Introduction
Influenza A virus (IAV) is an enveloped
negative-stranded RNA virus and its infection can result in both
respiratory and constitutional effects, such as chills, headache,
fever and general pain (1). In
total, there are >500,000 IAV-associated human deaths worldwide
each year, and several animal species with an elevated fatality
rate from the virus have emerged (2). High levels of genetic diversity are
the main cause of IAV pandemics, which represent a burden to human
health (3,4). Thus, it is important to explore new
strategies against viral replication.
Innate immunity is the first barrier to the invasion
of external pathogens. During viral infection, the innate immune
system recognizes various pattern recognition receptors and then
triggers downstream signal transduction, leading to the production
of cytokines, especially type-I IFNα/β (5–7).
Several studies have demonstrated that type-I IFNs effectively
protect the host from IAV infection (8,9).
However, IAV can utilize a number of strategies to escape host
innate immunity. For example, non-structural protein 1 (NS1) of IAV
can inhibit the transcriptional activity of virus-induced
interferon regulatory factor (IRF) 3, activator protein 1 and NF-κB
signaling, disrupting the host antiviral immune response (10–12).
In addition, Hayashi et al (13) demonstrated that a novel viral
protein expressed by ribosomal frameshifting, PA-X, contributes to
increased viral replication through the inhibition of host innate
and acquired immune responses in mice. In addition to these
proteins encoded by the virus itself, the virus uses host cell
components to escape from the antiviral response, which restricts
viral replication (13). However,
how IAV counteracts the antiviral activity of type-I IFN remains
poorly characterized.
MicroRNAs (miRNAs/miRs) are single-stranded
non-coding RNA molecules that negatively regulate gene expression
by binding to the 3′-untranslated region (UTR) of their target
genes at the post-transcriptional level (14). Increasing evidence has demonstrated
that miRNA suppresses type-I IFN production and inactivates the
JAK-STAT pathway during infections with various types of virus
(15–17). For example, infection with
enterovirus can induce miR-146a expression, which suppresses the
type-I IFN response of the host cell (18). Chen et al (19) reported that miR-21 was upregulated
during hepatitis C virus (HCV) infection, which promoted viral
replication by suppressing the type-I IFN-mediated antiviral
response in hepatocytes. For IAV, Zhang et al (20) demonstrated that miR-132-3p
suppressed the type-I IFN response by targeting IRF1 to facilitate
hemagglutinin (H)1 neuraminidase (N)1 IAV infection. Zhu et
al (21) demonstrated that
miR-30e could inhibit dengue virus replication by upregulating IFN
and IFN-stimulated gene (ISG) production. However, whether there
are more miRNAs involved in regulating the innate immune response
of host cells against IAV infection remains to be further
explored.
It has previously been shown that miR-221 influences
viral replication in several viruses, such as human cytomegalovirus
(HCMV) and human papillomavirus 16 E1-E2, and that miR-221
regulates innate antiviral immunity through IFNα/β (22,23).
In addition, Du et al (24)
also demonstrated that miR-221 negatively regulated the innate
immune response and promoted vesicular stomatitis virus and herpes
simplex virus type 1 replication. However, the role of miR-221 in
IAV infection remains unclear. Therefore, miR-221 was investigated
in the present study.
In the current study, the main research purpose was
to elucidate the role and molecular mechanism of miR-221 in H1N1
IAV replication in the host cells. The miRNA expression profile of
H1N1 IAV-infected A549 cells was investigated using a microarray
assay. Subsequently, the role of miR-221 on H1N1 IAV replication
was examined, and mechanisms underpinning the action of miR-221 in
the immune response to IAV infection were investigated. The present
findings may improve provide insight into the mechanism of IAV
immune escape and highlight miR-221 as a potential novel target for
the treatment of IAV infection.
Materials and methods
Cell culture
A549 cells were obtained from the American Type
Culture Collection and cultured in minimum Eagle's medium (MEM;
Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal
bovine serum, 100 U/ml penicillin and 100 µg/ml streptomycin at
37°C in a 5% CO2 atmosphere.
Viral infection and plaque assay
The IAV/Jingfang/01/1986 (H1N1) strain was obtained
from the Chinese Center for Disease Control and Prevention and was
propagated in A549 cells. A549 cells were infected with H1N1 at a
multiplicity of infection (MOI) of 0.1. After 1 h of infection, the
medium was discarded and the cells were washed with the free-serum
medium, and then MEM with 1 µg/ml tosylsulfonyl phenylalanyl
chloromethyl ketone (TPCK)-trypsin (Sigma-Aldrich; Merck KGaA) was
added into duplicate wells. The cells uninfected with H1N1 were
used as control (Mock) group. The copy number of virions were
determined using qPCR detection of the matrix protein 2 (M2) gene,
as later described. Viral titers in the supernatants were
determined using a standard plaque assay. Briefly, A549 cells were
seeded into 12-well plates (2×105 cells/well) and
infected with H1N1 in 1:10 dilutions for 60 min at 37°C. Then, 1%
low-melting-point agarose (Sigma-Aldrich; Merck KGaA) in 500 µl MEM
(Gibco; Thermo Fisher Scientific, Inc.) containing 1 µg/ml TPCK
trypsin were added to the wells. Plates were incubated at 37°C with
5% CO2 for 72 h and then were fixed for 2 h at room
temperature with 4% paraformaldehyde. Fixed cells were washed
extensively with PBS before staining with crystal violet (0.1% in
10% ethanol) for 30 min at room temperature.
Microarray analysis
RNA was extracted from A549 cells infected and
uninfected with H1N1 influenza virus using a mirVana™ miRNA
Isolation kit (Thermo Fisher Scientific, Inc.), then the RNA
concentration was analyzed using a NanoDrop™ 2000 spectrophotometer
(NanoDrop Technologies; Thermo Fisher Scientific, Inc.). Total RNA
(1 µg) was labeled using the miRCURY LNA™ Hy3™/Hy5™ Power labeling
kit (cat no. 208032-A; Exiqon A/S). Subsequently the samples were
hybridized on the miRCURY™ LNA Array (version 16.0; cat. no.
208040; Exiqon A/S) according to the manufacturer's protocol. The
procedure and imaging processes were performed as described
previously (20).
Reverse transcription-qPCR
(RT-qPCR)
Total RNA was extracted from cells using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.).
RNA was then reverse transcribed into cDNA using the PrimeScript™
RT reagent kit (cat no. RR047A; Takara Bio, Inc.) or the TaqMan™
MicroRNA Reverse Transcription kit (cat no. 4366596; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol,
respectively. For detection of miR-221 and mRNAs, qPCR was
conducted using SYBR® PrimeScript™ RT-PCR kit (Takara
Bio, Inc.) on ABI 7900HT Fast Real-Time PCR system (Applied
Biosystems; Thermo Fisher Scientific, Inc.). U6 and GAPDH were used
as internal controls for detecting miR-221 and mRNA targets,
respectively. The thermocycling conditions were as follows: Initial
denaturation at 95°C for 1 min, followed by 40 cycles of 95°C for
30 sec, 58°C for 30 sec and 68°C for 2 min/kb, followed by 68°C for
10 min. The primer sequences were listed as follows: i) miR-221
forward (F), 5′-GGGAAGCTACATTGTCTGC-3′ and reverse (R),
5′-CAGTGCGTGTCGTGGAGT-3′; ii) U6 F, 5′-GCTTCGGCAGCACATATACTAAAAT-3′
and R, 5′-CGCTTCAGAATTTGCGTGTCAT-3′; iii) suppressor of cytokine
signaling 1 (SOCS1) F, 5′-CTGCGGCTTCTATTGGGGAC-3′ and R,
5′-AAAAGGCAGTCGAAGGTCTCG-3′; iv) 2′-5′-oligoadenylate synthase 2
(OAS) F, 5′-AGGTGGTAAAGGGTGGCT-3′ and R, 5′-TGCTTGACTAGGCGGATG-3′;
v) interferon-inducible double-stranded RNA-dependent protein
kinase activator A (PKR) F, 5′-AGAGTAACCGTTGGTGACATAACCT-3′ and R,
5′-GCAGCCTCTGCAGCTCTATGTT-3′; vi) viperin, F,
5′-CAAGACCGGGGAGAATACCTG-3′ and R, 5′-GCGAGAATGTCCAAATACTCACC-3′;
vii) myxovirus resistance protein 1 (MxA) F,
5′-GGGAAGGTGAAGGTCGGAGT-3′ and R, 5′-TTGAGGTCAATGAAGGGGTCA-3′;
viii) M2 F, 5′-GACCGATCCTGTCACCTCTGAC-3′ and R,
5′-AGGGCATTCTGGACAAAGCGTCTA-3′; and ix) GAPDH F,
5′-AGCTTGTCATCAACGGGAAG-3′ and R, 5′-TTTGATGTTAGTGGGGTCTCG-3′.
