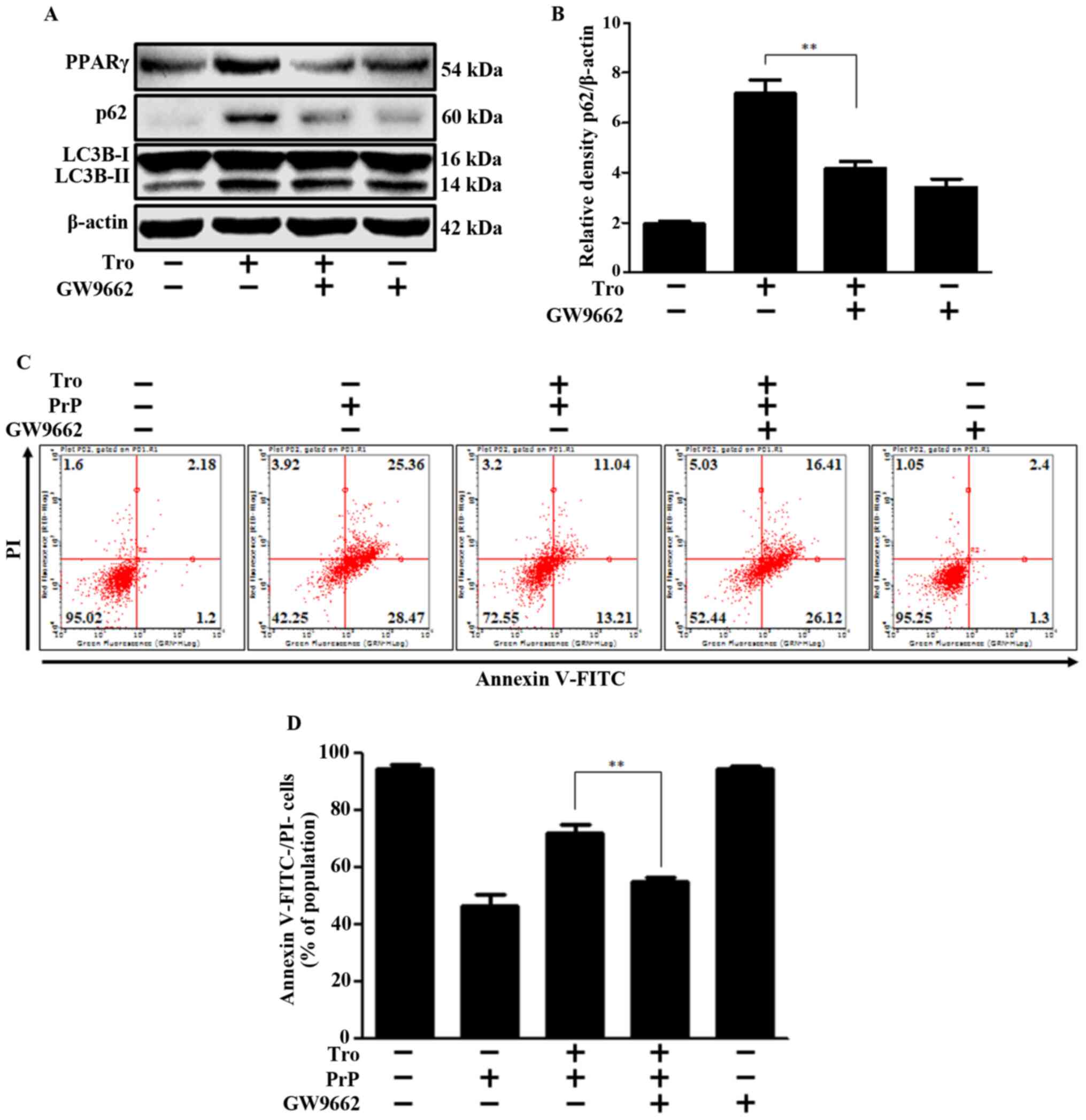
PPARγ activation by troglitazone
attenuates PrP (106–126)-mediated neuronal cell death via
inhibition of autophagy flux
We evaluated whether troglitazone-activated PPARγ
weakened prion peptide-mediated neuronal cell death by inhibiting
autophagy flux. Troglitazone dose-dependently increased the
activation of PPARγ, as indicated by the red fluorescence in
Fig. 5C and D. Treatment of the
cells with 80 µM troglitazone increased PPARγ protein levels by
3.74±0.60-fold compared to the control (Fig. 5A and B). PPARγ antagonist GW9662,
which was employed at a concentration consistent with previous
studies (37,38), suppressed PPARγ activation.