Changes in the expression of each gene were calculated using the
2−∆∆Cq method (25).
Transfection
A549 cells were cultured to 70% confluence, then
transfection of miR-221 mimics (100 nM), miR-221 inhibitor (100
nM), mimics negative control (NC; 100 nM), inhibitor NC (100 nM),
and small interfering RNA SOCS (si-SOCS1) (20 nM) or si-scramble
(20 nM) (Shanghai GenePharma Co., Ltd.) were performed at 37°C for
24 h using Lipofectamine® 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. The mimics NC, inhibitor NC and si-Scramble were
non-targeting. Non-transfected cells are used as the Blank group.
After 24 h the transfection, H1N1 virus were added in the cells at
an MOI of 0.1. The cells were harvested at 12 or 24 h
post-infection for testing.
ELISA
After 12 h or 24 h H1N1 virus infection, cell
culture supernatants were collected and the levels of IFN-α and
IFN-β were measured with human IFN-α kit (cat. no. 32400-1) kit and
a human IFN-β ELISA kit (cat. no. 32100-1) (both Pestka Biomedical
Laboratories, Inc.).
Luciferase reporter NF-κB activity
assays
PicTar (March 2007 release; http://pictar.mdc-berlin.de/) and TargetScan (version
7.2; http://www.targetscan.org) were used to
search for the putative targets of miR-221. The wild-type (wt)
3′-UTR of SOCS1 and the mutated (mut) sequence were inserted into
the pGL3 control vector (Promega Corporation) to construct the wt
and mut SOCS1-3′-UTR vectors, respectively. A549 cells were
transfected with miR-221 mimics/inhibitor, respective NCs and these
luciferase reporter plasmids using Lipofectamine. After 48 h,
luciferase activity was assessed using the
Dual-luciferase® Reporter Assay system (cat. no. E1910;
Promega Corporation). Renilla luciferase activity was used
to normalize firefly luciferase activity. The NF-κB activity was
assessed as previously described (26). Luciferase activity was quantified
using the aforementioned kit on a luminometer.
Western blotting
Total protein was obtained from A549 cells
transfected as aforementioned using RIPA lysis buffer (Beyotime
Institute of Biotechnology) and quantified using a BCA protein
assay kit (Pierce™; Thermo Fisher Scientific, Inc.). Next, protein
samples (40 µg/lane) in the lysates were separated by 12% SDS-PAGE
gels and transferred to PVDF membranes (GE Healthcare) followed by
incubation in a 5% skimmed milk solution for 1 h at room
temperature. Subsequently, the specific primary antibodies were
incubated in the membranes at 4°C overnight, including mouse
anti-M1 monoclonal antibody and rabbit anti-nucleoprotein (NP)
polyclonal antibody that were kindly provided by Dr Wenjun Liu
(Institute of Microbiology; Chinese Academy of Sciences), SOCS1
(cat. no. 68631; 1:1,000 dilution), NF-κB inhibitor α (IκB-α; cat.
no. 4814; 1:1,000 dilution), phosphorylated (p)-IκB-α (cat. no.
2859; 1:1,000 dilution), p-NF-κB p65 (cat. no. 3033; 1:1,000
dilution), NF-κB p65 (cat. no. 8242; 1:1,000 dilution) and β-actin
(cat. no. 3700; 1:1,000 dilution) (all Cell Signaling Technology,
Inc.). Subsequently, the corresponding anti-mouse or anti-rabbit
IgG HRP-conjugated secondary antibodies (cat no. 4409 and 3678,
respectively; both 1:2,000; Cell Signaling Technology, Inc.) were
added into the membranes for 2 h at room temperature. The protein
bands were detected using chemiluminescence with Pierce ECL kits
(Thermo Fisher Scientific, Inc.). Semi-quantification was performed
using ImageJ version 1.46 (National Institutes of Health).
Statistical analysis
Statistical analysis was performed using the SPSS
13.0 software package (SPSS Inc.). All data are presented as the
mean ± SD, and each experiment was repeated at least three times.
The two-group comparisons were conducted using unpaired Student's
t-test. Comparisons among multiple groups were performed using
one-way ANOVA followed by Tukey's post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
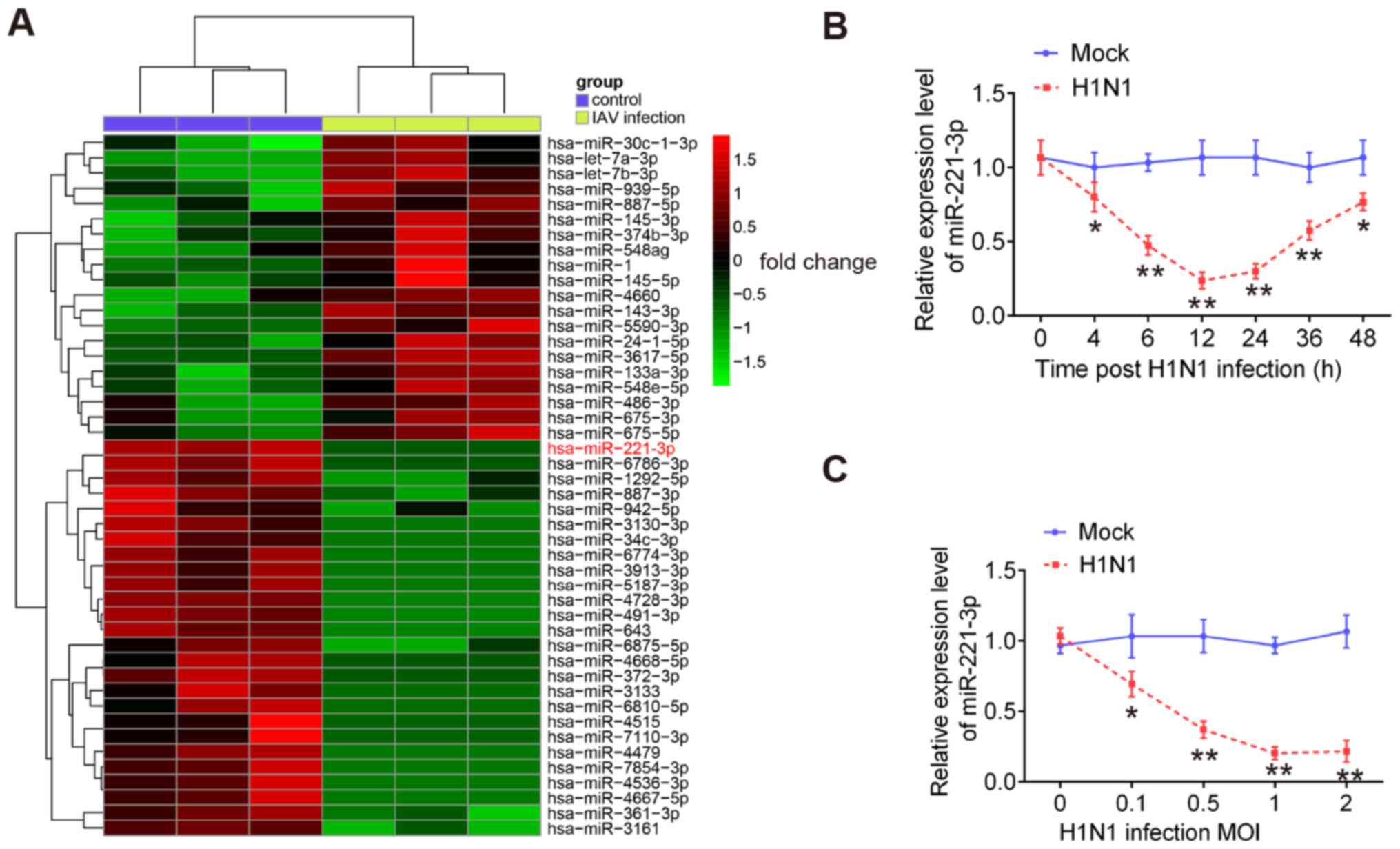
miR-221 is downregulated in H1N1
IAV-infected cells
To investigate the potential roles of miRNAs in
antiviral responses to IAV, a microarray assay was performed using
extracted RNA from H1N1-infected A549 cells or uninfected cells.
The results showed that 46 miRNAs had significantly altered
expression levels in H1N1 IAV-infected A549 cells compared with the
control cells. These miRNAs included 26 downregulated and 20
upregulated miRNAs (Fig. 1A). Among
the downregulated miRNAs, miR-221 was one of the most notably
downregulated miRNAs, which is consistent with a previous study
(27). However, the role of miR-221
in IAV infection remains unclear. Therefore, miR-221 was chosen for
subsequent experiments.
To validate the expression of miR-221 obtained from
the microarray assay, miR-221 expression in A549 cells was examined
at different time points of IAV infection. It was observed that
miR-221 expression was significantly downregulated in H1N1
IAV-infected A549 cells during the virus infection, especially in
the early stages of infection (Fig.
1B). Since the innate antiviral immune response occurs at early
stage of IAV infection (28), it
was hypothesized that miR-221 may affect IAV infection by
regulating the innate antiviral immune response. The miR-221 levels
in A549 cells infected with different MOIs of IAV were also
measured. miR-221 expression was downregulated in IAV-infected A549
cells in a dose-dependent manner (Fig.
1C). Collectively, these results suggested that miR-221 may be
involved in IAV infection.
miR-221 regulates IAV replication in
A549 cells
To investigate the biological function of miR-221 in
IAV infection, A549 cells were transfected with miR-221 mimics or
miR-221 inhibitor. As shown in Fig. 2A
and D, miR-221 expression significantly increased in A549 cells
after miR-221 mimics transfection, and significantly decreased
after miR-221 inhibitor transfection. Following by H1N1 infection,
plaque assay results showed that the viral titers and the viral
copy numbers of IAV were significantly decreased by miR-221 mimics
transfection, and increased by the miR-221 inhibitor (Fig. 2B, C, E and F). NP and M1 are two of
the most abundant proteins in the virion, which are relatively
well-conserved and display limited antigenic change (29,30).
Thus, it was investigated whether miR-221 affects the protein
levels of NP and M1 in IAV-infected A549 cells. The results showed
that the protein levels of NP and M1 were significantly reduced by
miR-221 upregulation, but increased by miR-221-knockdown (Fig. 2G and H). These results suggested
that miR-221 negatively regulated IAV replication.
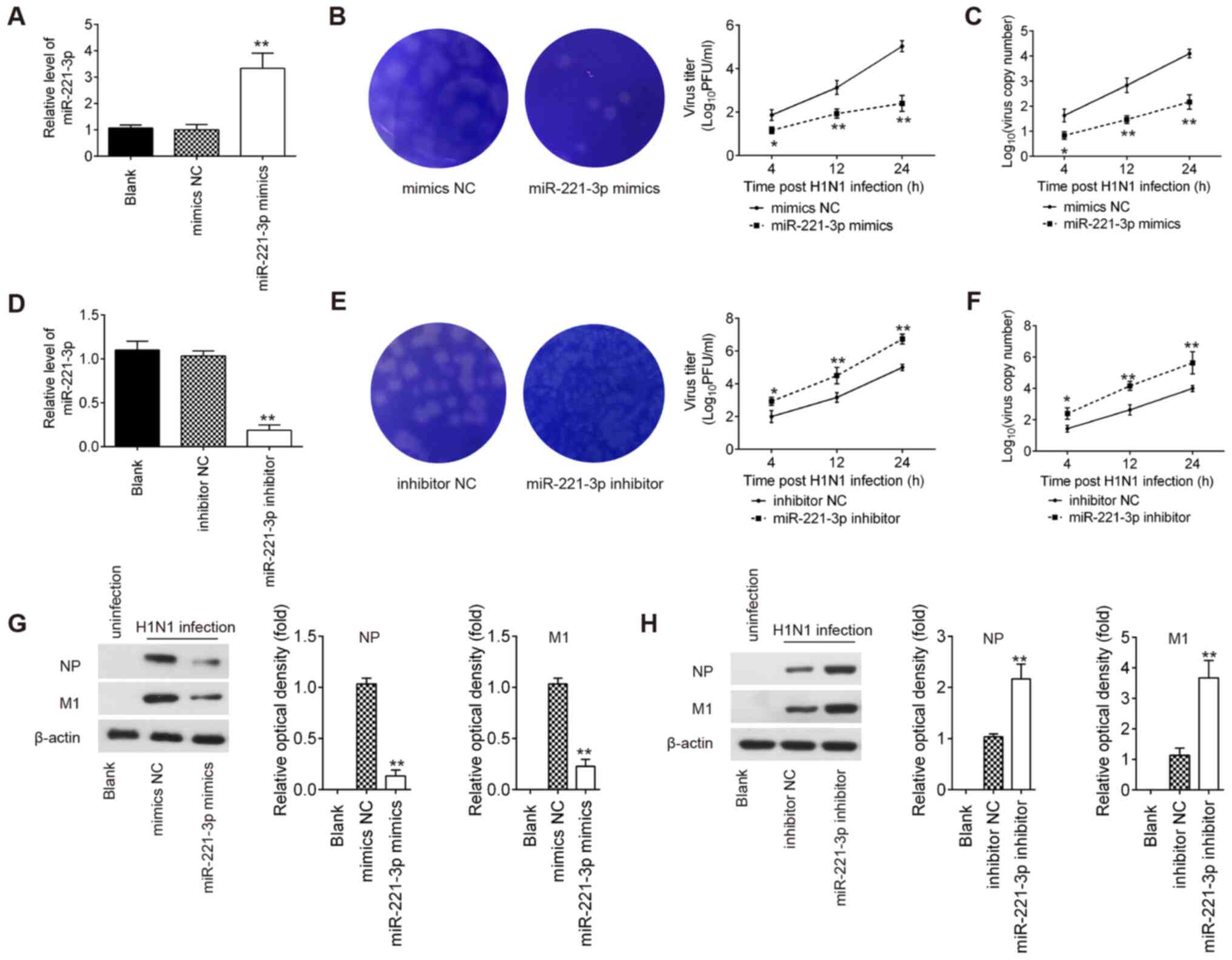 | Figure 2.miR-221 negatively regulates IAV
replication. (A) Transfection efficiency of miR-221 mimics was
determined using RT-qPCR. (B) After miR-221 mimics transfection,
viral titers in the cell cultures were determined by plaque assay
using 12-well plates. (C) After miR-221 mimics transfection, copy
number of virions of H1N1 was measured using qPCR. (D) Transfection
efficiency of miR-221 inhibitor was determined using RT-qPCR. (E)
After miR-221 inhibitor transfection, viral titers in the cell
cultures were determined via a plaque assay using 12-well plates.
(F) After miR-221 inhibitor transfection, copy number of virions of
H1N1 was measured using qPCR. (G and H) Levels of M1 and NP protein
expression levels were determined using western blotting.
*P<0.05 and **P<0.01 vs. mimics NC or inhibitor NC. NC,
negative control; IAV, influenza A virus; miR, microRNA; M1, matrix
protein 1; NP, nucleoprotein; H1N1, hemagglutinin 1 neuraminidase
1; RT-qPCR, reverse transcription-quantitative PCR; PFU,
plaque-forming unit. |
Overexpression of miR-221 increases
IAV-triggered type-I IFN production in A549 cells
It is recognized that the type-I IFN immune response
plays critical roles in the initial antiviral response (31). To test whether miR-221 regulates the
type-I IFN immune response, the expression of cytokines (IFN-α and
IFN-β) and ISGs (MxA, OAS, viperin and PKR) was measured. As shown
in Fig. 3A and B,
miR-221-overexpression enhanced the expression of IFN-α and IFN-β
at protein levels in A549 cells in response to IAV infection.
Moreover, similar results were observed for ISG mRNA expression
during IAV infection, as determined by RT-qPCR analysis (Fig. 3C-F). These data indicated that
overexpression of miR-221 facilitated the innate immune response
and subsequently inhibited IAV replication in A549 cells.