Moreover, increased LC3-II and p62 protein expression, which
indicates the inhibition of autophagy flux, was reversed by the
PPARγ antagonist (Fig. 6A and B).
These experiments demonstrate that troglitazone-induced inhibition
of autophagy flux is regulated by PPARγ. To investigate the effect
of PPARγ activation on PrP-mediated neurotoxicity, we performed
cell viability experiments with and without the PPARγ antagonist.
We determined that troglitazone's neuroprotective effect was
inhibited by the PPARγ antagonist (Fig.
6C and D). This data collectively provides a strong basis for
our conclusion that PPARγ activation by troglitazone prevents PrP
(106–126)-mediated neurotoxicity by inhibiting autophagy flux.
Discussion
The purpose of this study was to investigate whether
troglitazone could be a potential agent for targeting PPARγ and
autophagy flux, thereby decreasing prion protein-mediated neuronal
cell death. Our findings confirm troglitazone's potential as a
therapeutic agent in response to neurodegenerative diseases,
including prion diseases. We suggest that the PPARγ signaling and
autophagy flux affected by troglitazone might be a strategic
mechanism to target in prion disease.
Nah et al (39) reported that the Prnp gene
activates autophagy in response to the presence of amyloid-β in
neuronal cells. A number of studies have theorized that autophagy
generates cellular apoptosis (40–43).
These studies underlie our assumption that the amyloid plaque
formed by PrPsc accumulates and stimulates neuronal autophagic cell
death. In a previous study, we determined that human prion peptide
106–126 stimulated acute autophagy flux to promote autophagic cell
death in neurons and that CQ attenuated PrP-induced neurotoxicity
via autophagy inhibition (31). In
this study, we determined that autophagy inhibition by troglitazone
could protect neuronal cells against PrP-induced cell death.
Troglitazone inhibited PrP-mediated autophagic cell death, and the
addition of CQ had no change on the protective effect (Fig. 3). As co-treatment with troglitazone
and CQ resulted in no synergic effect, that the inference to be
drawn is that troglitazone inhibited autophagic signals. This
result was further affirmed in our tests using Atg5 siRNA tools
(Fig. 4).
We explored upstream of the troglitazone- induced
neuronal autophagy flux. Troglitazone, a PPARγ agonist, suppresses
the excretion of inflammatory cytokines and neurotoxic agents from
stimulated monocytes and microglia, and thus has anti-inflammatory
properties (44–46). We determined that troglitazone
stimulated PPARγ activation, which in turn regulated PrP-mediated
autophagic cell death.
The anti-inflammatory functions of PPARγ have
recently received a great deal of attention, as its agonists have
been shown to exert protective effects in neurologic diseases
(47–49). Some studies have reported that PPARγ
ligands protect against the oxidative stress in neuronal injury
(50–52), while others have suggested that
PPARγ activation may obstruct autophagy flux (53–55).
In this paper, we demonstrated that troglitazone activated PPARγ in
neurons and used a PPARγ inhibitor to prevent PrP-mediated
autophagic cell death (Figs. 5 and
6). These findings suggest that
troglitazone-activated PPARγ protected neuronal cells against
PrP-induced cell death via autophagy inhibition. Previous studies
suggested several materials, such as resveratrol, could prevent
prion-induced neurotoxicity (56,57).
This is the first study indicating that troglitazone-mediated PPARγ
may play a critical role in the neuroprotection against PrP
(106–126)-induced neurotoxicity.
Similar to PrPsc, a neurotoxic prion protein
segment, PrP (106–126), preserves biochemical substances, such as
protease resistance, β-sheet formation, and cytotoxicity (6,58–60).
Some studies have investigated prion pathogenesis and neurotoxic
pathways in vivo using Rocky Mountain Laboratory (RML)
infection and antibody-derived anti-PrP ligands (61–63).
In these studies, the neurodegenerative changes typical of prion
diseases were observed without detectable PrPsc, suggesting the
presence of additional mechanisms of neuronal death in prion
disorders, aside from those related to PrPsc (64,65).