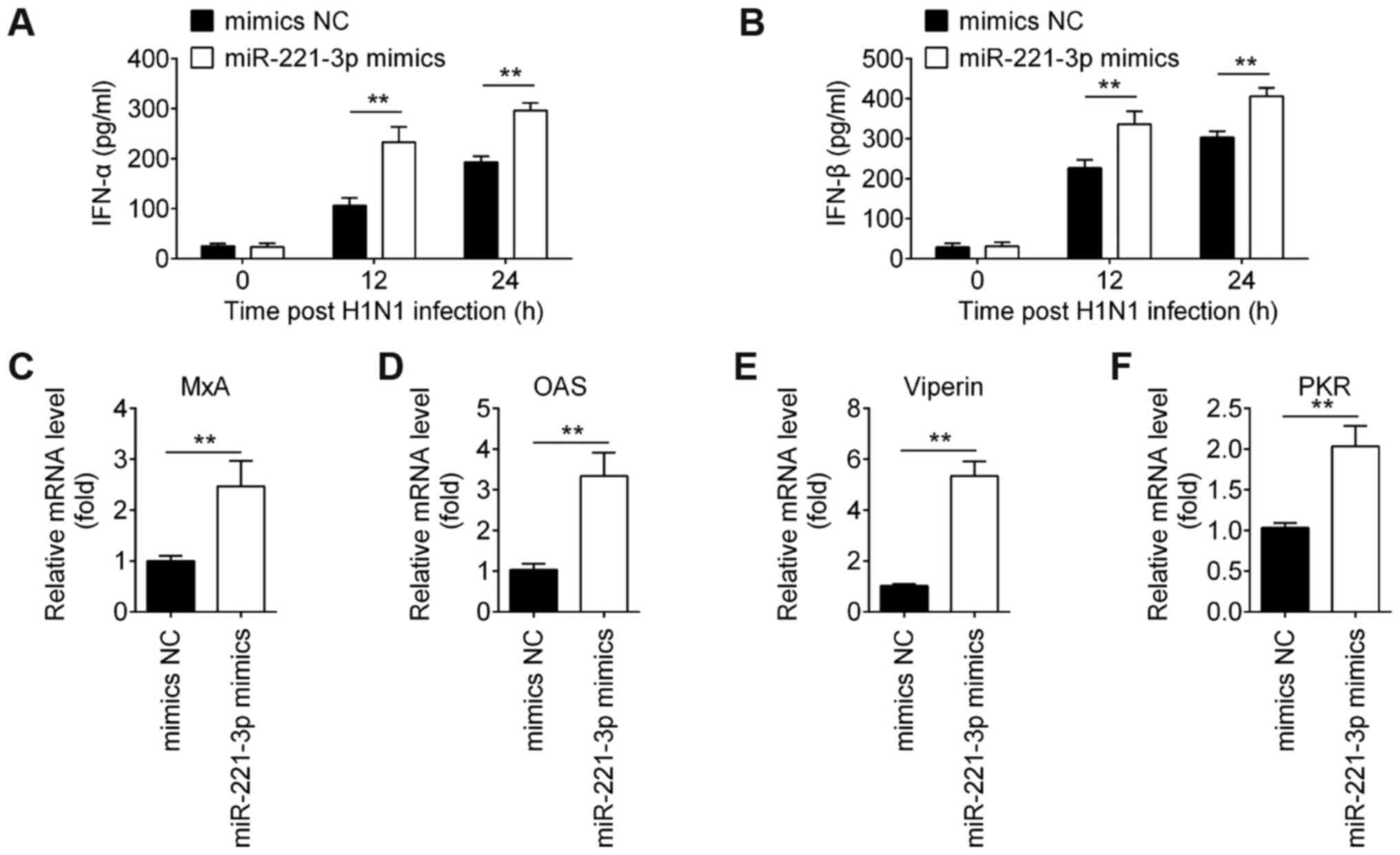 | Figure 3.miR-221 overexpression enhances
IAV-triggered type-I IFN production in A549 cells. (A and B) Cell
and supernatants were harvested at 0, 12 and 24 h post-infection,
and then ELISA assay was performed to measure IFN-α and IFN-β
expression. (C-F) Reverse transcription quantitative-PCR assay was
performed to measure the expression of IFN-stimulated genes (MxA,
OAS, viperin and PKR). Data are presented as the mean ± SD of three
individual experiments. **P<0.01 vs. mimics NC group. NC,
negative control; IAV, influenza A virus; miR, microRNA; OAS,
2′-5′-oligoadenylate synthase 2; MxA, myxovirus resistance protein
1; PKR, interferon-inducible double-stranded RNA-dependent protein
kinase activator A. |
Knockdown of miR-221 decreases
IAV-triggered type-I IFN production in A549 cells
To confirm whether miR-221 inhibition affects the
type-I IFN immune response during IAV infection, A549 cells were
transfected with miR-221 inhibitor, followed by IAV infection. As
shown in Fig. 4A and B, inhibition
of miR-221 significantly reduced IFN-α and IFN-β protein expression
levels in IAV-infected A549 cells. RT-qPCR analysis demonstrated
that transfection with the miR-221 inhibitor reduced the expression
levels of these ISGs in IAV-infected A549 cells (Fig. 4C-F). These data indicated that
miR-221 downregulation may promote IAV replication by inhibiting
the innate immune response in A549 cells.
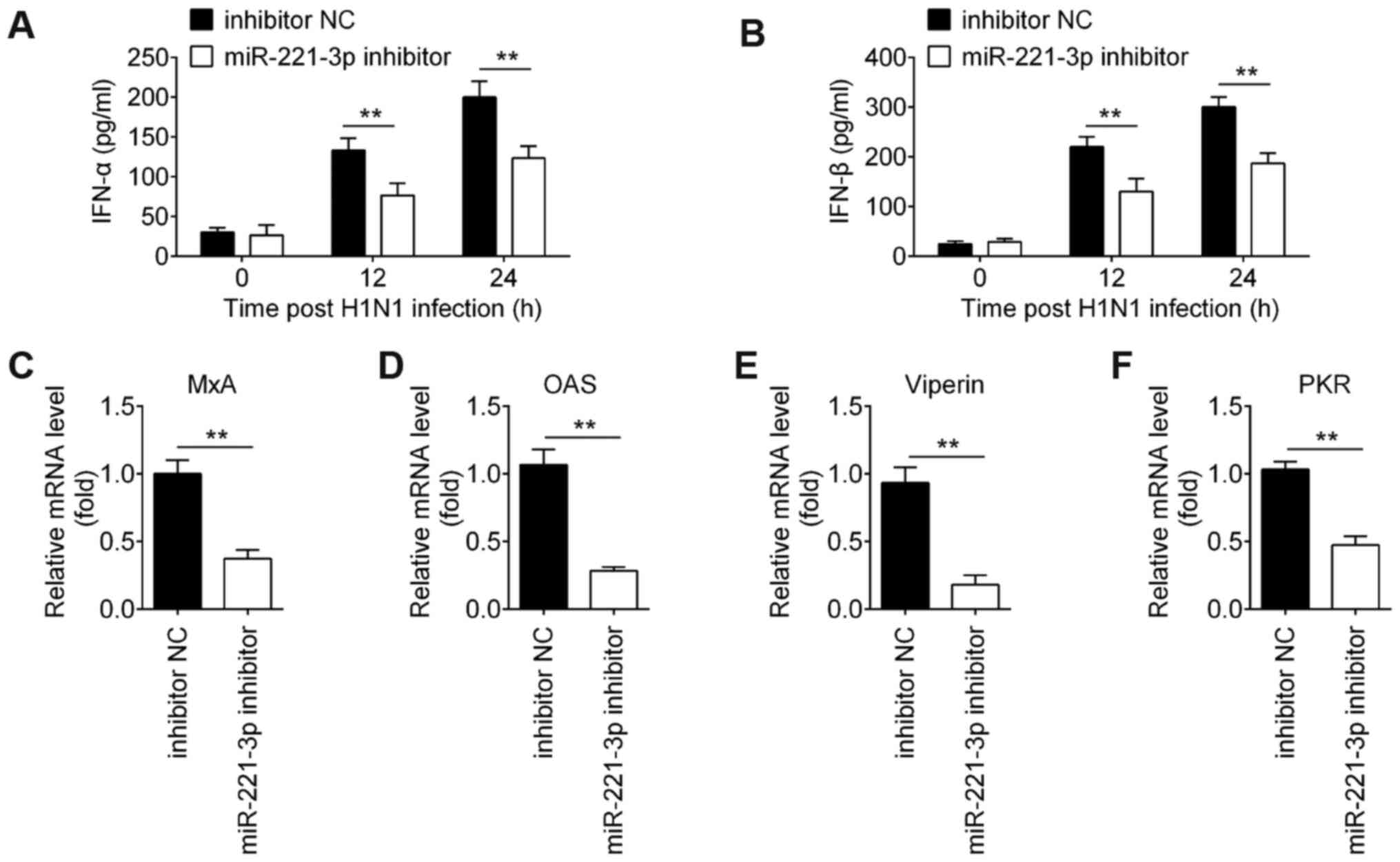 | Figure 4.miR-221-knockdown suppresses
IAV-triggered type-I IFN production in A549 cells. (A and B) Cell
and supernatant were harvested at 0, 12 and 24 h post-infection,
and then ELISA assay was performed to measure IFN-α and IFN-β
expression. (C-F) Reverse transcription quantitative-PCR was
performed to measure the expression of IFN-stimulated genes (MxA,
OAS, viperin and PKR). Data are presented as the mean ± SD of three
individual experiments. **P<0.01 vs. inhibitor NC group. NC,
negative control; IAV, influenza A virus; miR, microRNA; OAS,
2′-5′-oligoadenylate synthase 2; MxA, myxovirus influenza
resistance 1; PKR, interferon-inducible double-stranded
RNA-dependent protein kinase activator A. |
SOCS1 is a direct target of miR-221 in
IAV-infected A549 cells
To determine the possible mechanism of miR-221 in
the regulation of IAV replication, the target genes of miR-221 were
identified. Using the TargetScan and PicTar algorithms, the
complementary sequence of miR-221 was identified in the 3′-UTR of
SOCS1 mRNA (Fig. 5A). To verify
whether SOCS1 was directly regulated by miR-221, a dual-luciferase
reporter assay was performed. The results showed that miR-221
mimics significantly decreased the luciferase activity of wt-SOCS1
3′UTR compared with the mimics NC, while miR-221 inhibitor
increased the luciferase activity compared with inhibitor NC
treatment (Fig. 5B). However, the
effects produced by miR-221 were abrogated in the cells transfected
with the vector bearing the mut SOCS1 3′-UTR (Fig. 5B). To further validate this
conclusion, the mRNA and protein expression level of SOCS1 was
examined in A549 cells transfected with the miR-221 mimic or
inhibitor. SOCS1 expression was significantly decreased when
miR-221 was overexpressed, but increased at the mRNA and protein
expression levels when the miR-221 inhibitor was transfected
(Fig. 5C and D). Additionally, the
mRNA levels of SOCS1 in A549 cells during H1N1 infection was
verified. As shown in Fig. 5E,
SOCS1 mRNA expression levels were time-dependently increased and
reached the peak at 24 h after IAV infection. Subsequently, the
mRNA expression level of SOCS1 decreased slightly at 48 h. In
addition, its mRNA expression levels were also upregulated in a
dose-dependent manner (Fig. 5F).
Taken together, these data suggested that SOCS1 was a direct target
of miR-221 in A549 cells during infection of H1N1.
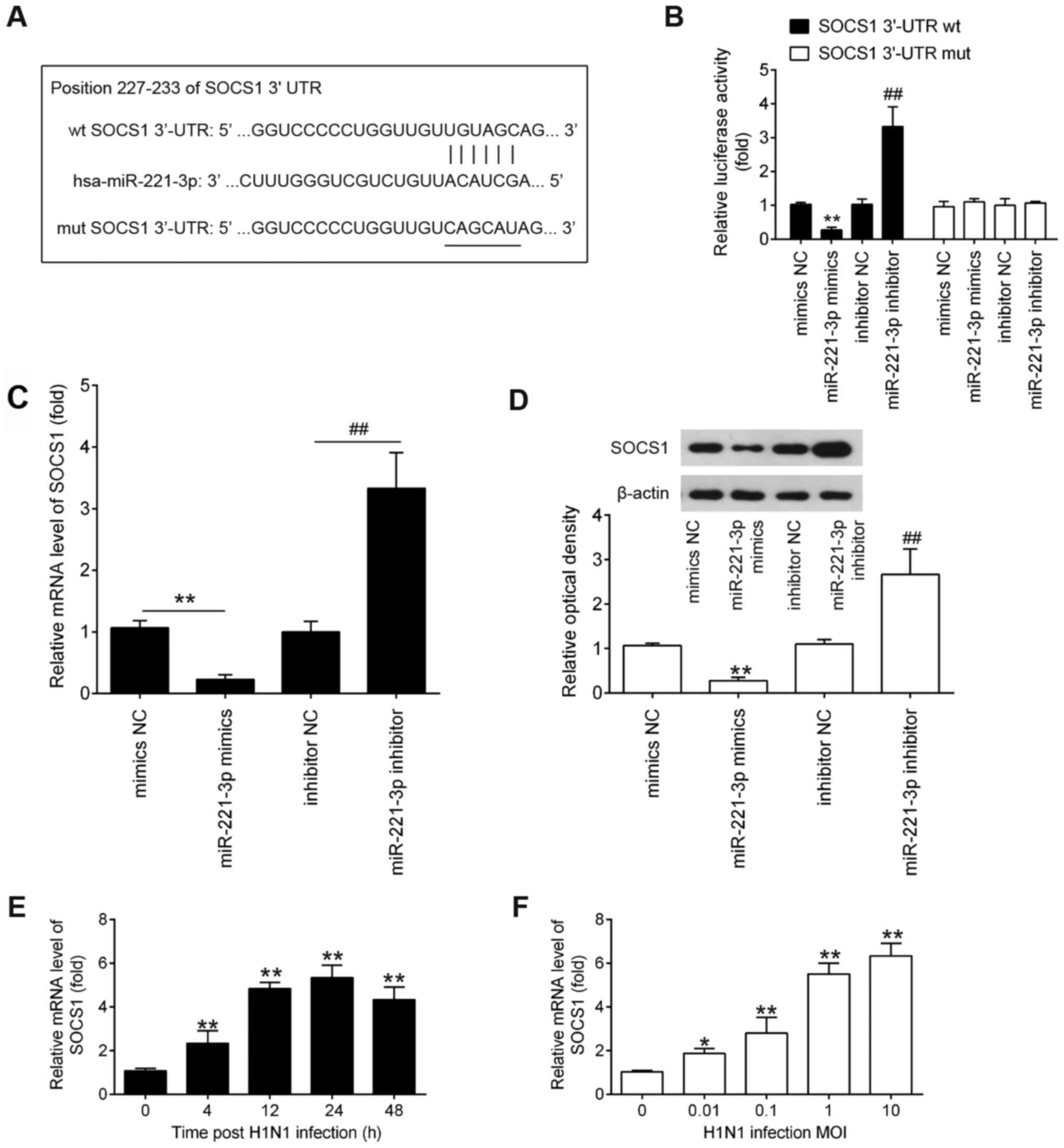 | Figure 5.SOCS1 is a direct target of miR-221.
(A) Putative binding site of miR-221 and SOCS1 with mut and wt 3′
UTRs. (B) Luciferase assay of A549 cells co-transfected with
firefly luciferase constructs containing the SOCS1 wt or mut 3′
UTRs and miR-221 mimics, mimics NC, miR-221 inhibitor or inhibitor
NC, as indicated (n=3). (C and D) mRNA and protein expression
levels of SOCS1 were measured using RT-qPCR and western blotting
after miR-221 mimics/inhibitor transfection in A549 cells. A549
cells were infected with IAV either at indicated time at (E) a MOI
of 1 or at (F) indicated MOIs for 24 h, and then the cells were
harvested for further RT-qPCR analysis of SOCS1 expression. Data
are presented as the mean ± SD of three individual experiments.
*P<0.05 and **P<0.01 vs. mock or mimics NC group;
##P<0.01 vs. inhibitor NC group. SOCS1, suppressor of
cytokine signaling 1; UTR, untranslated region; wt, wild-type; mut,
mutant; MOI, multiplicity of infection; NC, negative control; RT-q,
reverse transcription-quantitative; hsa, Homo sapiens. |
Knockdown of SOCS1 reverses the
promoting effects of miR-22-downregulation on IAV replication
Since SOCS1 is essential for the induction of type-I
IFNs during host defense (22,32),
it was hypothesized that miR-221 facilitates IAV replication by
regulating type-I IFN production by targeting SOCS1. Therefore,
miR-221 inhibitor and si-SOCS1 were co-transfected into A549 cells,
then infected with IAV. In A549 cells transfected with si-SOCS1,
SOCS1 protein expression levels were significantly reduced compared
with that in the si-Scramble-transfected cells (Fig. 6A). As shown in Fig. 6B, the viral titers of IAV were
significantly increased when miR-221 inhibitor was transfected,
compared with the inhibitor NC group. However, after
co-transfection of si-SOCS1, the viral titers were significantly
reduced compared with miR-221 inhibitor group. Subsequently, the
effects of si-SOCS1 on the expressions of IFNs and ISGs were
examined in IAV-infected A549 cells. The miR-221 inhibitor
significantly suppressed the expression levels of IFNs and ISGs,
while the inhibitory effects of miR-221 inhibitor against the
type-I IFN response were reversed by SOCS1-knockdown (Fig. 6C-H). All these data suggested that
miR-221 inhibition facilitated H1N1 IAV replication by targeting
SOCS1.