We suggest that prion peptide may have valuable role as a
therapeutic strategy for prion diseases though PrP (106–126) is not
equivalent to PrPsc. We found that the 106–126 sequence of the
prion protein is an efficient model for in vitro study of
prion-induced cell death, as it results in rapid depolarization of
mitochondrial membranes. Based on these observations, it was
suggested that PrP (106–126) is a major contributor to the
physicochemical and pathogenic properties of PrPsc. Although the
function and mechanism of prion protein are still obscure, numerous
studies have suggested that prior formation of prions are initiated
by accumulation of PrPsc in the brain, analogous to amyloid-β in
Alzheimer's disease (66–68).
Prior research has suggested that chemical drugs or
plant extracts protect neurons during autophagy in
neurodegenerative disease (69–73).
The biological role of autophagy in neurodegenerative diseases,
however, is debated. Nah et al (39) have suggested that abnormal autophagy
could be induced by amyloid-β or accumulation of misfolded prion
proteins in neuronal cells. In this study, we limited ourselves to
experiments with PrP (106–126) in neuronal cells, not animal
models. Accordingly, the prion peptide's autophagic effects have
yet to be demonstrated in vivo, and further studies will be
needed to discover whether prion peptide induces the neurotoxic
effect that occurs during autophagy in mice models.
Most experiments were conducted using primary
neuronal cells. In some experiments, SK-N-SH neuroblastoma cells
were used (as described in the figure legends) as it proved
difficult to observe fluorescent proteins in primary neuronal cells
were, the cells was crushed and their shape destroyed, and as cells
could no longer be propagated once they were terminally
differentiated into mature neurons (74). SK-N-SH neuroblastoma cells, as the
parental line of SH-SY5Y, have been widely used to characterize
neuron-like behavior when investigating potential neuroprotective
activity in in vitro models (74,75).
To obtain an understanding of their interplay, we
examined the impact of troglitazone treatment on PrP-induced cell
death, PPARγ, and autophagy flux in neuronal cells. This study
revealed that PrP-mediated autophagic cell death could be prevented
by troglitazone via neuronal PPARγ activation. Based on this study,
regulating autophagy flux may be an effective means of mitigating
the damage caused by prion diseases. Further studies could
determine optimal strategies for the modulation of ROS for
neurodegeneration treatment as ROS may be involved in both
autophagy flux and prion diseases.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by a Korean Grant from the
National Research Foundation (NRF) funded by the Ministry of
Education (grant no. 2019R1A2B5B02069765) and ‘Research Base
Construction Fund Support Program’ funded by Jeonbuk National
University in 2020.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JHM, JMH and SYP designed and executed the study,
analyzed the data and wrote the manuscript. JHM and SYP confirm the
authenticity of all the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
This project received approval from the
Institutional Review Board of Jeonbuk National University (approval
no. CBNU-2019-00113).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Peretz D, Williamson RA, Kaneko K, Vergara
J, Leclerc E, Schmitt-Ulms G, Mehlhorn IR, Legname G, Wormald MR,
Rudd PM, et al: Antibodies inhibit prion propagation and clear cell
cultures of prion infectivity. Nature. 412:739–743. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Aguzzi A: Prion diseases of humans and
farm animals: Epidemiology, genetics, and pathogenesis. J
Neurochem. 97:1726–1739. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Scheckel C and Aguzzi A: Prions, prionoids
and protein misfolding disorders. Nat Rev Genet. 19:405–418. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Aguzzi A and Heikenwalder M: Prion
diseases: Cannibals and garbage piles. Nature. 423:127–129. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Forloni G, Chiesa R, Bugiani O, Salmona M
and Tagliavini F: Review: PrP 106-126-25 years after. Neuropathol
Appl Neurobiol. 45:430–440. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fioriti L, Angeretti N, Colombo L, De
Luigi A, Colombo A, Manzoni C, Morbin M, Tagliavini F, Salmona M,
Chiesa R and Forloni G: Neurotoxic and gliotrophic activity of a
synthetic peptide homologous to Gerstmann-Sträussler-Scheinker
disease amyloid protein. J Neurosci. 27:1576–1583. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fioriti L, Quaglio E, Massignan T, Colombo
L, Stewart RS, Salmona M, Harris DA, Forloni G and Chiesa R: The
neurotoxicity of prion protein (PrP) peptide 106–126 is independent
of the expression level of PrP and is not mediated by abnormal PrP
species. Mol Cell Neurosci. 28:165–176. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Villa A, Mark AE, Saracino GA, Cosentino
U, Pitea D, Moro G and Salmona M: Conformational polymorphism of
the PrP106-126 peptide in different environments: A molecular
dynamics study. J Phys Chem B. 110:1423–1428. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Corsaro A, Thellung S, Villa V, Principe
DR, Paludi D, Arena S, Millo E, Schettini D, Damonte G, Aceto A, et
al: Prion protein fragment 106–126 induces a p38 MAP
kinase-dependent apoptosis in SH-SY5Y neuroblastoma cells
independently from the amyloid fibril formation. Ann NY Acad Sci.