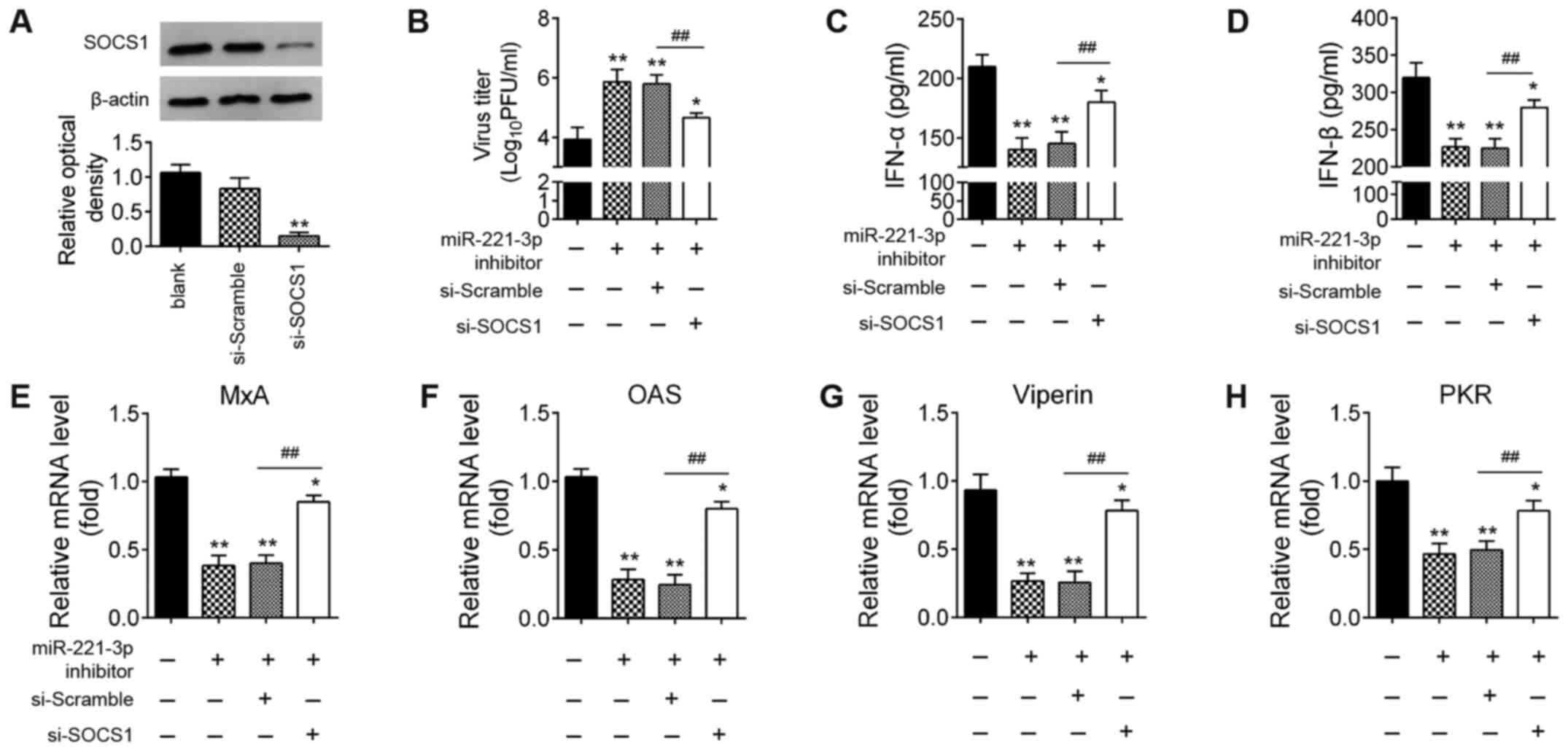 | Figure 6.miR-221 promotes IAV replication by
targeting SOCS1. A549 cells were transfected with miR-221
inhibitor, si-SOCS1, or both. (A) Expression levels of SOCS1
protein were measured using western blotting. (B) Viral titers in
the cell cultures were determined by plaque assay using 12-well
plates. (C and D) ELISA assay was performed to measure IFN-α and
IFNβ expression. (E-H) Reverse transcription-quantitative PCR assay
was performed to measure IFN-stimulated gene expression (MxA, OAS,
viperin and PKR). Data are presented as the mean ± SD of three
individual experiments. *P<0.05 and **P<0.01 vs. Blank group;
##P<0.01 vs. miR-221 inhibitor + si-scramble group.
NC, negative control; IAV, influenza A virus; miR, microRNA; OAS,
2′-5′-oligoadenylate synthase 2; SOCS1, suppressor of cytokine
signaling 1; si, small interfering; MxA, myxovirus influenza
resistance 1; PKR, interferon-inducible double-stranded
RNA-dependent protein kinase activator A; PFU, plaque-forming
unit. |
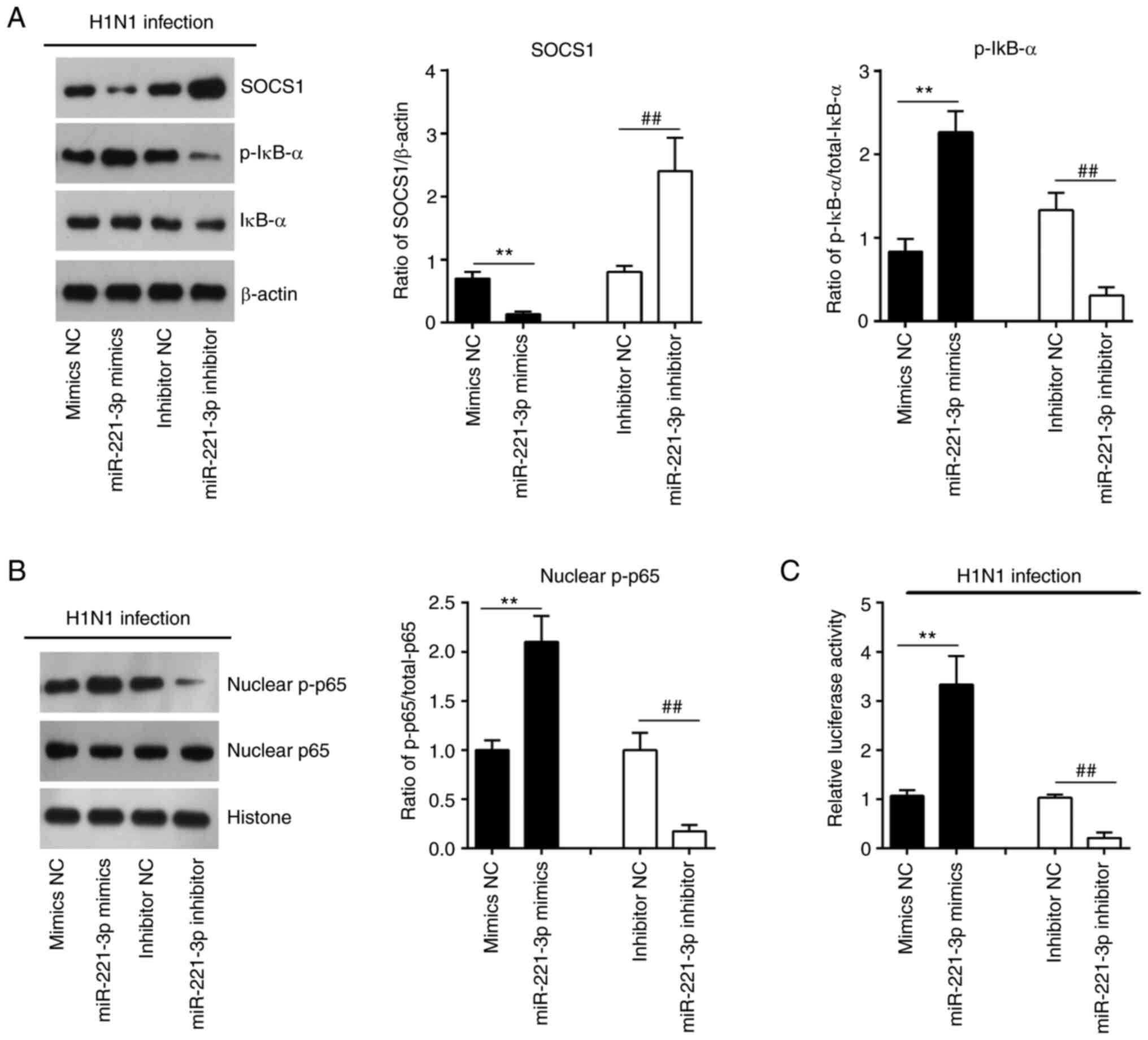
miR-221 positively regulates the
SOCS1-mediated activation of the NF-κB pathway
SOCS1 has been previously implicated in the
regulation of NF-κB pathway, which can induce the expression of
antiviral genes such as type I in IFNs and ISGs (33). To investigate the effect of miR-221
on the activity of the NF-κB signaling pathway, the expression
levels of downstream proteins in the NF-κB signaling pathway,
namely nuclear p-p65, p-IκB-α and IκB-α were evaluated. As shown in
Fig. 7A and B,
miR-221-overexpression significantly reduced the expression levels
of SOCS1, and increased the expression levels of nuclear p-p65 and
p-IκB-α, while miR-221 inhibition had opposite effects in
IAV-infected A549 cells. In addition, the NF-κB reporter luciferase
activity was significantly increased by miR-221-overexpression,
whereas decreased by miR-221 inhibition (Fig. 7C). Overall, these results
demonstrated that miR-221 positively regulated the SOCS1-mediated
activation of the NF-κB pathway in IAV-infected cells.
Discussion
In the present study, the expression of miR-221 was
significantly downregulated in A549 cells infected with H1N1.
Notably, miR-221 inhibition facilitated H1N1 replication by
alleviating the antiviral defense of host cells by targeting the
SOCS1/NF-κB pathway. The current findings identified a novel
strategy used by IAV to escape IFN-I-mediated antiviral immune
responses by downregulating miR-221 expression. This may improve
our understanding of IAV pathogenesis.
Innate immune responses to viral infection induce
the production of type-I IFN through a cascade of complex signaling
pathways that play critical roles in antiviral immunity (34). It has been demonstrated that the IAV
possesses multiple strategies to attenuate the type-I IFN-mediated
antiviral response for successful replication. Viral coding
proteins, including NS1, polymerase basic protein 2 (PB2) and
polymerase basic protein 1-frame 2 (PB1-F2), have been reported to
block the signaling pathways involved in type-I IFN synthesis
(35,36). For example, the IAV NS1 protein, the
major IFN antagonist of IAVs, can inhibit JAK/STAT signaling
activation by increasing SOCS1 and SOCS3 expression (37). The H protein of IAVs has been shown
to induce degradation of the type-I IFN receptor 1, thus
suppressing the expression of IFN-stimulated antiviral proteins
(38). In addition, PB2, another
non-structural protein of IAVs, interacts with the mitochondrial
antiviral signaling protein, a key component of the IFN synthesis
pathway, thus impairing IFN-β production without affecting viral
replication in vitro (39).
Therefore, it is important to explore how IAV escapes
IFN-I-mediated antiviral immune responses.
Increasing evidence has reported that IAV can change
the expression profiles of host miRNA, and some host miRNA
molecules participate in various types of viral infection by
modulating type-I IFNs. For example, Zhang et al (20) demonstrated that IAV infection could
upregulate miR-132-3p and that the miR-132-3p/IRF1 axis impaired
the type-I IFN-mediated antiviral defense, thus promoting IAV
replication. Zhang et al (40) showed that miR-146a was significantly
upregulated during IAV infection and miR-146a downregulation led to
a significant reduction in IAV replication by enhancing the type-I
IFN response. Shi et al (41) found that miR-21-3p downregulated
fibroblast growth factor 2 expression to accelerate IAV replication
by impairing the IFN response. The current study demonstrated that
IAV infection modulated the expression profile of miRNA in host
cells. In particular, miR-221 was one of the most notably
downregulated miRNA molecules during IAV infection. Moreover, IAV
infection regulated miR-221 expression in a time- and
dose-dependent manner. These data suggested that miR-221 may play
an important role in IAV infection. However, whether miR-221
participates in influenza virus-mediated inhibition of type-I
IFN-mediated antiviral responses is not clear. Previous studies
have shown that miR-221 can be induced by several viruses and
influence virus replication through the regulation of the host
antiviral innate immune response. Yan et al (22) found that miR-221 restricts HCMV
replication by promoting type-I IFN production. Another study
showed that miR-221 inhibits human papillomavirus 16E1-E2-mediated
DNA replication by regulating the type-I IFN signaling pathway
(23). In addition, Xu et al
(42) reported that miR-221 could
accentuate IFN anti-HCV effect by targeting SOCS1 and SOCS3. In the
present study, loss- and gain-of-function experiments demonstrated
that IAV replication was inhibited by overexpression of miR-221,
while promoted by miR-221 inhibition, as determined by virus
titers. Moreover, overexpression of miR-221 inhibited type-I
IFN-mediated antiviral defense of host cells, whereas miR-221
inhibition had an opposite effect, indicating that miR-221
contributes to IAV infection through negative modulation of the
type-I IFN response.
Following the identification of the roles of miR-221
in IAV infection, the underlying mechanism was further explored.
SOCS1 was identified as a direct target of miR-221. SOCS1, a
negative regulator of the IFN-I signaling pathway, has been
implicated in regulating immune response and viral pathogenesis
(43–45). For example, upregulation of SOCS1
precedes type-I IFN signaling activation and inhibits the
IFN-inducible antiviral response as well as chemokine induction
(46). Respiratory syncytial virus
infection upregulates SOCS1 expression in HEp-2 cells, and
suppression of SOCS1 inhibits viral replication through activating
type-I IFN signaling (47). Thus,
it was speculated that IAV escapes IFN-I antiviral activity via the
miR-221/SOCS1 axis. The current study found that the expression of
SOCS1 was increased during IAV infection in a dose- and
time-dependent manner, confirming that IAV-induced miR-221 is
responsible for the increased SOCS1. Moreover, the promoting
effects of miR-221-knockdown on IAV replication were abrogated by
SOCS1 inhibition.
It is well-known that NF-κB functions as an
important coordinator of immune responses (48). Some viruses utilize NF-κB modulation
to escape from host clearance, as well as to enhance viral
replication (49,50). Since the NF-κB signaling pathway
acts downstream of SOCS1, the present study sought to determine
whether miR-221 inhibition could influence the activation of the
NF-κB signaling pathway. Thus, the key kinases in the NF-κB pathway
were examined. miR-221 inhibition suppressed the activation of the
NF-κB signaling pathway by increasing SOCS1 expression, whilst
miR-221-overexpression had the opposite result in IAV-infected A549
cells. These findings suggested that IAV may escape innate immunity
through the miR-221/SOCS1/NF-κB pathway.
There are several limitations of the present study.
All these results were obtained from in vitro experiments;
thus, influenza virus challenge experiments should be performed
in vivo to test whether upregulation of miR-221 by
agomir-221 injection has a protective role during IAV infection in
mice. The role of miR-221 in the airway epithelial cells should
also be investigated, such as mouse alveolar macrophages (RAW264.7)
that serve as the primary target for virus infection and
replication (51,52). Thus, additional experiments should
be carried out to test how IAV attenuates the type-I IFN-mediated
antiviral response.
In conclusion, the current study demonstrated that
downregulation of miR-221 inhibited the type-I IFN-mediated immune
response by targeting the SOCS1/NF-κB pathway, and thereby
promoting IAV replication. These findings suggested that miR-221
may be an important therapy target for IAV control in future.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
NZ, YM, YTi, YZ, YTa and SH performed all the
experiments and collected the data. NZ conceived and designed the
study. NZ wrote the main manuscript and analyzed the data. NZ, YM
and YTi confirmed the authenticity of all raw data. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Smith DJ, Lapedes AS, de Jong JC,
Bestebroer TM, Rimmelzwaan GF, Osterhaus AD and Fouchier RA:
Mapping the antigenic and genetic evolution of influenza virus.
Science. 305:371–376. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cheung CY, Poon LL, Lau AS, Luk W, Lau YL,
Shortridge KF, Gordon S, Guan Y and Peiris JS: Induction of
proinflammatory cytokines in human macrophages by influenza A
(H5N1) viruses: A mechanism for the unusual severity of human
disease? Lancet. 360:1831–1837. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim H, Webster RG and Webby RJ: Influenza
virus: Dealing with a drifting and shifting pathogen. Viral
Immunol. 31:174–183. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Medina RA and Garcia-Sastre A: Influenza A
viruses: New research developments. Nat Rev Microbiol. 9:590–603.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hiscott J: Convergence of the NF-kappaB
and IRF pathways in the regulation of the innate antiviral
response. Cytokine Growth Factor Rev. 18:483–490. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Julkunen I, Sareneva T, Pirhonen J, Ronni
T, Melén K and Matikainen S: Molecular pathogenesis of influenza A
virus infection and virus-induced regulation of cytokine gene
expression. Cytokine Growth Factor Rev. 12:171–180. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Veckman V, Osterlund P, Fagerlund R, Melén
K, Matikainen S and Julkunen I: TNF-alpha and IFN-alpha enhance
influenza-A-virus-induced chemokine gene expression in human A549
lung epithelial cells. Virology. 345:96–104. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li R and Wang L: Baicalin inhibits
influenza virus A replication via activation of type I IFN
signaling by reducing miR-146a. Mol Med Rep. 20:5041–5049.
2019.PubMed/NCBI
|
|
9
|
Downey J, Pernet E, Coulombe F, Allard B,
Meunier I, Jaworska J, Qureshi S, Vinh DC, Martin JG, Joubert P and
Divangahi M: RIPK3 interacts with MAVS to regulate type I
IFN-mediated immunity to influenza A virus infection. PLoS Pathog.
13:e10063262017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Munir M, Zohari S and Berg M:
Non-structural protein 1 of avian influenza A viruses
differentially inhibit NF-κB promoter activation. Virol J.
8:3832011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ruckle A, Haasbach E, Julkunen I, Planz O,
Ehrhardt C and Ludwig S: The NS1 protein of influenza A virus
blocks RIG-I-mediated activation of the noncanonical NF-κB pathway
and p52/RelB-dependent gene expression in lung epithelial cells. J
Virol. 86:10211–10217. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gao S, Song L, Li J, Zhang Z, Peng H,
Jiang W, Wang Q, Kang T, Chen S and Huang W: Influenza A
virus-encoded NS1 virulence factor protein inhibits innate immune
response by targeting IKK. Cell Microbiol. 14:1849–1866. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hayashi T, MacDonald LA and Takimoto T:
Influenza A virus protein PA-X contributes to viral growth and
suppression of the host antiviral and immune responses. J Virol.
89:6442–6452. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kincaid RP and Sullivan CS: Virus-encoded
microRNAs: An overview and a look to the future. PLoS Pathog.