1010:610–622. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Troglitazone, . LiverTox: Clinical and
Research Information on Drug-Induced Liver Injury. National
Institute of Diabetes and Digestive and Kidney Diseases; Bethesda,
MD: 2012
|
|
11
|
Frias JP, Yu JG, Kruszynska YT and Olefsky
JM: Metabolic effects of troglitazone therapy in type 2 diabetic,
obese, and lean normal subjects. Diabetes care. 23:64–69. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chandra V, Huang P, Hamuro Y, Raghuram S,
Wang Y, Burris TP and Rastinejad F: Structure of the intact
PPAR-gamma-RXR-nuclear receptor complex on DNA. Nature.
456:350–356. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lehmann JM, Moore LB, Smith-Oliver TA,
Wilkison WO, Willson TM and Kliewer SA: An antidiabetic
thiazolidinedione is a high affinity ligand for peroxisome
proliferator-activated receptor gamma (PPAR gamma). J Biol Chem.
270:12953–12956. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Olefsky JM and Saltiel AR: PPAR gamma and
the treatment of insulin resistance. Trends Endocrinol Metab.
11:362–368. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hong OY, Youn HJ, Jang HY, Jung SH, Noh
EM, Chae HS, Jeong YJ, Kim W, Kim CH and Kim JS: Troglitazone
inhibits matrix metalloproteinase-9 expression and invasion of
breast cancer cell through a peroxisome proliferator-activated
receptor γ-dependent mechanism. J Breast Cancer. 21:28–36. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rosen ED and Spiegelman BM: PPARgamma: A
nuclear regulator of metabolism, differentiation, and cell growth.
J Biol Chem. 276:37731–37734. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tontonoz P, Hu E and Spiegelman BM:
Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a
lipid-activated transcription factor. Cell. 79:1147–1156. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jain MR, Giri SR, Trivedi C, Bhoi B, Rath
A, Vanage G, Vyas P, Ranvir R and Patel PR: Saroglitazar, a novel
PPARα/γ agonist with predominant PPARα activity, shows
lipid-lowering and insulin-sensitizing effects in preclinical
models. Pharmacol Res Perspect. 3:e001362015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Certo M, Endo Y, Ohta K, Sakurada S,
Bagetta G and Amantea D: Activation of RXR/PPARγ underlies
neuroprotection by bexarotene in ischemic stroke. Pharmacol Res.
102:298–307. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chiang MC, Cheng YC, Nicol CJ, Lin KH, Yen
CH, Chen SJ and Huang RN: Rosiglitazone activation of
PPARγ-dependent signaling is neuroprotective in mutant huntingtin
expressing cells. Exp Cell Res. 338:183–193. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lecca D, Nevin DK, Mulas G, Casu MA, Diana
A, Rossi D, Sacchetti G, Fayne D and Carta AR: Neuroprotective and
anti-inflammatory properties of a novel non-thiazolidinedione PPARγ
agonist in vitro and in MPTP-treated mice. Neuroscience. 302:23–35.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Thouennon E, Cheng Y, Falahatian V, Cawley
NX and Loh YP: Rosiglitazone-activated PPARγ induces neurotrophic
factor-α1 transcription contributing to neuroprotection. J
Neurochem. 134:463–470. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jiang P and Mizushima N: Autophagy and
human diseases. Cell Res. 24:69–79. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lum JJ, Bauer DE, Kong M, Harris MH, Li C,
Lindsten T and Thompson CB: Growth factor regulation of autophagy
and cell survival in the absence of apoptosis. Cell. 120:237–248.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mizushima N: Autophagy: Process and
function. Genes Dev. 21:2861–2873. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy (3rd edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shintani T and Klionsky DJ: Autophagy in
health and disease: A double-edged sword. Science. 306:990–995.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bjørkøy G, Lamark T, Brech A, Outzen H,
Perander M, Overvatn A, Stenmark H and Johansen T: p62/SQSTM1 forms
protein aggregates degraded by autophagy and has a protective
effect on huntingtin-induced cell death. J Cell Biol. 171:603–614.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nakai A, Yamaguchi O, Takeda T, Higuchi Y,
Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, et
al: The role of autophagy in cardiomyocytes in the basal state and
in response to hemodynamic stress. Nat Med. 13:619–624. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Su H, Li F, Ranek MJ, Wei N and Wang X:
COP9 signalosome regulates autophagosome maturation. Circulation.
124:2117–2128. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Moon JH, Lee JH, Nazim UM, Lee YJ, Seol
JW, Eo SK, Lee JH and Park SY: Human prion protein-induced
autophagy flux governs neuron cell damage in primary neuron cells.
Oncotarget. 7:29989–30002. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Beaudoin GM III, Lee SH, Singh D, Yuan Y,
Ng YG, Reichardt LF and Arikkath J: Culturing pyramidal neurons
from the early postnatal mouse hippocampus and cortex. Nat Protoc.
7:1741–1754. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cho DH, Lee EJ, Kwon KJ, Shin CY, Song KH,
Park JH, Jo I and Han SH: Troglitazone, a thiazolidinedione,
decreases tau phosphorylation through the inhibition of
cyclin-dependent kinase 5 activity in SH-SY5Y neuroblastoma cells
and primary neurons. J Neurochem. 126:685–695. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Uryu S, Harada J, Hisamoto M and Oda T:
Troglitazone inhibits both post-glutamate neurotoxicity and
low-potassium-induced apoptosis in cerebellar granule neurons.
Brain Res. 924:229–236. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Redmann M, Benavides GA, Berryhill TF,
Wani WY, Ouyang X, Johnson MS, Ravi S, Barnes S, Darley-Usmar VM
and Zhang J: Inhibition of autophagy with bafilomycin and
chloroquine decreases mitochondrial quality and bioenergetic
function in primary neurons. Redox Biol. 11:73–81. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shacka JJ, Klocke BJ, Shibata M, Uchiyama
Y, Datta G, Schmidt RE and Roth KA: Bafilomycin A1 inhibits
chloroquine-induced death of cerebellar granule neurons. Mol
Pharmacol. 69:1125–1136. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wojtowicz AK, Szychowski KA and Kajta M:
PPAR-γ agonist GW1929 but not antagonist GW9662 reduces
TBBPA-induced neurotoxicity in primary neocortical cells. Neurotox
Res. 25:311–322. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Martin HL, Mounsey RB, Mustafa S, Sathe K
and Teismann P: Pharmacological manipulation of peroxisome
proliferator-activated receptor γ (PPARγ) reveals a role for
anti-oxidant protection in a model of Parkinson's disease. Exp
Neurol. 235:528–538. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nah J, Pyo JO, Jung S, Yoo SM, Kam TI,
Chang J, Han J, Soo A, An S, Onodera T and Jung YK: BECN1/Beclin 1
is recruited into lipid rafts by prion to activate autophagy in
response to amyloid β 42. Autophagy. 9:2009–2021. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Saiki S, Sasazawa Y, Imamichi Y, Kawajiri
S, Fujimaki T, Tanida I, Kobayashi H, Sato F, Sato S, Ishikawa K,
et al: Caffeine induces apoptosis by enhancement of autophagy via
PI3K/Akt/mTOR/p70S6K inhibition. Autophagy. 7:176–187. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kumar D, Shankar S and Srivastava RK:
Rottlerin-induced autophagy leads to the apoptosis in breast cancer
stem cells: molecular mechanisms. Mol Cancer. 12:1712013.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Arsikin K, Kravic-Stevovic T, Jovanovic M,
Ristic B, Tovilovic G, Zogovic N, Bumbasirevic V, Trajkovic V and
Harhaji-Trajkovic L: Autophagy-dependent and -independent
involvement of AMP-activated protein kinase in 6-hydroxydopamine
toxicity to SH-SY5Y neuroblastoma cells. Biochim Biophys Acta.