8:e10030182012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cullen BR: Viruses and microRNAs. Nat
Genet. 38 (Suppl 1):S25–S30. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sullivan CS, Grundhoff A, Tevethia S,
Treisman R, Pipas JM and Ganem D: Expression and function of
microRNAs in viruses great and small. Cold Spring Harb Symp Quant
Biol. 71:351–356. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ho BC, Yu IS, Lu LF, Rudensky A, Chen HY,
Tsai CW, Chang YL, Wu CT, Chang LY, Shih SR, et al: Inhibition of
miR-146a prevents enterovirus-induced death by restoring the
production of type I interferon. Nat Commun. 5:33442014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen Y, Chen J, Wang H, Shi J, Wu K, Liu
S, Liu Y and Wu J: HCV-induced miR-21 contributes to evasion of
host immune system by targeting MyD88 and IRAK1. PLoS Pathog.
9:e10032482013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang F, Lin X, Yang X, Lu G, Zhang Q and
Zhang C: MicroRNA-132-3p suppresses type I IFN response through
targeting IRF1 to facilitate H1N1 influenza A virus infection.
Biosci Rep. 39:BSR201927692019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhu X, He Z, Hu Y, Wen W, Lin C, Yu J, Pan
J, Li R, Deng H, Liao S, et al: MicroRNA-30e* suppresses dengue
virus replication by promoting NF-κB-dependent IFN production. PLoS
Negl Trop Dis. 8:e30882014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yan B, Ma H, Jiang S, Shi J, Yang Z, Zhu
W, Kong C, Chen L, Yan H and Ma C: microRNA-221 restricts human
cytomegalovirus replication via promoting type I IFN production by
targeting SOCS1/NF-κB pathway. Cell Cycle. 18:3072–3084. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lu H and Gu X: MicroRNA-221 inhibits human
papillomavirus 16 E1-E2 mediated DNA replication through activating
SOCS1/Type I IFN signaling pathway. Int J Clin Exp Pathol.
12:1518–1528. 2019.PubMed/NCBI
|
|
24
|
Du H, Cui S, Li Y, Yang G, Wang P, Fikrig
E and You F: MiR-221 negatively regulates innate anti-viral
response. PLoS One. 13:e02003852018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ding Y, Wang L, Zhao Q, Wu Z and Kong L:
MicroRNA-93 inhibits chondrocyte apoptosis and inflammation in
osteoarthritis by targeting the TLR4/NF-κB signaling pathway. Int J
Mol Med. 43:779–790. 2019.PubMed/NCBI
|
|
27
|
Nakamura S, Horie M, Daidoji T, Honda T,
Yasugi M, Kuno A, Komori T, Okuzaki D, Narimatsu H, Nakaya T and
Tomonaga K: Influenza A virus-induced expression of a GalNAc
transferase, GALNT3, via MicroRNAs is required for enhanced viral
replication. J Virol. 90:1788–1801. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Terán-Cabanillas E, Montalvo-Corral M,
Silva-Campa E, Caire-Juvera G, Moya-Camarena SY and Hernández J:
Production of interferon α and β, pro-inflammatory cytokines and
the expression of suppressor of cytokine signaling (SOCS) in obese
subjects infected with influenza A/H1N1. Clin Nutr. 33:922–926.
2014. View Article : Google Scholar
|
|
29
|
Subbarao K and Joseph T: Scientific
barriers to developing vaccines against avian influenza viruses.
Nat Rev Immunol. 7:267–278. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chiang C, Chen GW and Shih SR: Mutations
at alternative 5′ splice sites of M1 mRNA negatively affect
influenza A virus viability and growth rate. J Virol.
82:10873–10886. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pichlmair A, Schulz O, Tan CP, Näslund TI,
Liljeström P, Weber F and Reis e Sousa C: RIG-I-mediated antiviral
responses to single-stranded RNA bearing 5′-phosphates. Science.
314:997–1001. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Prêle CM, Woodward EA, Bisley J,
Keith-Magee A, Nicholson SE and Hart PH: SOCS1 regulates the IFN
but not NFkappaB pathway in TLR-stimulated human monocytes and
macrophages. J Immunol. 181:8018–8026. 2008. View Article : Google Scholar
|
|
33
|
Nakahara T, Tanaka K, Ohno S, Egawa N,
Yugawa T and Kiyono T: Activation of NF-κB by human papillomavirus
16 E1 limits E1-dependent viral replication through degradation of
E1. J Virol. 89:5040–5059. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Akira S, Uematsu S and Takeuchi O:
Pathogen recognition and innate immunity. Cell. 124:783–801. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gack MU, Albrecht RA, Urano T, Inn KS,
Huang IC, Carnero E, Farzan M, Inoue S, Jung JU and García-Sastre
A: Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to
evade recognition by the host viral RNA sensor RIG-I. Cell Host
Microbe. 5:439–449. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Varga ZT, Ramos I, Hai R, Schmolke M,
García-Sastre A, Fernandez-Sesma A and Palese P: The influenza
virus protein PB1-F2 inhibits the induction of type I interferon at
the level of the MAVS adaptor protein. PLoS Pathog. 7:e10020672011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Stranden AM, Staeheli P and Pavlovic J:
Function of the mouse Mx1 protein is inhibited by overexpression of
the PB2 protein of influenza virus. Virology. 197:642–651. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xia C, Vijayan M, Pritzl CJ, Fuchs SY,
McDermott AB and Hahm B: Hemagglutinin of influenza A virus
antagonizes type I interferon (IFN) responses by inducing
degradation of type I IFN receptor 1. J Virol. 90:2403–2417. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Graef KM, Vreede FT, Lau YF, McCall AW,
Carr SM, Subbarao K and Fodor E: The PB2 subunit of the influenza
virus RNA polymerase affects virulence by interacting with the
mitochondrial antiviral signaling protein and inhibiting expression
of beta interferon. J Virol. 84:8433–8445. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang F, Sun X, Zhu Y and Qin W:
Downregulation of miR-146a inhibits influenza A virus replication
by enhancing the type I interferon response in vitro and in vivo.
Biomed Pharmacother. 111:740–750. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shi J, Feng P and Gu T: MicroRNA-21-3p
modulates FGF2 to facilitate influenza A virus H5N1 replication by
refraining type I interferon response. Biosci Rep. May 19–2020.doi:
10.1042/BSR20200158. View Article : Google Scholar
|
|
42
|
Xu G, Yang F, Ding CL, Wang J, Zhao P,
Wang W and Ren H: MiR-221 accentuates IFN's anti-HCV effect by
downregulating SOCS1 and SOCS3. Virology 462–463. 343–350. 2014.
View Article : Google Scholar
|
|
43
|
Kawai T and Akira S: Toll-like receptor
and RIG-I-like receptor signaling. Ann N Y Acad Sci. 1143:1–20.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Alexander WS and Hilton DJ: The role of
suppressors of cytokine signaling (SOCS) proteins in regulation of
the immune response. Annu Rev Immunol. 22:503–529. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Akhtar LN and Benveniste EN: Viral
exploitation of host SOCS protein functions. J Virol. 85:1912–1921.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zheng J, Yang P, Tang Y, Pan Z and Zhao D:
Respiratory syncytial virus nonstructural proteins upregulate SOCS1
and SOCS3 in the different manner from endogenous IFN signaling. J
Immunol Res. 2015:7385472015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hashimoto K, Ishibashi K, Ishioka K, Zhao
D, Sato M, Ohara S, Abe Y, Kawasaki Y, Sato Y, Yokota S, et al: RSV
replication is attenuated by counteracting expression of the
suppressor of cytokine signaling (SOCS) molecules. Virology.
391:162–170. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Deng L, Zeng Q, Wang M, Cheng A, Jia R,
Chen S, Zhu D, Liu M, Yang Q, Wu Y, et al: Suppression of NF-κB
activity: A viral immune evasion mechanism. Viruses. 10:4092018.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yeh JX, Park E, Schultz KLW and Griffin
DE: NF-κB Activation promotes alphavirus replication in mature
neurons. J Virol. 93:e01071–19. 2019. View Article : Google Scholar
|
|
50
|
Wei F, Jiang Z, Sun H, Pu J, Sun Y, Wang
M, Tong Q, Bi Y, Ma X, Gao GF and Liu J: Induction of PGRN by
influenza virus inhibits the antiviral immune responses through
downregulation of type I interferons signaling. PLoS Pathog.
15:e10080622019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kumagai Y, Takeuchi O, Kato H, Kumar H,
Matsui K, Morii E, Aozasa K, Kawai T and Akira S: Alveolar
macrophages are the primary interferon-alpha producer in pulmonary
infection with RNA viruses. Immunity. 27:240–252. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Newby CM, Sabin L and Pekosz A: The RNA
binding domain of influenza A virus NS1 protein affects secretion
of tumor necrosis factor alpha, interleukin-6, and interferon in
primary murine tracheal epithelial cells. J Virol. 81:9469–9480.
2007. View Article : Google Scholar : PubMed/NCBI
|