1822:1826–1836. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lee JH, Yoon YM, Han YS, Jung SK and Lee
SH: Melatonin protects mesenchymal stem cells from
autophagy-mediated death under ischaemic ER-stress conditions by
increasing prion protein expression. Cell Prolif. 52:e125452019.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jiang C, Ting AT and Seed B: PPAR-gamma
agonists inhibit production of monocyte inflammatory cytokines.
Nature. 391:82–86. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ricote M, Li AC, Willson TM, Kelly CJ and
Glass CK: The peroxisome proliferator-activated receptor-gamma is a
negative regulator of macrophage activation. Nature. 391:79–82.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Combs CK, Johnson DE, Karlo JC, Cannady SB
and Landreth GE: Inflammatory mechanisms in Alzheimer's disease:
Inhibition of beta-amyloid-stimulated proinflammatory responses and
neurotoxicity by PPARgamma agonists. J Neurosci. 20:558–567. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Beheshti F, Hosseini M, Hashemzehi M,
Soukhtanloo M, Khazaei M and Shafei MN: The effects of PPAR-γ
agonist pioglitazone on hippocampal cytokines, brain-derived
neurotrophic factor, memory impairment, and oxidative stress status
in lipopolysaccharide-treated rats. Iran J Basic Med Sci.
22:940–948. 2019.PubMed/NCBI
|
|
48
|
Zhang H, Gong M and Luo X:
Methoxytetrahydro-2H-pyran-2-yl)methyl benzoate inhibits spinal
cord injury in the rat model via PPAR-γ/PI3K/p-Akt activation.
Environ Toxicol. 35:714–721. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
de Brito TV, Júnior GJD, da Cruz Júnior
JS, Silva RO, da Silva Monteiro CE, Franco AX, Vasconcelos DFP, de
Oliveira JS, da Silva Costa DV, Carneiro TB, et al: Gabapentin
attenuates intestinal inflammation: Role of PPAR-gamma receptor.
Eur J Pharmacol. 873:1729742020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Khasabova IA, Khasabov SG, Olson JK,
Uhelski ML, Kim AH, Albino-Ramírez AM, Wagner CL, Seybold VS and
Simone DA: Pioglitazone, a PPARγ agonist, reduces cisplatin-evoked
neuropathic pain by protecting against oxidative stress. Pain.
160:688–701. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Aoun P, Watson DG and Simpkins JW:
Neuroprotective effects of PPARgamma agonists against oxidative
insults in HT-22 cells. Eur J Pharmacol. 472:65–71. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Jodeiri Farshbaf M, Forouzanfar M, Ghaedi
K, Kiani-Esfahani A, Peymani M, Shoaraye Nejati A, Izadi T,
Karbalaie K, Noorbakhshnia M, Rahgozar S, et al: Nurr1 and PPARγ
protect PC12 cells against MPP(+) toxicity: Involvement of
selective genes, anti-inflammatory, ROS generation, and
antimitochondrial impairment. Mol Cell Biochem. 420:29–42. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Mahmood DFD, Jguirim-Souissi I, Khadija
EH, Blondeau N, Diderot V, Amrani S, Slimane MN, Syrovets T, Simmet
T and Rouis M: Peroxisome proliferator-activated receptor gamma
induces apoptosis and inhibits autophagy of human monocyte-derived
macrophages via induction of cathepsin L: Potential role in
atherosclerosis. J Biol Chem. 286:28858–28866. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yao J, Zheng K and Zhang X: Rosiglitazone
exerts neuroprotective effects via the suppression of neuronal
autophagy and apoptosis in the cortex following traumatic brain
injury. Mol Med Rep. 12:6591–6597. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Gao N, Yao X, Jiang L, Yang L, Qiu T, Wang
Z, Pei P, Yang G, Liu X and Sun X: Taurine improves low-level
inorganic arsenic-induced insulin resistance by activating
PPARγ-mTORC2 signalling and inhibiting hepatic autophagy. J Cell
Physiol. 234:5143–5152. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hu C, Chen C, Chen J, Xiao K, Wang J, Shi
Q, Ma Y, Gao LP, Wu YZ, Liu L, et al: The low levels of nerve
growth factor and its upstream regulatory kinases in prion
infection is reversed by resveratrol. Neurosci Res. 162:52–62.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Jeong JK, Moon MH, Bae BC, Lee YJ, Seol
JW, Kang HS, Kim JS, Kang SJ and Park SY: Autophagy induced by
resveratrol prevents human prion protein-mediated neurotoxicity.
Neurosci Res. 73:99–105. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Tagliavini F, Forloni G, D'Ursi P, Bugiani
O and Salmona M: Studies on peptide fragments of prion proteins.
Adv Protein Chem. 57:171–201. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ilitchev AI, Giammona MJ, Olivas C, Claud
SL, Lazar Cantrell KL, Wu C, Buratto SK and Bowers MT:
Hetero-oligomeric amyloid assembly and mechanism: Prion fragment
PrP(106–126) catalyzes the islet amyloid polypeptide β-hairpin. J
Am Chem Soc. 140:9685–9695. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Singh N, Gu Y, Bose S, Kalepu S, Mishra RS
and Verghese S: Prion peptide 106–126 as a model for prion
replication and neurotoxicity. Front Biosci. 7:a60–a71. 2002.
View Article : Google Scholar
|
|
61
|
Herrmann US, Sonati T, Falsig J, Reimann
RR, Dametto P, O'Connor T, Li B, Lau A, Hornemann S, Sorce S, et
al: Prion infections and anti-PrP antibodies trigger converging
neurotoxic pathways. PLoS Pathog. 11:e10046622015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Tremblay P, Ball HL, Kaneko K, Groth D,
Hegde RS, Cohen FE, DeArmond SJ, Prusiner SB and Safar JG: Mutant
PrPSc conformers induced by a synthetic peptide and several prion
strains. J Virol. 78:2088–2099. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zhu C, Herrmann US, Li B, Abakumova I,
Moos R, Schwarz P, Rushing EJ, Colonna M and Aguzzi A: Triggering
receptor expressed on myeloid cells-2 is involved in prion-induced
microglial activation but does not contribute to prion pathogenesis
in mouse brains. Neurobiol Aging. 36:1994–2003. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Giaccone G and Moda F: PMCA applications
for prion detection in peripheral tissues of patients with variant
creutzfeldt-jakob disease. Biomolecules. 10:4052020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Haley NJ and Hoover EA: Chronic wasting
disease of cervids: Current knowledge and future perspectives. Annu
Rev Anim Biosci. 3:305–325. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kaufman SK and Diamond MI: Prion-like
propagation of protein aggregation and related therapeutic
strategies. Neurotherapeutics. 10:371–382. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Aguzzi A and Calella AM: Prions: Protein
aggregation and infectious diseases. Physiol Rev. 89:1105–1152.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zhu C, Li B, Frontzek K, Liu Y and Aguzzi
A: SARM1 deficiency up-regulates XAF1, promotes neuronal apoptosis,
and accelerates prion disease. J Exp Med. 216:743–756. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Jeong JK and Park SY: Melatonin regulates
the autophagic flux via activation of alpha-7 nicotinic
acetylcholine receptors. J Pineal Res. 59:24–37. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Damme M, Suntio T, Saftig P and Eskelinen
EL: Autophagy in neuronal cells: General principles and
physiological and pathological functions. Acta Neuropathol.
129:337–362. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Lee JH, Jeong JK and Park SY:
Sulforaphane-induced autophagy flux prevents prion protein-mediated
neurotoxicity through AMPK pathway. Neuroscience. 278:31–39. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Ciechanover A and Kwon YT: Degradation of
misfolded proteins in neurodegenerative diseases: Therapeutic
targets and strategies. Exp Mol Med. 47:e1472015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Nakagaki T, Satoh K, Ishibashi D, Fuse T,
Sano K, Kamatari YO, Kuwata K, Shigematsu K, Iwamaru Y, Takenouchi
T, et al: FK506 reduces abnormal prion protein through the
activation of autolysosomal degradation and prolongs survival in
prion-infected mice. Autophagy. 9:1386–1394. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Kovalevich J and Langford D:
Considerations for the use of SH-SY5Y neuroblastoma cells in
neurobiology. Methods Mol Biol. 1078:9–21. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Forster JI, Köglsberger S, Trefois C, Boyd
O, Baumuratov AS, Buck L, Balling R and Antony PM: Characterization
of differentiated SH-SY5Y as neuronal screening model reveals
increased oxidative vulnerability. J Biomol Screen. 21:496–509.
2016. View Article : Google Scholar : PubMed/NCBI
|